
Abstract
Background
Plasmodium falciparum is the most virulent parasite of the five Plasmodium species that cause human malaria, and biological analysis of the parasite is critical for the development of novel strategies for disease control. DNA endonucleases are important for maintaining the biological activity, gene stability of the parasite and interaction with host immune systems. In this study, ten sequences of DNA endonucleases were found in the genome of P. falciparum 3D7 clone, seven of them were predicted to contain an endonuclease/exonuclease/phosphatase (IPR005135) domain which plays an important role in DNA catalytic activity. The seven DNA endonucleases of P. falciparum were systematically investigated.
Methods
Plasmodium falciparum 3D7 clone was cultured in human O+ RBCs, RNA was extracted at 8, 16, 24, 32, 40, and 48 h post invasion and real-time quantitative PCR was carried out to analyse the transcription of the seven DNA endonuclease genes in asexual stages. Immunofluorescence assay was carried out to confirm the location of the encoded proteins expressed in the erythrocytic stages. Finally, the catalytic activity of the DNA nucleases were tested.
Results
Of the seven proteins analysed, two proteins were not soluble. Fragments derived from the rest five endonuclease sequences were successfully expressed as soluble proteins, and which were used to generate antisera for protein localization. The proteins were all located in the nucleus at ring and trophozoite stages. While at schizont stage, proteins encoded by PF3D7_1238600, PF3D7_0107200 and PF3D7_0319200 were in the punctuated forms in the parasite most likely around nuclei of the merozoites. But the proteins encoded by PF3D7_0305600 and PF3D7_1363500 were distributed around the infected erythrocyte membrane. The enzymatic activity of the recombinant GST-PF3D7_1238600 was very efficient without divalent iron, while the activity of the rest four enzymes was iron dependent. Further, divalent irons did not show any specific enhancement on the activity of GST-PF3D7_1238600, but the activity of GST-PF3D7_0107200, GST-PF3D7_1363500 and GST-PF3D7_0319200 were Cu2+ dependent. The activity of GST-PF3D7_0305600 was dependent on Mg2+ and Mn2+. Except GST-PF3D7_1363500, four of the GST tagged recombinant proteins hydrolysed the supercoiled circular plasmid DNA with or without divalent metal ions. The GST-PF3D7_1363500 protein only changed the supercoiled circular plasmid DNA into nicked plasmids, even with Cu2+.
Conclusions
Fragments derived from five of the endonuclease sequences of P. falciparum 3D7 clone were successfully expressed. The proteins displayed diverse cell distribution, biochemical and enzymatic activities, which indicated that they carried different biological function in the development of the parasite in the erythrocytes. The DNA repair and DNA degradation capacity of the DNA endonucleases in the biology of the parasite remained further studied.
Similar content being viewed by others

Background
Plasmodium falciparum is the most virulent parasite of all five Plasmodium species that cause human malaria, an estimated 3.3 billion people are at risk of malaria, and 1.2 billion are at high risk [1]. The main pathophysiological symptoms of malaria are caused by repeated merozoite invasion into RBCs and exponential parasite proliferation in the blood stage.
DNA endonucleases are a type of enzymes that hydrolyse internal phosphodiester bonds, which exist in DNA strands. DNase I is a DNA-specific enzyme that was discovered in the cells of spleen, liver and digestive tracts of mammalian hosts [2]. Some pathogens successfully survive from the killing of the host cells by the expression of DNases which can degrade the neutrophil extracellular traps (NETs) [3,4,5]. While NETs are mainly composed of DNA and proteases which released from neutrophils and contributed to the innate immune response by capturing pathogens [6, 7]. Further, it was reported that hosts infected with Plasmodium malariae, was accompanied by increased DNase and RNase activities in the sera [8]. During the necrocytosis, DNase I and the plasma fibrinolysis system concentrate at the nucleus of the dead cell and degrade chromosomal DNA, which prevents the appearance of anti-nucleus antibodies [9]. DNase II is a type of acid endonuclease that is independent of divalent metal ions. In mouse fetal development, a deficiency of DNase II leads to the accumulation of large DNA-containing bodies that were resulted from engulfed, but undigested cell corpses in tissues, such as thymus, kidney, spleen, and liver, which could result in dyserythropoietic anaemia and death of the fetus [10]. Deficiency of DNase II in adult mice results in chronic polyarthritis [11]. Apoptotic DNA leads to cell cycle arrest of fibroblasts and epithelial cells. Degraded apoptotic DNA by DNase II activated p53 and p21 pathways, which protected normal cells from apoptotic DNA [12].
The function of DNases is mostly determined by endonuclease/exonuclease/phosphatase (EEP) domain [13,14,15,16,17,18,19,20]. EEP hydrolyses the phosphodiester bond in nucleic acids, proteins and phospholipids. The EEP domain exists in a large number of enzymes, including AP endonuclease, DNase I, inositol-polyphosphate 5-phosphatase and sphingomyelinase, and these enzymes participate in DNA metabolic processes and intracellular signalling [14, 15].
Plasmodium falciparum contains a 23 Mb nuclear genome encoding 5400 genes on 14 linear chromosomes [21], a 35 Kb apicoplast genome [22] and a 6 Kb mitochondrion genome [23]. Over 50% of the genes’ encoded proteins have not been well studied [21, 24, 25]. Here, proteins with EEP domains that may encompass DNA hydrolytic ability of P. falciparum 3D7 clone were identified and characterized. This study combined a bioinformatics assessment, protein localization and DNA catalytic activity tests. The data generated will facilitate a better understanding of the biology of P. falciparum.
Methods
Parasites and culture
Parasites of P. falciparum 3D7 clone [26] were cultured in human O+ RBCs as previously described [27] and synchronized [28] with 5% sorbitol at early ring stage post-invasion. Parasites were harvested at 8, 16, 24, 32, 40 and 48 h post-invasion.
Sequence and bioinformatic analysis
Ten sequences of DNA endonucleases were found in the genome of P. falciparum 3D7 clone (http://plasmodb.org/plasmo/), seven of them contain an EEP domain respectively. The amino acid and nucleotide sequences of the seven genes of P. falciparum 3D7 clone were downloaded at PlasmoDB (http://plasmodb.org/plasmo/). Domain prediction and classification were conducted with InterPro (http://www.ebi.ac.uk/interpro/) and SCOP (http://scop.mrc-lmb.cam.ac.uk/scop/index.html). Homologous sequences of other species were downloaded at National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/). DNAMAN (version 6.0.3.99) was used for molecular weight calculation and analysis of recombinant protein solubility. Multiple sequence alignments and phylogenetic analysis were conducted with COBALT (http://www.st-va.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi) and MEGA 6.
Preparation of cDNA and real-time quantitative PCR
Parasite RNA at six time points post invasion was extracted by TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. DNA was removed by DNase I (TaKaRa, Dalian, China), and AMV reverse transcriptase (TaKaRa, Dalian, China) and oligo(dT) primer (TaKaRa, Dalian, China) were used to obtain first-strand cDNA. Real-time quantitative PCR was carried out as previously described [29]. The seryl-tRNA synthetase gene (PF3D7_1205100) is stably transcribed in blood stage, and was used as the internal control [30]. The primers for real-time quantitative PCR are listed in Table 1. Real-time quantitative PCR was conducted on an ABI PRISM® 7500 Real-Time PCR System (Applied Biosystems, CA, USA) with SYBR® Premix Ex Taq™ (TaKaRa). Transcription changes were calculated as 2−ΔΔCt [31]. The mean and standard error were determined using three biological and technical replicates.
Expression and purification of His-tagged and GST-tagged recombinant proteins
Specific primers were designed for amplification of the genes and expression of His-tagged and GST-tagged recombinant proteins (Tables 2 and 3) in the plasmids pET-28a and pGEX-4T-1, respectively. Escherichia coli BL21 (DE3) strain was used for the generation of the recombinant proteins which were purified with His-Trap purification kit (GE, USA) and glutathione-Sepharose, respectively [32].
Generation of specific antibodies and detection of native proteins in Western blots
To obtain a specific antiserum, 300 μg of His-tagged recombinant protein emulsified with Freund’s Adjuvants was injected into female New Zealand white rabbits every 2 weeks. After four injections, the antiserum and purified total IgG were collected with Protein A Sepharose™ 4 Fast Flow (GE Healthcare) according to the manufacturer’s protocol. Western blot was carried out for detection of native proteins. Erythrocytes infected with parasites were isolated by centrifugation with gradient Percoll (GE health) as described [33] and then lysed in the loading buffer containing 250 mM Tris, 1.92 M glycine and 1% SDS. The proteins were resolved in SDS-PAGE gel and transferred on a nylon membrane. The rabbit anti-His-tagged recombinant protein IgG (1 mg/ml) was used as a primary antibody (1:500). Alkaline phosphatase conjugated goat anti-rabbit IgG (Sigma, 1:10,000) was used as a secondary antibody. The membrane was developed with BCIP/NPT substrate (sigma) to reveal native proteins.
Immunofluorescence assay
Indirect immunofluorescence assays (IFA) were carried out to localize the proteins inside the parasites. Thin smears with parasites at ring, trophozoite and schizont stages were made and then fixed with 4% paraformaldehyde containing 1 × PBS, 0.8 M NaOH and 0.0075% glutaraldehyde for 15 min. Next, parasite membrane was permeabilized with 0.01% TritonX-100 for 15 min. The slides were blocked with TBST containing 5% non-fat milk (Sigma, St. Louis, USA) for 1 h, and incubated with the protein-specific antibody mentioned above (with a dilution of 1:100 for anti-PF3D7_0305600 IgG and anti-PF3D7_1363500 IgG; 1:50 for anti-PF3D7_1238600 IgG; 1:25 for anti-PF3D7_0107200 IgG and anti-PF3D7_0319200 IgG). The secondary antibody [1:1000, Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen)] and Hoechst 33342 (Invitrogen, USA) were added. A fluorescence microscope (Olympus, BX 53) was used for capturing high resolution images.
DNA catalytic assay
A DNA catalytic assay was carried out in a 10 µl volume solution in 1 × PBS, with 10 ng genomic DNA extracted from P. falciparum 3D7 clone as previously described [33] and 1.6 μg recombinant protein. The GST protein was included as a negative control. The reaction was conducted at 37 °C for 5, 10, 15, 30, 45, and 60 min. Then, the hydrolysed DNA was detected via agarose gel electrophoresis.
To test the dependency of ion on the enzymatic activity, divalent metal ions (Cu2+, Mn2+, Ca2+, Ni2+, Mg2+, Co2+, and Zn2+) were added to the reaction respectively with DNA in linear or circular form as described above. Agarose gel electrophoresis was used for detection of the digested DNA.
Results
Sequence and EEP domain identification
Seven genes encoding proteins with an endonuclease/exonuclease/phosphatase (IPR005135) (EEP) were identified in the genome of P. falciparum 3D7 clone (Fig. 1). All identified proteins belong to the DNase I-like superfamily according to structure identification in proteins (SCOP). The homologous sequences of PF3D7_1363500 were found in Theileria orientalis strain Shintoku, Theileria parva and Babesia microti strain RI. The homologous sequence of protein PF3D7_0519500 was found in Cryptosporidium muris RN66. Homologous sequences of protein PF3D7_1430600 were in Trypanosoma vivax Y486 and Vitrella brassicaformis CCMP3155 (see Additional file 1).

Schematic map of EEP domain within seven DNA endonucleases of P. falciparum 3D7 clone. Grey indicates full length of proteins, and red indicates the EEP domain analysed with InterPro
Transcription analysis
In qPCR, all seven genes were found transcribed at the six time points post erythrocyte invasion. Gene PF3D7_1238600 showed the highest transcriptional level and gene PF3D7_0519500 showed the lowest transcriptional level of the seven genes at the time points of 24, 32, 40 and 48 h. The transcription is generally higher when the parasites reach more mature stage, after 16 h post erythrocyte invasion (Fig. 2).

Transcription of the seven DNA endonucleases genes of the P. falciparum 3D7 clone. The transcriptions of the seven DNA endonuclease genes at 8, 16, 24, 32, 40 and 48 h post invasion are shown. Transcript levels relative to that of gene PF3D7_0305600 at 16 h post invasion were calculated as 2−∆∆Ct. A log-scale was calculated and used on the y-axis
Expression and purification of His-tagged and GST-tagged recombinant proteins
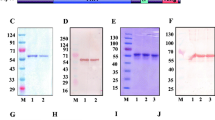
Of the seven protein analysed, two proteins encoded respectively by PF3D7_1430600 and PF3D7_0519500 were not soluble. His-tagged and GST-tagged recombinant proteins (see Additional files 2 and 3) were generated and verified by SDS-PAGE and Western blot.
Detection of native proteins by Western blot and IFA
Western blot was carried out for the detection of the native proteins in the blood stage of P. falciparum 3D7 clone. Protein specific IgGs generated from rabbits were used as primary antibodies. The molecular weight of the protein displayed in the Western blot was consistent with bioinformatic prediction (Fig. 3).

Western blot analysis of the native DNA endonucleases in the P. falciparum 3D7 clone. Lane 1 represents infected erythrocytes, and lane 2 represents uninfected erythrocytes. A single band was detected in infected erythrocytes. Protein specific IgG was used as a primary antibody. Alkaline phosphatase conjugated goat anti-rabbit IgG was used as a secondary antibody
The proteins were further localized by immunofluorescence assay (IFA) in the ring, trophozoite and schizont developmental stages with protein-specific IgG. The proteins were all located in the nucleus at ring and trophozoite stages. While at schizont stage, proteins encoded by PF3D7_1238600, PF3D7_0107200 and PF3D7_0319200 (Fig. 4a, b, e) were in the punctuated forms in the parasite most likely around nuclei of the merozoites. But the proteins encoded by PF3D7_0305600 and PF3D7_1363500 (Fig. 4c, d) were distributed around the infected erythrocyte membrane.

Localization of the five DNA endonucleases in the P. falciparum 3D7 clone by immunofluorescence assays. Immunofluorescence assay (IFA) was performed with protein-specific IgG as the primary antibody. Alexa Fluor 488-conjugated goat anti-rabbit IgG was used as a secondary antibody. Hoechst 33342 stained the nuclei blue. a Rabbit anti-PF3D7_1238600 IgG was used as a primary antibody. b Rabbit anti-PF3D7_0107200 IgG was used as a primary antibody. c Rabbit anti-PF3D7_0305600 IgG was used as a primary antibody. d Rabbit anti-PF3D7_1363500 IgG was used as a primary antibody. e Rabbit anti-PF3D7_0319200 IgG was used as a primary antibody
DNA nuclease activity test
The enzymatic activity of the recombinant GST-PF3D7_1238600 was very efficient without divalent iron (Fig. 5a), while the activity of the rest four enzymes were iron dependent (Fig. 5b–e). Further, divalent irons did not show any specific enhancement on the activity of GST-PF3D7_1238600 (Fig. 6a), but the activity of GST-PF3D7_0107200, GST-PF3D7_1363500 and GST-PF3D7_0319200 were Cu2+ dependent (Fig. 6b, d, e). The activity of GST-PF3D7_0305600 was dependent on Mg2+ and Mn2+ (Fig. 6c). Except GST-PF3D7_1363500, four of the GST tagged recombinant proteins hydrolysed the supercoiled circular plasmid DNA with or without divalent metal ions (Fig. 7a–c, e). The GST-PF3D7_1363500 protein only changed the supercoiled circular plasmid DNA into nicked plasmids, even with Cu2+ (Fig. 7d).

DNA hydrolytic test of the five GST-tagged recombinant proteins without divalent ions. Lane 1 is blank with only genomic DNA incubated at 37 °C for 60 min. Lane 2 is the negative control with genomic DNA and GST protein incubated at 37 °C for 60 min. Lanes 3–8 are genomic DNA and GST-tagged recombinant proteins incubating at 37 °C for 5, 10, 15, 30, 45 and 60 min. a GST-PF3D7_1238600 (0.1 mg/ml); b GST-PF3D7_0107200 (0.1 mg/ml). c. GST-PF3D7_1363500 (0.07 mg/ml); d GST-PF3D7_0319200 (0.1 mg/ml); e GST-PF3D7_0305600—(0.2 mg/ml)

DNA hydrolytic test of the five GST-tagged recombinant proteins with different divalent metals. Divalent metal ions at different concentrations were added into the reaction and incubated for 30 min. The lane with no protein is the blank control with only genomic DNA incubated for 30 min. a No specific effect of divalent metal ions on the activity of GST-PF3D7_1238600 was observed. b The activity of GST-PF3D7_0107200 was dependent on Cu2+ with an optimal concentration of 2 mM. c The activity of GST-PF3D7_0305600 was dependent on Mn2+ and Mg2+. d The activity of GST-PF3D7_1363500 was dependent on Cu2+ with an optimal concentration of 2 mM. e The activity of GST-PF3D7_0319200 was dependent on Cu2+ with an optimal concentration of 10 mM

The catalytic effect of the five GST-tagged recombinant proteins on linear DNA and supercoiled circular plasmid DNA. Linear genomic DNA and supercoiled plasmid were used as substrates in a DNA digestion assay. a GST-PF3D7_1238600 digestion of linear genomic DNA and supercoiled circular plasmid with or without Mn2+ and Mg2+. b GST-PF3D7_0305600 digestion of linear genomic DNA and supercoiled circular plasmid with Mn2+ or Mg2+. c GST-PF3D7_0107200 digestion of linear genomic DNA and supercoiled circular plasmid with or without Cu2+. d GST-PF3D7_1363500 digestion linear genomic DNA with Cu2+. e GST-PF3D7_0319200 digestion of linear genomic DNA and supercoiled circular plasmid with Cu2+
Discussion
The function of a protein is closely related to its captured domains. Proteins with the same function share similar domains. In this study, a common domain, EEP domain with activity of hydrolysis of phosphodiester bonds in nucleic acids, proteins and phospholipids was identified in 7 protein sequences of DNases in P. falciparum. The EEP domain exists in a large number of enzymes, including AP endonuclease, DNase I, inositol-polyphosphate 5-phosphatase and sphingomyelinase, and these enzymes participate in DNA metabolism processes and intracellular signalling [14, 15].
The DNase I-like superfamily is a member of SCOP 1.75, which groups protein structural domains hierarchically into class, fold, superfamily and family. This superfamily contains three families: DNase I-like, inositol polyphosphate 5-phosphatase and sphingomyelin phosphodiesterase-like. Except the protein PF3D7_1238600, which belongs to the sphingomyelin phosphodiesterase-like family, six of the identified proteins belong to the DNase I-like family. Proteins PF3D7_0305600 and PF3D7_1430600 were AP endonuclease 1 family members in InterPro analysis, and they specifically create a nick at the AP site in the DNA base excision repair pathway. In eukaryotes, there is only one AP endonuclease. However, in E. coli, endonuclease IV and exonuclease III are the AP endonucleases [34].
In transcriptional analysis, the lowest transcription level relative to the internal control gene was used for normalization; the fold changes of the gene PF3D7_0305600 relative to the control at 16 h post invasion was set as one. The transcription levels of the genes PF3D7_1238600 and PF3D7_1363500 were respectively a thousand times and a hundred times higher than that of PF3D7_0305600, and the results were consistent with that obtained by microarray assays recorded in PlasmoDB. Peak transcript levels may represent the main stages of activity of the encoded proteins. All seven genes reached their peak transcription at the late trophozoite and early schizont stages, which was further confirmed by Western blot assays (Fig. 3).
The distribution of the proteins inside the infected erythrocytes were mainly in two patterns. The proteins were all located in the nucleus at ring and trophozoite stages. While at schizont stage, proteins encoded by PF3D7_1238600, PF3D7_0107200 and PF3D7_0319200 were in the punctuated forms in the parasite cytoplasm around nuclei of the merozoites (Fig. 4a, b, e). But the proteins encoded by PF3D7_0305600 and PF3D7_1363500 were distributed around the infected erythrocyte membrane (Fig. 4c, d). The phylogenetic analysis indicated that the genes were grouped in separated clusters implying that they perform different function in the development of the parasite.
The DNA catalytic activity of five proteins containing the EEP domain was investigated, and all of the proteins displayed DNA hydrolytic activity with different dependency in divalent irons (Figs. 5, 6 and 7). Thus the proteins with EEP domains encoded by the genes identified in the P. falciparum genome indeed could catalyse DNA in a similar manner as observed in other organisms. However, the function of these enzymes in the biology of the parasite remained further investigation.
Conclusions
Seven genes encoding potential DNA hydrolytic activity were identified in the P. falciparum genome and their transcription was analysed by qPCR. The expression of five proteins containing an EEP domain were confirmed by Western blot and IFA, and their DNA catalysis activity were analysed. The proteins displayed diverse cell distribution, biochemical and enzymatic activities, which indicated that they carried different biological function in the development of the parasite in the erythrocytes.
References
Kantele A, Jokiranta S. Plasmodium knowlesi—the fifth species causing human malaria. Duodecim. 2010;126:427–34.
Takeshita H, Mogi K, Yasuda T, Nakajima T, Nakashima Y, Mori S, et al. Mammalian deoxyribonucleases I are classified into three types: pancreas, parotid, and pancreas-parotid (mixed), based on differences in their tissue concentrations. Biochem Biophys Res Commun. 2000;269:481–4.
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396.
Walker MJ, Hollands A, Sandersonsmith ML, Cole JN, Kirk JK, Henningham A, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–5.
Chang Z, Jiang N, Zhang Y, Lu H, Yin J, Mats W, et al. The TatD-like DNase of Plasmodium is a virulence factor and a potential malaria vaccine candidate. Nat Commun. 2016;7:11537.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Urban CF, Ermert D, Schmid M, Abuabed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
Kovalenko GA, Gal’Vita AV. Time course of serum nuclease activity in mice infected with Plasmodium malariae. Bull Exp Biol Med. 2000;130:877–8.
Napirei M, Wulf S, Mannherz HG. Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum. 2004;50:1873–83.
Krieser RJ, MacLea KS, Longnecker DS, Fields JL, Fiering S, Eastman A. Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ. 2002;9:956–62.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002.
Holmgren L, Bergsmedh A, Spetz AL. Horizontal transfer of DNA by the uptake of apoptotic bodies. Vox Sang. 2002;83(Suppl 1):305–6.
Lahm A, Suck D. DNase I-induced DNA conformation. 2 A structure of a DNase I–octamer complex. J Mol Biol. 1991;222:645–67.
Mol CD, Kuo CF, Thayer MM, Cunningham RP, Tainer JA. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature. 1995;374:381–6.
Dlakić M. Functionally unrelated signalling proteins contain a fold similar to Mg2+-dependent endonucleases. Trends Biochem Sci. 2000;25:272–3.
Lee SH, Kerff F, Chereau D, Ferron F, Klug A, Dominguez R. Structural basis for the actin-binding function of missing-in-metastasis. Structure. 2007;15:145–55.
Repanas K, Zingler N, Layer LE, Schumann GG, Perrakis A, Weichenrieder O. Determinants for DNA target structure selectivity of the human LINE-1 retrotransposon endonuclease. Nucleic Acids Res. 2007;35:4914–26.
Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63.
Kudryashov DS, Durer ZA, Ytterberg AJ, Sawaya MR, Pashkov I, Prochazkova K, et al. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc Natl Acad Sci USA. 2008;105:18537.
Schmiedel R, Kuettner EB, Keim A, Sträter N, Greiner-Stöffele T. Structure and function of the abasic site specificity pocket of an AP endonuclease from Archaeoglobus fulgidus. DNA Repair. 2009;8:219–31.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511.
Wilson RJ, Denny PW, Preiser PR, Rangachari K, Roberts K, Roy A, et al. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J Mol Biol. 1996;261:155–72.
Preston MD, Campino S, Assefa SA, Echeverry DF, Ocholla H, Amambua-Ngwa A, et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun. 2014;5:4052.
Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–6.
Waters AP. Genome-informed contributions to malaria therapies: feeding somewhere down the (pipe)line. Cell Host Microbe. 2008;3:280–3.
Walliker D, Quakyi IA, Wellems TE, McCutchan TF, Szarfman A, London WT, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–6.
Trager W, Jensen JB. Human malaria parasites in continuous culture. 1976. J Parasitol. 2005;91:484–6.
Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–20.
Zhang Y, Jiang N, Chang Z, Wang H, Lu H, Wahlgren M, et al. The var3 genes of Plasmodium falciparum 3D7 strain are differentially expressed in infected erythrocytes. Parasite. 2014;21:19.
Moll K, Chene A, Ribacke U, Kaneko O, Nilsson S, Winter G, et al. A novel DBL-domain of the P. falciparum 332 molecule possibly involved in erythrocyte adhesion. PLoS ONE. 2007;2:e477.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8.
Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40.
Ljungström I, Perlmann H, Schlichtherle M, Scherf A, Wahlgren M. Methods in malaria research. 4th ed. Manassas: MR4/ATCC; 2004.
Nishino T, Morikawa K. Structure and function of nucleases in DNA repair: shape, grip and blade of the DNA scissors. Oncogene. 2002;21:9022–32.
Authors’ contributions
QC designed the study. NJ and ZT, YZ performed most laboratory work. NJ, YZ, JL, YF, NY and XS performed laboratory analyses. QC and NJ analysed and interpreted the data. NJ, ZT, YZ and QC wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by grants of the National Key Research and Development Program of China (2017YFD0500400, 2017YFD0501200) and the National Natural Science Foundation of China (Grant Numbers 81420108023, 81772219).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed in the current study are available from the corresponding author on reasonable request.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1.
Phylogenetic analysis of seven P. falciparum 3D7 DNA endonucleases with homologous proteins of other species. Amino acid sequences were aligned using MEGA 6, and a phylogenetic tree was generated by the neighbour-joining method. The scale bar represents amino acid substitutions in the sequences and evolutionary distances. P. falciparum DNA endonucleases are highlighted in red.
Additional file 2.
Purification of His-tagged recombinant proteins. A. SDS-PAGE analysis of purified His-tagged recombinant proteins. His-tagged proteins of PF3D7_1238600 (Lane 1), PF3D7_0107200 (Lane 2), PF3D7_0305600 (Lane 3), PF3D7_1363500 (Lane 4) and PF3D7_0319200 (Lane 5) were separated on a 12% SDS-PAGE gel and stained with Coomassie brilliant blue R-250. B. Western blot analysis of purified His-tagged recombinant protein with mouse anti His-tag IgG.
Additional file 3.
Purification of GST-tagged recombinant proteins. A. SDS-PAGE analysis of purified GST-tagged recombinant proteins. GST-tagged proteins of PF3D7_1238600 (Lane 1), PF3D7_0107200 (Lane 2), PF3D7_0305600 (Lane 3), PF3D7_1363500 (Lane 4) and PF3D7_0319200 (Lane 5) were separated on a 12% SDS-PAGE gel and stained with Coomassie brilliant blue R-250. B. Western blot analysis of purified GST-tagged recombinant proteins with an anti-GST tag IgG.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, N., Tu, Z., Zhang, Y. et al. Identification and characterization of DNA endonucleases in Plasmodium falciparum 3D7 clone. Malar J 17, 232 (2018). https://doi.org/10.1186/s12936-018-2388-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2388-0