
Abstract
Background
Plasmodium falciparum is the aetiological agent for malaria, a deadly infectious disease for which no vaccine has yet been licensed. The proteins displayed on the merozoite cell surface have long been considered attractive vaccine targets because of their direct exposure to host antibodies; however, progress in understanding the functional role of these targets has been hindered by technical challenges associated with expressing these proteins in a functionally active recombinant form. To address this, a method that enables the systematic expression of functional extracellular Plasmodium proteins was previously developed, and used to create a library of 42 merozoite proteins.
Methods
To compile a more comprehensive library of recombinant proteins representing the repertoire of P. falciparum merozoite extracellular proteins for systematic vaccine and functional studies, genome-wide expression profiling was used to identify additional candidates. Candidate proteins were recombinantly produced and their integrity and expression levels were tested by Western blotting and ELISA.
Results
Twenty-five additional genes that were upregulated during late schizogony, and predicted to encode secreted and cell surface proteins, were identified and expressed as soluble recombinant proteins. A band consistent with the entire ectodomain was observed by immunoblotting for the majority of the proteins and their expression levels were quantified. By using sera from malaria-exposed immune adults, the immunoreactivity of 20 recombinant proteins was assessed, and most of the merozoite ligands were found to carry heat-labile epitopes. To facilitate systematic comparative studies across the entire library, multiple Plasmodium proteins were simultaneously purified using a custom-made platform.
Conclusions
A library of recombinant P. falciparum secreted and cell surface proteins was expanded by 20 additional proteins, which were shown to express at usable levels and contain conformational epitopes. This resource of extracellular P. falciparum merozoite proteins, which now contains 62 full-length ectodomains, will be a valuable tool in elucidating the function of these proteins during the blood stages of infection, and facilitate the comparative assessment of blood stage vaccine candidates.
Similar content being viewed by others
Background
Plasmodium falciparum is the aetiological agent of the most deadly form of malaria, an infectious tropical disease that accounts for up to one million deaths annually[1, 2]. The vast majority of malaria fatalities (85-90%) occur in sub-Saharan Africa, primarily in pregnant women and children under the age of five[2, 3]. While anti-malarial drugs exist, the emergence of drug-resistant parasite strains remains a global health concern and no vaccine has been licensed to date.
The asexual blood stages of malaria are initiated when a form of the parasite, called a merozoite, invades, replicates and synchronously ruptures host erythrocytes[4] releasing up to 32 progeny merozoites that can invade new erythrocytes. This cyclical phase causes the recurrent fevers and chills that are characteristic of malaria infection[5]. Merozoites are ovoid cells containing apically located secretory organelles that release proteins which are required for the invasion of new erythrocytes[6, 7]. While erythrocyte invasion is a rapid process, the brief extracellular exposure of merozoites outside of their intra-erythrocytic niche places them in direct contact with host antibodies, which contribute to naturally acquired immunity to malaria[8, 9]; therefore, merozoite cell surface and secreted proteins have long been considered attractive targets for rational vaccine development.
The publication of the P. falciparum genome project in 2002[10] identified the full complement of parasite proteins but progress in understanding the function of these proteins, including those displayed on the merozoite cell surface, has been hindered by the technical difficulties in expressing Plasmodium proteins in a functionally active form[11]. Although the reasons why Plasmodium proteins are difficult to express in heterologous expression systems are not clear, several protein characteristics, such as high molecular mass (>60 kDa), presence of export motifs, and atypical signal peptide sequences negatively impact recombinant expression[12]. In addition, the remarkably high (~80%) A + T content of parasite genes can result in long stretches of repetitive amino acids[13], and codons that are not frequently used by organisms popular for heterologous protein expression. Extracellular vaccine candidates, in particular, present an additional challenge because they often require structurally critical disulfide bonds for correct folding and contain transmembrane domains that make them difficult to solubilize in detergents that retain their native conformation[14–16].
Despite these challenges, recombinant expression of Plasmodium proteins has been attempted in a number of expression systems[12, 17] ranging from bacteria[18], yeast[14, 19], Dictyostelium discoideum[20], plants and algae[21, 22] to mammalian cells[23, 24] and cell-free systems[13]. Among them, Escherichia coli is the most popular[17], but the systematic expression of functional P. falciparum proteins remains difficult, with success rates as low as just 6%[25], and often requires subsequent laborious and complex refolding procedures with uncertain outcomes[26]. Consequently, the functional characterization of extracellular parasite proteins has typically been restricted to smaller subfragments that can be expressed rather than the full-length protein or entire ectodomain, which is more likely to be representative of the native protein.
The development of a standardized method to express large panels of P. falciparum cell surface and secreted proteins in their native conformation would enable comprehensive protein libraries to be systematically screened in parallel so that direct comparisons between antigens can be made in functional assays such as vaccine screening and immuno-epidemiology studies. To achieve this, Crosnier and colleagues recently developed a method of expressing the entire ectodomains of functional recombinant Plasmodium proteins and used it to compile a large library of 42 proteins[27]. Working towards a comprehensive library of cell surface and secreted P. falciparum merozoite proteins, this manuscript describes the identification and characterization of an additional 20 proteins.
Methods
Recombinant protein design and expression
Proteins were expressed essentially as described previously[27]. Briefly, full-length secreted molecules and the entire ectodomains of membrane-embedded proteins were identified using transmembrane[28], GPI-anchor[29], and signal peptide[30] prediction software. To prevent the inappropriate addition of glycans, which are absent from Plasmodium proteins[31], all potential N-linked glycosylation sites (N-X-S/T, where X is not proline) were systematically mutated by substituting alanine for serine/threonine at these sites. Gene constructs were made by gene synthesis (GeneartAG) using sequences that were codon-optimized for expression in human cells. Protein coding sequences were flanked with unique NotI and AscI restriction sites and subcloned into a derivative of the pTT3 expression plasmid between a 5’ mouse variable κ light chain signal peptide[32], and a 3’ tag consisting of the rat Cd4 domains 3 and 4 followed by an enzymatic biotinylation sequence and a hexahistidine tag[33]. Proteins were expressed as soluble monobiotinylated proteins by transient cotransfection of HEK293E cells with the BirA biotin ligase[34] and harvested six days post transfection[35]. All expression plasmids are openly available from Addgene[36].
Parallel protein purification
His-tagged recombinant merozoite proteins were purified from spent tissue culture supernatants by using a custom-built, piston-driven, sample-loading apparatus as described previously[33]. Briefly, a 96-well His MultiTrap HP filter plate (GE Healthcare) was pre-equilibrated with binding buffer (20 mM sodium phosphate, 40 mM imidazole, 0.5 M NaCl, pH 7.4) at a flow rate of 1 mL/min. The harvested tissue culture supernatants (~200 mL for each protein) were supplemented with imidazole (10 mM) before loading each well at 1 mL/min. The plate was washed with 1.6 mL of binding buffer and proteins eluted with 0.2 mL of elution buffer (20 mM sodium phosphate, 0.4 M imidazole, 0.5 M NaCl, pH 7.4).
Enzyme-linked immunosorbent assay (ELISA)
ELISAs were performed as previously described[37]. Briefly, purified recombinant biotinylated proteins were serially diluted in PBS-T (PBS, 0.1% Tween-20) with 2% BSA, and captured on streptavidin-coated, 96-well plates (NUNC) for one hour before washing and incubating with the anti-Cd4 monoclonal antibody OX68 (1 μg/mL in PBS-T, 2% BSA) for another hour. Plates were washed and incubated with an anti-mouse IgG (Sigma) secondary antibody conjugated to alkaline phosphatase for one hour before further washes and incubation with p-nitrophenyl phosphate (Substrate 104; Sigma) at 1 mg/mL. Absorbance was measured at 405 nm on a PHERAstar plus (BMG Labtech). Concentrations were calculated by comparison to known standards.
Western blotting
Each purified recombinant protein was resolved by SDS-PAGE under reducing conditions before blotting onto Hybond-P PVDF membrane (GE Healthcare) for one hour at 30 V. Membranes were blocked with 2% BSA, in PBS-T and incubated with 0.02 μg/mL of streptavidin-HRP (Jackson Immunoresearch) diluted in PBS-T, 0.2% BSA and detected with the Supersignal West pico chemiluminescent substrate (Pierce).
Immunogenicity study
Proteins were heat treated at 80°C for 10 minutes or left untreated and immobilized on streptavidin-coated, 96-well plates (NUNC), at concentrations sufficient for complete saturation of the available binding surface/well (as determined by ELISA). Following three washes in PBS-T, plates were incubated with pooled sera from malaria-exposed Malawian adults or malaria-naïve UK individuals at a 1:1,000 dilution in PBS-T, 2% BSA followed by an alkaline phosphatase-conjugated anti-human IgG secondary antibody (Sigma) and detected as above.
Results
Identification of candidate Plasmodium falciparum merozoite cell surface and secreted proteins
With the aim of expanding an existing recombinant P. falciparum merozoite cell surface and secreted protein library, publicly available, genome-wide transcription microarray data of P. falciparum intra-erythrocytic stages were analysed[38, 39]. To compile a list of merozoite extracellular proteins with possible roles in erythrocyte invasion, the transcription profiles of four well-established P. falciparum merozoite ligands (RH5, AMA1, EBA140, EBA175) that have all been previously implicated in erythrocyte invasion were examined[11, 38–41]. It was observed that they all follow a similar expression pattern, passing through a minimum 20 ± 6 hours post invasion and peaking at approximately 42 ± 6 hours after invasion[38, 39]. Of the 465 candidate blood stage genes that showed similar expression time windows, 207 encoded a predicted signal peptide and/or a single transmembrane domain/GPI anchor[28–30], suggesting they are likely cell surface or secreted proteins. Multi-pass membrane proteins were excluded from this list because these proteins are unlikely to be expressed in a soluble, secreted form. Within the 207 candidates, 31 were already represented in the existing merozoite library[27] and another 120 were excluded because of their predicted function (e g, involvement in lipid metabolism or nuclear localization) or protein domain content (e g, RNA or DNA binding motif) following gene ontology analysis using PlasmoDB[42], and protein domain mapping using Pfam[43]; also, 42 proteins were excluded due to their large size (>1,400 amino acids). In total, this bioinformatics analysis identified 14 putative secreted or membrane-tethered merozoite proteins. To this list, other members of the MSP3[44, 45], MSP7-like[46–48] and SERA paralogous protein families[49] were added, which were not present in the existing P. falciparum merozoite recombinant protein library[27]. Finally, the available literature was scanned to identify an additional seven merozoite cell surface and secreted proteins[50, 51]. In total, 25 putative merozoite secreted or cell surface proteins were chosen for recombinant expression, two of which contained putative GPI anchors, and 23 contained no predicted membrane anchor (Table 1).
To design the expression constructs, the entire predicted extracellular domain was selected between the signal sequence and the GPI-anchor, if present. Any predicted N-linked glycosylation sites were systematically removed by substituting alanine for serine/threonine at these sites. Expression constructs were made by gene synthesis and codon optimized for expression in human cells. All expression plasmids are publicly available through Addgene, a not-for-profit, open access plasmid repository[36].
Expression and purification of an expanded Plasmodium falciparum merozoite protein library
All proteins were expressed in HEK293E cells as soluble fusion proteins that contained a C-terminal rat Cd4(d3 + 4)-hexahistidine tag for purification and could be optionally monobiotinylated by cotransfecting a secreted version of the E. coli BirA enzyme[33]. To purify many proteins in parallel for comparative screening, a custom-built protein purification system was employed, that enables the simultaneous purification of up to 96 His-tagged proteins, even from large (>50 mL) tissue culture volumes[33].
Following purification, recombinant proteins were quantitated by ELISA and while expression levels varied significantly between individual proteins, most were purified to micromolar levels (Table 1). Overall, detectable expression was obtained for 22 out of 25 (88%) proteins. Both GLURP and SERA6, although detected by ELISA, were expressed at levels that were too low to include in further analysis and three proteins (PF14_0044, PFA0445w, SERA1) remained undetectable by ELISA despite repeated transfections.
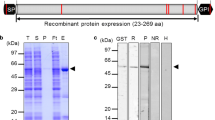
To assess their integrity, the recombinant proteins were resolved by SDS-PAGE, and the presence of the C-terminal biotin tag detected by Western blotting (Figure 1). A band consistent with the full-length ectodomain was obtained for 20 out of 25 proteins (80%); in some cases, smaller bands were also evident, most likely due to proteolytic processing. For SERA7 and PF08_0006, bands of ~30 and ~25 kDa were detected, respectively, suggesting complete cleavage at the C-terminus of the protein. Collectively, these observations establish the successful expression of 18/25 (72%) recombinant P. falciparum merozoite proteins at a size consistent with a full-length recombinant protein at usable amounts.

The majority of recombinant merozoite extracellular proteins are expressed at their expected size. One microgram of each purified biotinylated merozoite protein (as estimated by the absorbance at 280 nm) was resolved under reducing conditions by SDS-PAGE, blotted, and probed using streptavidin-HRP. The expected molecular mass of each recombinant protein is indicated in brackets above each lane, including the Cd4-6xHis tag (25 kDa).
Members of the recombinant merozoite protein library proteins are immunoreactive and carry heat-labile epitopes
In nature, protective antibodies largely recognize proteins in their native conformation; therefore, to examine whether the recombinant merozoite library proteins were correctly folded, their immunoreactivity against hyperimmune sera from adults living in malaria-endemic regions was tested. All 20 proteins from the library expressed at useable levels were arrayed on a streptavidin-coated, microtitre plate and their relative immunoreactivity to pooled sera from Malawian adults was compared to that from malaria-naïve individuals[52]. All but one (MSRP4) of the proteins were immunoreactive (Figure 2). Strikingly, strong immunoreactivity was observed for most of the SERA proteins, consistent with previous observations[49].

The merozoite recombinant proteins are immunoreactive against hyperimmune sera. The immunoreactivity of the recombinant P. falciparum merozoite proteins was tested using pooled sera from malaria-exposed Malawian adults (red bar) or malaria-naïve adults (green bar). The reduced immunoreactivity of immune sera to heat-denatured antigens (blue bar) demonstrates the presence of heat-labile (conformational) epitopes. All proteins except one (MSRP4) were identified as being immunoreactive as assessed by immunoreactivity >3 SD above negative control (green bar). AMA-1 and Cd4 were used as the positive and negative control, respectively. Data points are shown as mean ± s.d.; n = 3.
To demonstrate that serum antibodies were recognizing conformational epitopes within the protein library, all recombinant proteins were denatured by heat treatment before being captured via their biotin tag. For 16 of the 20 proteins (all but MSRP4, SERA7, PFA0210c, and PFB0475c) the immunoreactivity decreased significantly when the proteins were heat inactivated, establishing that the antigens contain heat-labile epitopes. These data show that the merozoite recombinant proteins are correctly folded and at least in part mimic the native protein conformation.
Discussion
The technical difficulties in expressing Plasmodium proteins in a recombinant functional form has presented difficulties both for basic malaria research and vaccine development[12]. Here, an approach based on a mammalian expression system has been utilized to significantly expand a recombinant library consisting of recombinant P. falciparum merozoite extracellular proteins from 42 to 62 members[27]. A high-throughput, custom-made purification platform was successfully used to purify recombinant proteins from large volumes of tissue culture supernatants to permit systematic comparative studies of purified antigens[33].
While the overall success rate of expression was high, consistent with previous experience of the HEK293 system[27, 53–57], a few merozoite proteins still failed to express, or were expressed at low levels. Although the genes were codon-optimized for human cells, P. falciparum proteins are unusual because they are enriched in asparagine, glutamic acid and lysine, and often contain homopolymeric stretches of amino acids. Indeed, it has been recently demonstrated that the stability of several Plasmodium proteins depends upon their association with heat shock proteins which act as molecular chaperones[58]. Therefore, recombinant protein expression in the HEK293 system could be further enhanced by the presence of Pf Hsp110c, which has been proposed to be a protein-stabilizing chaperone[58].
Using sera from malaria-immune adults, it has been shown that the expressed recombinant proteins contained heat-labile epitopes suggesting that they adopt their native conformation and are likely to be biochemically active. A number of proteins (e g, PF10_0166, PFA0135w) showed little serological response in comparison to the negative control. While these proteins may not contain epitopes present in the native protein, the possibility that the native proteins are poorly immunogenic and do not induce a strong antibody response in vivo, cannot be excluded. For example, previous studies reported that only 23% of immune sera examined contained specific serum IgG antibodies against PFA0135w, suggesting that this protein does not normally elicit strong humoral responses[59]. Interestingly, RIPR, which binds to Pf RH5[60], is also among the group of generally low responders. Pf RH5 is a high priority vaccine target as it plays an essential and universal role in erythrocyte invasion[53, 57, 61–63], yet it is poorly immunogenic in vivo[61], possibly due to its late release onto the merozoite surface during erythrocyte invasion. RIPR may be similarly masked from the host immune system, but further work with this antigen is clearly required.
Conclusion
A library of recombinant P. falciparum proteins has been expanded and characterized with the eventual aim of compiling a set that is representative of the merozoite surface. These plasmids, which are freely available to the global research community through Addgene[36], will be a valuable resource for basic research and aid the efforts to develop an effective malaria vaccine.
References
Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD: Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012, 379: 413-431. 10.1016/S0140-6736(12)60034-8.
World Health Organization: World Malaria Report: 2012. 2012, Geneva: Switzerland: Global Malaria Programme
Geels MJ, Imoukhuede EB, Imbault N, van Schooten H, McWade T, Troye-Blomberg M, Dobbelaer R, Craig AG, Leroy O: European vaccine initiative: lessons from developing malaria vaccines. Expert Rev Vaccines. 2011, 10: 1697-1708. 10.1586/erv.11.158.
Miller LH, Baruch DI, Marsh K, Doumbo OK: The pathogenic basis of malaria. Nature. 2002, 415: 673-679. 10.1038/415673a.
Chen Q, Schlichtherle M, Wahlgren M: Molecular aspects of severe malaria. Clin Microbiol Rev. 2000, 13: 439-450. 10.1128/CMR.13.3.439-450.2000.
Garcia CRS, de Azevedo MF, Wunderlich G, Budu A, Young JA, Bannister L: Plasmodium in the postgenomic era: new insights into the molecular cell biology of malaria parasites. Int Rev Cell Mol Biol. 2008, 266: 85-156.
Dvorak JA, Miller LH, Whitehouse WC, Shiroishi T: Invasion of erythrocytes by malaria merozoites. Science. 1975, 187: 748-750. 10.1126/science.803712.
Cohen S, McGregor IA, Carrington S: Gamma-globulin and acquired immunity to human malaria. Nature. 1961, 192: 733-737. 10.1038/192733a0.
Cohen S, Butcher GA, Crandall RB: Action of malarial antibody in vitro. Nature. 1969, 223: 368-371. 10.1038/223368a0.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan M-S, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Bartholdson SJ, Crosnier C, Bustamante LY, Rayner JC, Wright GJ: Identifying novel Plasmodium falciparum erythrocyte invasion receptors using systematic extracellular protein interaction screens. Cell Microbiol. 2013, 15: 1304-1312. 10.1111/cmi.12151.
Birkholtz L-M, Blatch G, Coetzer TL, Hoppe HC, Human E, Morris EJ, Ngcete Z, Oldfield L, Roth R, Shonhai A, Stephens L, Louw AI: Heterologous expression of plasmodial proteins for structural studies and functional annotation. Malar J. 2008, 7: 197-10.1186/1475-2875-7-197.
Tsuboi T, Takeo S, Iriko H, Jin L, Tsuchimochi M, Matsuda S, Han E-T, Otsuki H, Kaneko O, Sattabongkot J, Udomsangpetch R, Sawasaki T, Torii M, Endo Y: Wheat germ cell-free system-based production of malaria proteins for discovery of novel vaccine candidates. Infect Immun. 2008, 76: 1702-1708. 10.1128/IAI.01539-07.
Stowers AW, Zhang Y, Shimp RL, Kaslow DC: Structural conformers produced during malaria vaccine production in yeast. Yeast. 2001, 18: 137-150. 10.1002/1097-0061(20010130)18:2<137::AID-YEA657>3.0.CO;2-X.
Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong A, Wasney G, Gao M, Hills T, Brokx S, Qiu W, Sharma S, Diassiti A, Alam Z, Melone M, Mulichak A, Wernimont A, Bray J, Loppnau P, Plotnikova O, Newberry K, Sundararajan E, Houston S, Walker J, Tempel W, Bochkarev A, Kozieradzki I, Edwards A, Arrowsmith C, Roos D: Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol. 2007, 151: 100-110. 10.1016/j.molbiopara.2006.10.011.
Wright GJ: Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Mol Biosyst. 2009, 5: 1405-1412. 10.1039/b903580j.
Fernández-Robledo JA, Vasta GR: Production of recombinant proteins from protozoan parasites. Trends Parasitol. 2010, 26: 244-254. 10.1016/j.pt.2010.02.004.
Outchkourov NS, Roeffen W, Kaan A, Jansen J, Luty A, Schuiffel D, van Gemert GJ, van de Vegte-Bolmer M, Sauerwein RW, Stunnenberg HG: Correctly folded Pf s48/45 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in mice. Proc Natl Acad Sci U S A. 2008, 105: 4301-4305. 10.1073/pnas.0800459105.
Tsai CW, Duggan PF, Shimp RL, Miller LH, Narum DL: Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J Biotechnol. 2006, 121: 458-470. 10.1016/j.jbiotec.2005.08.025.
Van Bemmelen MX, Beghdadi-Rais C, Desponds C, Vargas E, Herrera S, Reymond CD, Fasel N: Expression and one-step purification of Plasmodium proteins in Dictyostelium. Mol Biochem Parasitol. 2000, 111: 377-390. 10.1016/S0166-6851(00)00330-3.
Ghosh S, Malhotra P, Lalitha P: Expression of Plasmodium falciparum C-terminal region of merozoite surface protein (Pf MSP1 19), a potential malaria vaccine candidate, in. Plant Sci. 2002, 162: 335-343. 10.1016/S0168-9452(01)00555-6.
Gregory J a, Li F, Tomosada LM, Cox CJ, Topol AB, Vinetz JM, Mayfield S: Algae-produced Pf s25 elicits antibodies that inhibit malaria transmission. PLoS One. 2012, 7: e37179-10.1371/journal.pone.0037179.
VanBuskirk KM, Sevova E, Adams JH: Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc Natl Acad Sci U S A. 2004, 101: 15754-15759. 10.1073/pnas.0405421101.
Tolia NH, Enemark EJ, Sim BKL, Joshua-Tor L: Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005, 122: 183-193. 10.1016/j.cell.2005.05.033.
Mehlin C, Boni E, Buckner FS, Engel L, Feist T, Gelb MH, Haji L, Kim D, Liu C, Mueller N, Myler PJ, Reddy JT, Sampson JN, Subramanian E, Van Voorhis WC, Worthey E, Zucker F, Hol WGJ: Heterologous expression of proteins from Plasmodium falciparum: results from 1000 genes. Mol Biochem Parasitol. 2006, 148: 144-160. 10.1016/j.molbiopara.2006.03.011.
De Marco A: Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact. 2009, 8: 26-10.1186/1475-2859-8-26.
Crosnier C, Wanaguru M, McDade B, Osier FH, Marsh K, Rayner JC, Wright GJ: A library of functional recombinant cell-surface and secreted P. falciparum merozoite proteins. Mol Cell Proteomics. 2013, 12: 3976-3986. 10.1074/mcp.O113.028357.
Sonnhammer EL, von Heijne G, Krogh A: A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998, 6: 175-182.
Eisenhaber B, Bork P, Eisenhaber F: Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999, 292: 741-758. 10.1006/jmbi.1999.3069.
Petersen TN, Brunak S, von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011, 8: 785-786. 10.1038/nmeth.1701.
Dieckmann-Schuppert A, Bender S, Odenthal-Schnittler M, Bause E, Schwarz RT: Apparent lack of N-glycosylation in the asexual intraerythrocytic stage of Plasmodium falciparum. Eur J Biochem. 1992, 205: 815-825. 10.1111/j.1432-1033.1992.tb16846.x.
Crosnier C, Staudt N, Wright GJ: A rapid and scalable method for selecting recombinant mouse monoclonal antibodies. BMC Biol. 2010, 8: 76-10.1186/1741-7007-8-76.
Sun Y, Gallagher-Jones M, Barker C, Wright GJ: A benchmarked protein microarray-based platform for the identification of novel low-affinity extracellular protein interactions. Anal Biochem. 2012, 424: 45-53. 10.1016/j.ab.2012.01.034.
Durocher Y, Perret S, Kamen A: High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30: E9-10.1093/nar/30.2.e9.
Kerr JS, Wright GJ: Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. J Vis Exp. 2012, 7: e3881-
Herscovitch M, Perkins E, Baltus A, Fan M: Addgene provides an open forum for plasmid sharing. Nat Biotechnol. 2012, 30: 316-317. 10.1038/nbt.2177.
Bushell KM, Söllner C, Schuster-Boeckler B, Bateman A, Wright GJ: Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 2008, 18: 622-630. 10.1101/gr.7187808.
Llinás M, Bozdech Z, Wong ED, Adai AT, DeRisi JL: Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006, 34: 1166-1173. 10.1093/nar/gkj517.
Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL: The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1: E5-
Tham W-H, Healer J, Cowman AF: Erythrocyte and reticulocyte binding-like proteins of Plasmodium falciparum. Trends Parasitol. 2012, 28: 23-30. 10.1016/j.pt.2011.10.002.
Cowman AF, Crabb BS: Invasion of red blood cells by malaria parasites. Cell. 2006, 124: 755-766. 10.1016/j.cell.2006.02.006.
Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Treatman C, Wang H: PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37 (Database issue): D539-D543.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD: The Pfam protein families database. Nucleic Acids Res. 2012, 40 (Database issue): D290-D301.
Singh S, Soe S, Weisman S, Barnwell JW, Pérignon JL, Druilhe P: A conserved multi-gene family induces cross-reactive antibodies effective in defense against Plasmodium falciparum. PLoS One. 2009, 4: e5410-10.1371/journal.pone.0005410.
Burgess BR, Schuck P, Garboczi DN: Dissection of merozoite surface protein 3, a representative of a family of Plasmodium falciparum surface proteins, reveals an oligomeric and highly elongated molecule. J Biol Chem. 2005, 280: 37236-37245. 10.1074/jbc.M506753200.
Kadekoppala M, Holder AA: Merozoite surface proteins of the malaria parasite: the MSP1 complex and the MSP7 family. Int J Parasitol. 2010, 40: 1155-1161. 10.1016/j.ijpara.2010.04.008.
Kadekoppala M, Ogun SA, Howell S, Gunaratne RS, Holder AA: Systematic genetic analysis of the Plasmodium falciparum MSP7-like family reveals differences in protein expression, location, and importance in asexual growth of the blood-stage parasite. Eukaryot Cell. 2010, 9: 1064-1074. 10.1128/EC.00048-10.
Kadekoppala M, O’Donnell RA, Grainger M, Crabb BS, Holder AA: Deletion of the Plasmodium falciparum merozoite surface protein 7 gene impairs parasite invasion of erythrocytes. Eukaryot Cell. 2008, 7: 2123-2132. 10.1128/EC.00274-08.
Miller SK, Good RT, Drew DR, Delorenzi M, Sanders PR, Hodder AN, Speed TP, Cowman AF, de Koning-Ward TF, Crabb BS: A subset of Plasmodium falciparum SERA genes are expressed and appear to play an important role in the erythrocytic cycle. J Biol Chem. 2002, 277: 47524-47532. 10.1074/jbc.M206974200.
Hu G, Cabrera A, Kono M, Mok S, Chaal BK, Haase S, Engelberg K, Cheemadan S, Spielmann T, Preiser PR, Gilberger T-W, Bozdech Z: Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat Biotechnol. 2010, 28: 91-98. 10.1038/nbt.1597.
Haase S, Cabrera A, Langer C, Treeck M, Struck N, Herrmann S, Jansen PW, Bruchhaus I, Bachmann A, Dias S, Cowman AF, Stunnenberg HG, Spielmann T, Gilberger T-W: Characterization of a conserved rhoptry-associated leucine zipper-like protein in the malaria parasite Plasmodium falciparum. Infect Immun. 2008, 76: 879-887. 10.1128/IAI.00144-07.
Taylor TE, Molyneux ME, Wirima JJ, Borgstein A, Goldring JD, Hommel M: Intravenous immunoglobulin in the treatment of paediatric cerebral malaria. Clin Exp Immunol. 1992, 90: 357-362.
Bustamante LY, Bartholdson SJ, Crosnier C, Campos MG, Wanaguru M, Nguon C, Kwiatkowski DP, Wright GJ, Rayner JC: A full-length recombinant Plasmodium falciparum Pf RH5 protein induces inhibitory antibodies that are effective across common Pf RH5 genetic variants. Vaccine. 2013, 31: 373-379. 10.1016/j.vaccine.2012.10.106.
Wanaguru M, Crosnier C, Johnson S, Rayner JC, Wright GJ: Biochemical analysis of the Plasmodium falciparum Erythrocyte-Binding Antigen-175 (EBA175)-Glycophorin-A interaction: implications for vaccine design. J Biol Chem. 2013, 288: 32106-32117. 10.1074/jbc.M113.484840.
Bartholdson SJ, Bustamante LY, Crosnier C, Johnson S, Lea S, Rayner JC, Wright GJ: Semaphorin-7A is an erythrocyte receptor for P. falciparum merozoite-specific TRAP homolog, MTRAP. PLoS Pathog. 2012, 8: e1003031-10.1371/journal.ppat.1003031.
Taechalertpaisarn T, Crosnier C, Bartholdson SJ, Hodder AN, Thompson J, Bustamante LY, Wilson DW, Sanders PR, Wright GJ, Rayner JC, Cowman AF, Gilson PR, Crabb BS: Biochemical and functional analysis of two Plasmodium falciparum blood-stage 6-cys proteins: P12 and P41. PLoS One. 2012, 7: e41937-10.1371/journal.pone.0041937.
Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, Mboup S, Ndir O, Kwiatkowski DP, Duraisingh MT, Rayner JC, Wright GJ: Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011, 480: 534-537.
Muralidharan V, Oksman A, Pal P, Lindquist S, Goldberg DE: Plasmodium falciparum heat shock protein 110 stabilizes the asparagine repeat-rich parasite proteome during malarial fevers. Nat Commun. 2012, 3: 1310-
Ntumngia FB, Bouyou-Akotet MK, Uhlemann A-C, Mordmüller B, Kremsner PG, Kun JFJ: Characterisation of a tryptophan-rich Plasmodium falciparum antigen associated with merozoites. Mol Biochem Parasitol. 2004, 137: 349-353. 10.1016/j.molbiopara.2004.06.008.
Chen L, Lopaticki S, Riglar DT, Dekiwadia C, Uboldi AD, Tham W-H, O’Neill MT, Richard D, Baum J, Ralph SA, Cowman AF: An EGF-like Protein Forms a Complex with Pf Rh5 and Is Required for Invasion of Human Erythrocytes by Plasmodium falciparum. PLoS Pathog. 2011, 7: e1002199-10.1371/journal.ppat.1002199.
Douglas AD, Williams AR, Illingworth JJ, Kamuyu G, Biswas S, Goodman AL, Wyllie DH, Crosnier C, Miura K, Wright GJ, Long CA, Osier FH, Marsh K, Turner AV, Hill AVS, Draper SJ: The blood-stage malaria antigen Pf RH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nat Commun. 2011, 2 (May): 601-
Hayton K, Gaur D, Liu A, Takahashi J, Henschen B, Singh S, Lambert L, Furuya T, Bouttenot R, Doll M, Nawaz F, Mu J, Jiang L, Miller LH, Wellems TE: Erythrocyte binding protein Pf RH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008, 4: 40-51. 10.1016/j.chom.2008.06.001.
Baum J, Chen L, Healer J, Lopaticki S, Boyle M, Triglia T, Ehlgen F, Ralph SA, Beeson JG, Cowman AF: Reticulocyte-binding protein homologue 5 - an essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. Int J Parasitol. 2009, 39: 371-380. 10.1016/j.ijpara.2008.10.006.
Acknowledgements
This work was supported by the Wellcome Trust grant number [098051]. We thank Cécile Crosnier for helpful comments on the manuscript and Faith Osier and Kevin Marsh for malaria immune sera.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZAZ, JCR and GJW conceived the study and ZAZ performed the experiments. ZAZ wrote the paper with contributions from JCR and GJW. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zenonos, Z.A., Rayner, J.C. & Wright, G.J. Towards a comprehensive Plasmodium falciparum merozoite cell surface and secreted recombinant protein library. Malar J 13, 93 (2014). https://doi.org/10.1186/1475-2875-13-93
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-13-93