
Abstract
Malaria remains the world's most devastating tropical infectious disease with as many as 40% of the world population living in risk areas. The widespread resistance of Plasmodium parasites to the cost-effective chloroquine and antifolates has forced the introduction of more costly drug combinations, such as Coartem®. In the absence of a vaccine in the foreseeable future, one strategy to address the growing malaria problem is to identify and characterize new and durable antimalarial drug targets, the majority of which are parasite proteins. Biochemical and structure-activity analysis of these proteins is ultimately essential in the characterization of such targets but requires large amounts of functional protein. Even though heterologous protein production has now become a relatively routine endeavour for most proteins of diverse origins, the functional expression of soluble plasmodial proteins is highly problematic and slows the progress of antimalarial drug target discovery. Here the status quo of heterologous production of plasmodial proteins is presented, constraints are highlighted and alternative strategies and hosts for functional expression and annotation of plasmodial proteins are reviewed.
Similar content being viewed by others

Background
Malaria is a devastating disease and its long-term control and eradication are still a long way off. There is no licensed vaccine and our dispensary of affordable, effective drugs is in danger of being depleted in the short term owing to increased drug resistance. The only solution currently is to resort to drug combinations coupled to insecticide spraying and/or the distribution of insecticide-treated bed nets. A number of novel drug targets have been identified and/or validated, and are being pursued for the development of new drugs. However, it is obvious from the nearly 60% of hypothetical proteins in the parasite genome that have not yet been annotated, that relatively little of the biology of the parasite is known compared to the biology of for example, humans, yeast or plants [1]. The potential for the discovery of new and better drug targets is thus considerable. Bioinformatics tools are useful in characterizing specific properties of selected proteins and can even suggest possible functions of hypothetical proteins. However, protein function and structural properties can only be confidently inferred from biochemical and cell biological experimentation, whilst the design of inhibitors and evaluation of their interaction with the protein of interest are ultimately dependent on the availability of soluble and functional proteins. It is widely accepted that a specific protein of interest can only in rare instances be isolated in sufficient quantities from the natural host cell for downstream studies. Heterologous expression of the selected protein is therefore of paramount importance. This review highlights the properties that have been associated with difficult-to-express plasmodial proteins and provides insights into the variety of options available for the heterologous expression of malaria parasite proteins for structural and functional annotation studies.
Escherichia coli as workhorse for plasmodial protein expression
Escherichia coli is still the preferred host for the heterologous expression of recombinant proteins due to cost considerations, speed, ease of use and genetic manipulation. However, for many proteins [2], including plasmodial proteins, this approach is fraught with difficulties as reflected by the paucity of plasmodial protein structures in the Protein Data Bank (PDB, ~250), when compared to the approximately 50,000 protein structures available in the PDB. One of the major limitations of this system is the expression of insoluble recombinant proteins sequestered in inclusion bodies [3, 4], as evidenced by the results from several large scale protein expression endeavours [5–7]. The Structural Genomics of Pathogenic Protozoa group (SGPP, University of Washington), selected 1,000 Plasmodium falciparum open reading frames for expression from a modified pET vector in E. coli BL21 Star cells [5]. Of these, expression was obtained for approximately one third with only 63 proteins expressed in soluble form. Another large scale attempt at expressing plasmodial proteins yielded only nine soluble, purified proteins out of 95 [6]. Lastly, the Structural Genomics Consortium (SGC, Toronto, Canada) selected 400 distinct P. falciparum target genes representing different cellular classes, along with orthologues from four other Plasmodium species as well as Cryptosporidium parvum and Toxoplasma gondii [7]. From the selected 1008 genes, 30% produced soluble proteins from pET vectors and E. coli BL21 CodonPlus (DE3)-RIL cells of which ~100 crystallized, culminating in 36 crystal structures. The most significant conclusion from this study was that the percentage of pure protein, crystals and structures could be doubled when orthologues from all six genomes were included compared to those obtained from P. falciparum alone.
The E. coli host expression system can be optimized to ensure expression plasmid stability, the absence of harmful natural proteases and availability of the relevant genetic elements, e.g. DE3 [8]. The most popular expression vector/E. coli strain combination seems to be based on pET vectors and the BL21 (DE3) E. coli strain as evidenced by the experiences of structural genomics consortia with the expression and purification of more than 100 000 proteins from Eubacteria, Archea and Eukarya. These include the recA negative strains (BLR strain) for the stabilization of plasmids containing repetitive sequences, lacY mutants (Tuner series), which enable adjustable levels of protein expression and the trzb/gor negative mutants (Origami) for improved cytoplasmic disulphide bond formation [8]. Gene expression is also controlled by the decay rate of mRNA and host strains that are deficient in specific RNases may enhance expression by protecting the mRNA from degradation [8]. In addition, several counter strategies can be employed to overcome codon bias and to assist disulphide bond formation, protein phosphorylation and protein refolding (reviewed in [9]). Furthermore, fusion tags such as hexahistidine, Glutathione-S-transferase (GST) and others (reviewed in [10]), often improve solubility of the recombinant protein and their effect can be predicted bioinformatically, which is useful to guide the investigator in the selection of an appropriate vector. New technologies include the Gateway System, which uses the araBAD promoter designed for tight control of background expression and L-arabinose-dependent graded expression of the target protein. It also allows flexibility in terms of a rapid and highly efficient route to multiple expression and functional analysis options and has been successfully applied for high-throughput recombinatorial cloning of P. falciparum functional molecules (especially vaccine candidates) [6] and as a transfection vector set [11].
Overexpression of proteins in E. coli frequently leads to insoluble protein in inclusion bodies due to metabolic stress and it is worthwhile to explore various expression conditions such as lower incubation temperatures and enriched media to promote expression of soluble recombinant proteins [9, 12, 13]. Interesting new advances, which have thus far not been applied to plasmodial protein expression include the use of bacterial bioreactors utilizing a combination of the above-mentioned attributes, as well as low temperature, and the induction of an mRNA-specific endoribonuclease causing host cell growth arrest and culture condensation. These bioreactors facilitate expression of heterologous proteins to ~30% of total cellular protein in a stable, condensed fashion without toxic effects due to the arrested state of the E. coli [14]. Recent experiments indicated that in response to expression of insoluble proteins, direct targets of the σ32 heat shock sigma factor are highly expressed [15, 16]. Manipulation of this functional response could provide a wide-ranging mechanism for improving recombinant protein solubility. Thus the σ32-regulon would be induced before the accumulation of insoluble proteins, producing a larger pool of chaperones and a simultaneous increase in protein folding [15].
Protein characteristics that jeopardized soluble expression of recombinant plasmodial proteins were found to include higher molecular weight (> 56 kDa), greater protein disorder, more basic pI (> 6) and lack of homology to E. coli proteins. In addition, the presence of low complexity regions (SEG > 29%) and Plasmodium- specific inserts, transmembrane regions, as well as signal peptides, transit peptides and export motifs, have a negative impact on protein expression [5–7]. The presence of conserved sequences for prokaryotic promoters and ribosome binding sites within the plasmodial gene of interest, as well as unusual mRNA and/or stable secondary structures should also be avoided [17]. The difficulties of expressing plasmodial proteins in E. coli have also been attributed to toxicity of plasmodial proteins [7] and the difference in amino acid content between proteins of the two species, which imposes a metabolic burden on the bacterial host. Ribosome stalling is triggered by the limited availability of specific amino acids, which causes errors in and/or truncated forms of the recombinant protein. The prevalence of codons rare in E. coli, especially Arg codons (AGA and AGG), can also cause the ribosome to pause and bring about the early termination of expression due to tRNA exhaustion [18]. The marked A+T bias in the P. falciparum genome [19, 20] has also been implicated in low level expression of proteins, although two large scale studies revealed that A+T richness per se does not appear to affect expression [5–7].
It is clear that the ability to predict soluble protein expression is limited, but bioinformatics analyses can guide the design of strategies to deal with features associated with difficult-to-express proteins in E. coli. Proteins of the apicomplexan species have unusually high contents of long, disordered regions compared to other eukaryotes. In addition, the mammalian malaria parasites are particularly enriched in intrinsically unstructured proteins when compared to those of the rodent species [21]. Intrinsically unstructured and low complexity regions overlap with each other to some extent but often coexist discretely in the P. falciparum genome [22]. These regions, their extraordinary lengths as well as the occurrence of bifunctional plasmodial proteins expressed from one open-reading frame, impose significant constraints on the probability of obtaining soluble expression of these proteins [19, 20, 23]. The intrinsically disordered regions in proteins have distinct functions [24] and their complete removal is not necessarily a viable option even if it occurs at the N- or C-terminal ends of proteins [25] without prior consideration to functional motifs that can be identified by bioinformatics methods and retained in gene constructs [26]. Alternatively, systematic N- and C-terminal deletions can be conducted to promote soluble expression [27]. It is also worthwhile taking note of the application of isolation methods specifically designed for isolation of proteins with high structural disorder in soluble form [28, 29] and newer methods for co-expression of proteins with interacting protein partners to improve soluble expression of largely unstructured proteins [30–32].
Beyond the obvious removal of e.g. signal peptides and/or minor genetic engineering of other interfering traits [8, 19, 20, 23] other options include searching for orthologous genes in other plasmodial species and apicomplexan parasites with attributes more amenable to soluble expression (see e.g. [23]) or expression of single domains of bifunctional or unstructured proteins [33–35].
Faced with the myriad of possibilities described above it is gratifying to note that newer methods are available that are especially applicable in academic laboratories for the efficient parallel screening of multiple constructs with minimal cost. These methods include novel expression vectors, micro-expression in 96-well format, a colony filtration blot procedure and new shakers or fermentors allowing for the simultaneous expression analyses of several different protein samples [13].
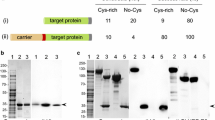
A survey was performed on the expression of potential antimalarial drug targets from the WHO TDR Targets database [36]. From the 57 potential drug targets with druggability evidence indices above 0.7, 32 were chosen. Hypothetical or putative proteins were excluded (Table 1). From these, no evidence for the expression of 19% were obtained whereas all of the remaining proteins could be expressed in E. coli, one in yeast and two proteins in cell-free systems. His-tagging for isolation of the protein had preference over other tags. Special conditions utilized to express these plasmodial proteins include truncations, codon optimization and tRNA additions. The following sections introduce alternative strategies and expression hosts worth exploiting.
Codon-optimization vs -harmonization of plasmodial proteins
Codon bias refers to the high frequency preferential use of a particular codon coding for an amino acid within an organism. There is a high level of codon mismatch between P. falciparum and expression hosts, such as E. coli and two different strategies have been used to minimize codon bias. Firstly, the intracellular tRNA pool can be expanded by using plasmids which encode rare tRNAs used in certain organisms, for example E. coli [23]. Secondly, altering the codons of the target genes to suit the codon preference of the non-natural expression host is a process referred to as 'codon-optimization' [18, 37–39]. This approach has met with some success, especially in antimalarial vaccine targets [17, 18, 40, 41], but unfortunately the expression of several other malaria proteins was not improved through codon-optimization alone [42]. As an example, SGPP [5] reported that nine out of the twelve P. falciparum genes that had been synthesized with optimized codons could still not be expressed in E. coli, and the three proteins that were expressed were in inclusion bodies. This led them to conclude that codon usage did not have a significant impact on protein expression.
During translation, ribosomes often encounter low frequency codons that may cause translational pausing. It is the stalling at these essential pause sites that halts protein expression and allows a predetermined interval which permits correct folding of the protein [38, 43]. The technique of codon-harmonization or translational attenuation refers to the process of recognizing the low usage frequency codons within the natural host (P. falciparum) and altering the code of the gene to be expressed to codons recognized as low usage frequency codons by the non-natural host [38, 39]. These changes ensure the positional codon frequency of low/intermediate and high usage codons to remain the same in the non-natural host, which allows for the translational processes to match that of the natural host. By allowing the translational machinery to move and pause at the correct sites, the folding of the secondary and tertiary structures occur as in the natural host [39, 43–45]. This approach has been remarkably successful in the heterologous production of a liver-stage antigen of P. falciparum (LSA-1) previously refractory to recombinant expression even with codon-optimization as well as co-expression with a rare frequency tRNA plasmid [45].
Molecular chaperones to aid plasmodial protein expression
One of the difficulties in heterologous expression of plasmodial proteins is incorrect folding of the proteins in the non-natural host. Molecular chaperones of the heat shock protein family (Hsp) ensure that proteins in a cell are properly folded and functional (reviewed in [46, 47]). Of particular interest is the demonstration that the co-transformation of target genes and chaperones from the same species cloned into different plasmids resulted in an increased yield of soluble protein in E. coli [32, 48].
Hsp70, a 70 kDa heat shock protein, is an important class of molecular chaperones involved in protein folding by ATP-controlled cycles of substrate binding and release. At least six genes for Hsp70-type proteins have been identified in P. falciparum [49, 50] and Pf Hsp70-1 has features to justify its candidature as co-expression partner, including successful overexpression in E. coli [51]. Pf Hsp70-1 was able to reverse the thermosensitivity of E. coli cells in an in vivo assay, which suggests that it interacted with E. coli protein substrates, protecting them against heat stress [50, 52]. Pf Hsp70-1 is able to recognize typical Hsp70 peptide substrates [53], due to its ability to suppress protein aggregation in vitro [54]. A study of the interactome of PfHsp70-1 showed that it potentially recognizes a wide range of P. falciparum substrates, including apicoplast proteins [50, 55, 56] and mutating these residues on an apicoplast transit peptide led to the protein being mistargeted [57].
Like other Hsp70 proteins, Pf Hsp70-1 needs an Hsp40 co-chaperone partner to function as a refoldase, otherwise its role during co-expression with a target protein in E. coli would be a mere suppression of protein aggregation. Pf Hsp70 and Pf Hsp40 have been identified in the parasitophorous vacuole and Maurer's clefts, implicating them in erythrocyte quality control of parasitic proteins and their possible export into the erythrocyte [49, 58, 59]. More than forty Hsp40 proteins have been identified and the Pf Hsp40 for Pf Hsp70 is predicted to be PF14_0359 [56, 60]. It is therefore conceivable that molecular chaperones from P. falciparum could be harnessed as co-expression partners during the overproduction of target proteins in E. coli to provide a specialized folding pathway.
Refolding of incorrectly folded, insoluble plasmodial proteins
The immense drawback during high level protein expression in E. coli is the production of insoluble proteins in inclusion bodies [4, 61]. Due to the reversibility of protein aggregation in bacteria, several strategies are employed to isolate and refold inclusion bodies since they are a good source of reasonably pure polypeptides, which could provide protein in the native conformation after refolding [62]. Inclusion bodies almost exclusively contain the over-expressed recombinant protein and very little host protein, ribosomal components or DNA/RNA fragments [63]. Protein refolding is a complicated procedure requiring several optimization steps [64]. Solubilization of inclusion bodies results in soluble protein in its non-native conformation, which needs to be refolded [64]. The inclusion of chaotropic reagents ensures the successful formation of the native structure [64, 65] by allowing intra-molecular interactions and/or preventing inter-molecular interactions.
Sirawaraporn et al showed that disaggregated and refolded P. falciparum dihydrofolate reductase (DHFR) from inclusion bodies yielded active protein in sufficient amounts for enzyme function determinations [34]. Optimal refolding of P. falciparum cysteine protease falcipain-1 resulted in an enzymatically active monomeric form of this protein, which exhibited high immunogenic activity [66]. Refolding procedures were also used to obtain high levels of P. falciparum cysteine protease falcipain-2, resulting in the crystallization of this protein [67]. Immunologically active P. falciparum 42 kDa MSP142 was obtained by refolding of MSP142 as a fusion protein from inclusion bodies [68]. Due to the undesirability of fusion tags in clinical trials, refolding protocols were utilized to obtain the first recombinant form of P. falciparum MSP142 without a 6× His affinity tag [69].
Extensive optimization is required to produce functional refolded plasmodial proteins, but commercial kits are now available, and refolding represents a valuable, under-utilized tool to improve the yield of soluble recombinant P. falciparum proteins.
Alternative systems for expression of plasmodial proteins
Yeasts
The commonly-used yeast species Saccharomyces cerevisiae and Pichia pastoris have been exploited extensively for the heterologous expression of plasmodial proteins, both for preparative purposes and functional characterization. S. cerevisiae is a popular choice as host owing to the ease with which it can be manipulated genetically and to the extraordinary amount of information accumulated about its molecular biology and physiology [70]. The use of this model organism varies from vaccine production [71] to functional characterization, drug discovery and resistance studies and elucidating parasite protein interaction networks ("interactomes") through high-throughput 2-hybrid analyses (e.g. [55, 72]).
Expression in S. cerevisiae is typically controlled by inducible promoters, e.g. ADH2 (a glucose-repressed alcohol dehydrogenase promoter), while N-terminal fusion to a pheromone results in efficient secretion of recombinant protein from the yeast cells [73, 74]. Coupled with carefully controlled growth in fermenters, impressive yields of 10 – 75 mg/L have been reported [75, 76]. Secretion into the culture medium represents a major advantage of the yeast system over E. coli, as it circumvents formation of inclusion body precipitates and simplifies subsequent purification. Furthermore, trafficking through the yeast secretory compartments facilitates appropriate disulphide bond formation and protein folding, which is especially important for malaria vaccine production [76–78]. However, the latter advantage may have a sting in its tail: disulphide bonding in individual proteins may be heterogeneous, resulting in a population of structural conformers, not all of which contain the conformationally correct epitopes [76]. In addition, the yeast secretory pathway may result in spurious O- and N-linked glycosylation of the heterologous proteins [79]. The latter is especially relevant for plasmodial proteins, since these are rarely (if ever) glycosylated in their native state. The problem may be circumvented to some extent by altering the coding sequence to remove potential N-glycosylation motifs or inclusion of N-glycosylation inhibitors [80]. Limitations in yeast systems that can reduce the final yield include the codon usage of the expressed gene, the gene copy number, efficient transcription by using strong promoters leading to the depletion of precursors and energy, translocation, processing and folding in the endoplasmic reticulum and Golgi, as well as protein turnover by proteolysis [81]. Cells which produce high levels of proteins can accumulate unfolded protein in the ER, which aggregates, overwhelms and eventually shuts down the secretory pathway [82]. The yeast proteins that assist in folding and disulphide bond formation differ from their counterparts in higher eukaryotes, which may affect folding of foreign proteins [83]. Saccharomyces cerevisiae moreover recognizes distinct A+T containing codons as termination signals which could lead to the truncation of encoded proteins especially of the P. falciparum genome. Due to this codon incompatibility, synthetic genes incorporating a yeast codon bias are often used [84].
Recombinant vaccine production in S. cerevisiae has focused largely on the preparation of transmission-blocking vaccines [76]. The principal components of these experimental vaccines that block transmission of malaria parasites to Anopheles are the epidermal growth factor (EGF)-like domains of Pfs 25 (of P. falciparum) or Pvs 25 (of P. vivax), a glycosylphosphatidyl inositol (GPI)-anchored surface protein of the mosquito-stage zygotes and ookinetes of the parasite [85–87]. Additional vaccine expression studies in S. cerevisiae have centred on the C-terminal domain of the major GPI-anchored surface protein of merozoites, merozoite surface protein 1 (MSP119) [88, 89]. Despite these promising efforts, S. cerevisiae has largely been superseded by Pichia pastoris as the host cell of choice for vaccine production (see discussion below). Nonetheless, S. cerevisiae has remained relevant to malaria research through its highly characterized biology and amenability to genetic manipulation, which allows it to be exploited for functional characterization through complementation experiments. In these studies, the ability of the heterologously expressed plasmodial proteins to rectify or modify aberrant phenotypes of conditional yeast deletion mutants is typically assessed. The studies are aided by the fact that relevant yeast mutants may be obtained from extensive repositories e.g. EUROSCARF [90], the Saccharomyces Genome Deletion Project [91] and the ATCC [92]. Often, the plasmodial coding sequence is modified to increase G+C content and improve yeast codon compatibility, single-copy yeast episomal plasmids are used to keep heterologous protein expression to physiologically relevant levels and expression is driven by constitutive (e.g. ADH1) or inducible (e.g. GAL1) promoters. Several classes of plasmodial proteins have been characterized in this fashion including integral membrane transporters [93–95]; phospholipid synthesis and modifying enzymes [96–99]; cell cycle, transcriptional and translational regulators [100–103]; and possible drug targets e.g. Vitamin B6 synthesis enzymes (Pf Pdx1 and Pf Pdx2, [104]) and folate metabolism proteins, dihydrofolate reductase-thymidylate synthase, (DHFR-TS) and dihydrofolate synthase-folylpolyglutamate synthase (DHFS-FPGS, [105]). A highly informative series of experiments focused on DHFR-TS demonstrated how yeast complementation may be used as a versatile alternative to preparing soluble purified protein for the characterization and exploitation of parasite drug targets. The complementation system was employed to determine the effects of DHFR mutations in parasite field isolates on sensitivity/resistance to antifolate drugs [106, 107], the contribution of various DHFR mutations to antifolate resistance and predict future resistance trends by using a PCR-based random mutagenesis approach [108], and to screen novel antifolate compounds for activity against sensitive and resistant alleles of DHFR [109, 110].
Lastly, S. cerevisiae is widely used to determine protein binding partners by yeast two-hybrid analysis which has resulted in a characterization of the P. falciparum interactome and identification of a network of 2,846 interactions between 1,312 plasmodial proteins which have minimal similarity to interaction networks of other model organisms [55, 72].
Some of the non-conventional yeasts, such as Kluyveromyces lactis and the methylotrophs Pichia pastoris and Hansenula polymorpha (now known as Pichia angusta, [111]) have also been developed as expression systems. Pichia pastoris is now the most frequently used yeast species for heterologous protein expression in general. It shares the underlying features and methodology of S. cerevisiae, with one notable exception: its biology is considerably less well characterized. Hence, it is used almost exclusively for preparative expression as opposed to the large number of functional studies that have been carried out in S. cerevisiae. However, the same principles apply: plasmodial sequences are altered to incorporate a yeast codon bias; potential N-glycosylation sites in the plasmodial sequences are removed by mutation; expression is driven by an inducible promoter (the methanol-inducible alcohol oxidase promoter, AOX1); efficient secretion into the culture medium is afforded by N-terminal fusion to the S. cerevisiae prepro-α-factor leader peptide and C-terminal tags are incorporated to facilitate purification from the medium. The redox environment and co-factors in the yeast secretory pathway should enhance correct folding, solubility and intra-molecular disulphide bonding of vaccine proteins that rely critically on conformational epitopes to generate a protective immune response; the latter may be compromized by spurious and unpredictable O-glycosylation of the heterologous protein, as well as the formation of disulphide bond conformers with reduced protective immunogenicity [112, 113]. It appears that the use of Pichia pastoris may improve heterologous protein yield, compared with S. cerevisiae, and levels of 30 mg/L to more than 1 g/L of purified protein have consistently been reported, especially when combined with batch-fed fermentation [113–121]. In addition, the ratio of desirable/undesirable conformers may be improved in Pichia pastoris [113]. In one study, this was further enhanced by co-expressing the heterologous protein with P. falciparum or Pichia pastoris protein disulphide isomerase (PDI), which improved yield as well as reducing O-glycosylation [121]. Exoprotease truncation of heterologous proteins may also be encountered in Pichia pastoris, but was circumvented in one case by alteration of the N-terminal sequence of a P. falciparum circumsporozoite protein (CSP)-derived vaccine product prone to digestion [122].
A number of plasmodial vaccine proteins have been successfully expressed in Pichia pastoris, notably merozoite proteins required for erythrocyte invasion (AMA-1, EBA-175, MSP-1, MSP-3), the sporozoite-stage antigen CSP, and transmission-blocking mosquito-stage antigens (Pfs 25) [41, 112–120, 123–127]. A notable exception to the vaccine preparation studies in which Pichia pastoris has been used almost exclusively in malaria research is the functional characterization of P. falciparum transporters thought to be involved in quinoline drug resistance, Pf CRT (P. falciparum chloroquine resistance transporter) and a P-glycoprotein homologue, Pgh-1 [128–131]. In addition to optimizing the codon usage for yeast expression, the plasmodial sequences were altered to remove poly(A) stretches, potential yeast-specific premature termination signals and predicted stem-loop structures. A 5' Kozak sequence was added, as well as C-terminal fusions to bacterial biotin acceptor domains to enable subsequent detection and/or purification. Successful membrane-localized expression of the transporters was followed by functional characterization of these proteins.
Baculovirus
The use of baculovirus-infected insect cells to express recombinant proteins is well established [132]. A lytic virus, Autographa californica nuclear polyhedrosis virus, is most commonly used. The gene of interest is cloned into a transfer vector, which is co-transfected with viral DNA into Spodoptera frugiperda (usually Sf 9 or Sf 21) insect cells. In a homologous recombination event, the foreign gene, under the control of a strong viral promoter such as polh or p10, is integrated into the viral genome and recombinant protein is produced in the infected cells. A C-terminal or N-terminal fusion tag is incorporated into the protein to facilitate purification. The large size of the viral genome makes it amenable to the insertion of large segments of foreign DNA. Numerous baculovirus systems, including a variety of transfer vectors, are now commercially available and technological improvements have simplified the production and isolation of recombinant baculovirus.
This approach has been successfully applied to several P. falciparum proteins. In one large study, 17 proteins that were expressed as insoluble inclusion bodies in E. coli were transferred to a baculovirus/Sf 21 system, which resulted in high levels of soluble protein in one case (11.7 mg/500 ml culture) and lower levels of six other proteins, ranging from 0.6 to 7.2 mg/500 ml culture [5]. The low yield can easily be overcome by scaling up the procedure.
The baculovirus system uses eukaryotic insect cells to express recombinant proteins and thus the folding and assembly of newly synthesized polypeptides may be similar to the natural process in Plasmodium. As a result, soluble recombinant baculovirus P. falciparum proteins are likely to fold correctly to produce immunologically active proteins that are recognized by conformation-specific monoclonal antibodies. This feature is vital when assessing the immunogenic response of the host to a recombinant Plasmodium protein that is considered to be a vaccine target and several studies on P. falciparum antigens illustrate this aspect. Recombinant domains of merozoite surface protein MSP-1 [133, 134], CSP [135, 136] and a erythrocyte membrane protein Pf EMP1 variant [137] retained their native conformational epitopes and elicited antibody responses. In contrast, recombinant Pf MSP-1 expressed in yeast had a different conformation to the native protein and was immunologically inactive [133]. Erythrocyte binding antigen (Pf EBA-175) has also been expressed in a baculovirus system in soluble form that was functionally active [138, 139]. Additional examples are apical membrane antigen (Pf AMA-1), which was successfully purified from baculovirus-infected insect cells [140] and recombinant serine repeat antigen (Pf SERA), which was subsequently processed into several fragments that reflect the in vivo situation in the parasite [141].
The genes coding for two P. falciparum aminopeptidases were chemically synthesized using codons optimized for the yeast Pichia pastoris, but expression in yeast was unsuccessful. In contrast, when these constructs were transfected into Sf 9 insect cells, soluble recombinant proteins that were enzymatically active were produced in sufficient quantities to perform kinetic and biochemical analyses [142, 143]. A subtilisin-like protease (Pf SUB-1), involved in merozoite invasion, was expressed and secreted in a baculovirus system and used in functional studies to assess the interaction with its cognate propeptide [144]. A micronemal protein present in Plasmodium berghei ookinetes (von Willebrand factor A domain-related protein, Pb WARP) was produced in a baculovirus system and retained its function and oligomeric structure [145]. This protein plays a role in the motility and attachment of the ookinete in the mosquito midgut and homologues of this protein exist in P. falciparum and P. vivax [145].
An important aspect of the baculovirus/insect cell system is that it recognizes eukaryotic targeting signals, which allows expression and processing of different classes of proteins, including those that are secreted, localized to the plasma membrane or nucleus, and cytoplasmic proteins [135, 146]. The host insect cell can also perform most post-translational modifications, such as phosphorylation, acylation and glycosylation. The high expression levels of recombinant proteins in the late stages of the viral life cycle can however lead to incomplete post-translational processing, resulting in, for example, hypophosphorylation of some proteins. Glycosylation can also be problematic [132], especially since N- and O-glycosylation only seem to occur at very low levels in P falciparum [147]. Native Pf EBA-175 is essentially unglycosylated, despite the presence of putative sites, whereas 20% of the recombinant baculovirus protein was N-glycosylated, although this did not affect its immunogenicity [139]. The major carbohydrate modification in Plasmodium is the addition of a glycosylphosphatidylinositol (GPI) anchor to the C-terminal amino acid of a protein [147]. GPI-anchored proteins play an important role in numerous cellular functions and are essential for parasite survival. Baculovirus may not be ideal for expressing GPI-anchors since recombinant CSP was devoid of the GPI moiety, despite the presence of a potential GPI cleavage/attachment site in the DNA.
A modification of the baculovirus system is to use a virus specific for silkworm larvae, as opposed to cultured insect cells. The yield of biologically and immunologically active Pf MSP-142 recombinant protein from these larvae was > 100-fold higher than from Sf cells [148]. However, expertise in handling silkworm larvae is currently centred mainly in China.
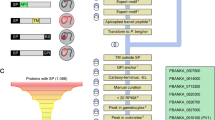
The baculovirus system is a valuable tool for the expression of Plasmodium proteins and represents a viable alternative to explore, especially since it is difficult to predict which heterologous host will be optimal for a specific protein and also because a universally applicable method to express recombinant malaria proteins is not available (Figure 1). In addition, the system lends itself to automation and scaling up of protein production. However, it requires a considerable investment in time and financial resources and it is technically more demanding than manipulating E. coli.

Generalized strategy for the successful expression of plasmodial proteins. Extensive sequence analysis of a gene could facilitate genetic manipulation of the gene if needed. E. coli should remain the starting point for the expression of soluble proteins due to its ease of use and Xenopus remains the preferred choice for membrane protein expression. Several optimizations including codon adaptation, plasmodial chaperone co-expression and small sequence changes might be particularly necessary for the expression of the plasmodial protein of interest. Various other expression hosts including mammalian, baculovirus, yeasts, Toxoplasma, Dictyostelium and cell free systems have been used with various levels of success and might include an iterative process incorporating necessary optimizations.
Dictyostelium discoideum
The single-celled amoeba Dictyostelium discoideum has become an important and accessible model organism for studying numerous aspects of eukaryotic cell biology, such as cell division, locomotion, signal transduction and vesicular trafficking, [149]. From the point of view of plasmodial expression, D. discoideum offers several attractive features: 1) The relative ease with which it can be cultured and manipulated genetically may be second only to yeast amongst eukaryotic systems, hence its popularity as a model organism [149]; 2) It contains the canonical organelles and cell biological features of eukaryotic cells, and could be used to infer the functional attributes of heterologously expressed plasmodial proteins by their interaction with endogenous proteins. It may be particularly useful for integral membrane protein expression. For example, expression and targeting of Pf CRT to the digestive compartments of D. discoideum provided evidence for the role of mutations in this membrane protein in reducing chloroquine (CQ) accumulation and hence mediating CQ resistance in parasites [150]; 3) The relative ease with which endogenous D. discoideum genes may be disrupted by homologous recombination or silencing offers the potential to create mutants for use in the functional characterization of heterologous proteins by complementation. For example, adenylate cyclase null mutants were used to demonstrate the properties of Pf ACα, an integral membrane protein possibly involved in gametocytogenesis [151]; 4) Plasma membrane anchoring via endogenous signal sequences and GPI-anchoring motifs could be used to study the adhesion properties of malaria proteins, e.g. CSP [152]. Additionally, such recombinant D. discoideum strains may be explored as potential live, non-toxic, non-pathogenic "sporozoite-mimicking" vaccines [152]; 5) Dictyostelium discoideum originally became an attractive vehicle for heterologous plasmodial protein expression due to the unusual A+T bias of its gene coding sequences: a feature it shares with P. falciparum [153]. The organism was first used for the preparative expression of malaria vaccine candidates including P. falciparum CSP and, subsequently, the C-terminal portions of CSP from P. yoelii and P. falciparum and a fragment of MSP1 of P. vivax [152, 154, 155]. Although codon optimization was not performed, it was noted that expression was considerably improved by the N-terminal addition of D. discoideum leader sequences or fusion partners. Unfortunately, the paucity of reports on the use of D. discoideum for the preparative expression of plasmodial proteins prohibits an assessment of the advantages conferred by the organism's A+T bias and its utility in this regard over the more conventional yeast expression systems. Nonetheless, it is possible that D. discoideum is underexplored as an alternative to mammalian cells and Xenopus oocytes for the functional expression of plasmodial membrane proteins and transporters (see discussions below).
Toxoplasma gondii
Toxoplasma gondii is an important parasite pathogen in immuno-compromized individuals and during pregnancy. Like malaria parasites, it belongs to the phylum Apicomplexa, with the advantage that it is considerably more susceptible to experimental manipulation by standard molecular genetic approaches [156]. Phylogenetic relatedness notwithstanding, the differences in host cell niche, pathogenesis and life-cycles (in addition to a G+C vs. A+T codon bias) makes it debatable to what extent the cell biology and metabolic pathways of T. gondii are related to those of the various life-cycle stages of malaria parasites. Nonetheless, Toxoplasma and Plasmodium share the same mechanism of locomotion as well as the specialized secretory organelles and mechanisms required for host cell invasion [157]. Consequently, T. gondii may be a useful host for the heterologous expression and characterization of malaria proteins involved in locomotion, invasion and biogenesis of the specialized secretory organelles (rhoptries, micronemes and dense granules). Per illustration, expression of a dominant negative mutant form of Pf Vps4 led to the identification of a multi-vesicular body endosomal compartment involved in rhoptry biogenesis in Toxoplasma and suggested the existence of similar organelles and their functions in malaria parasites [158]. A further potential use of T. gondii expressing plasmodial sequences is as an attenuated live vaccine. Experiments in Rhesus monkeys and mice have demonstrated the promising utility of T. gondii strains expressing P. knowlesi and P. yoelii CSP in this regard [159, 160]. From a preparative point of view, the P. falciparum cGMP-dependent protein kinase (Pf PKG) was expressed and purified from T. gondii to characterize its enzymatic properties and susceptibility to inhibitors [161]. Nonetheless, it is unlikely that T. gondii will challenge E. coli or yeast systems as a first choice for this type of application.
Mammalian cells
To a significant extent, mammalian cells share with S. cerevisiae the features of being well characterized, having versatile reagents and protocols for transgene expression and analysis as well as potentially aiding correct folding of heterologous proteins in secretory compartments and imparting post-translational modifications (e.g. lipid modifications, GPI-anchoring, etc.). However, the substantially lower yields of recombinant proteins minimize their use for preparative expression. An advantage of mammalian cells is the ability to target heterologous membrane proteins to subcellular compartments and the cell surface. As a result, the large majority of studies reporting the expression of plasmodial proteins in mammalian cells have focused on the identification and characterization of parasite surface ligands that mediate host cell binding. These experiments have significantly contributed to our understanding of erythrocyte invasion by helping to identify merozoite proteins and their domains that mediate erythrocyte binding, notably EBA-175, AMA-1, Duffy-binding protein, JESEBL, MAEBL, MSA-1 and Pf AARP (e.g [162–170]). In addition, the studies have characterized regions of Pf EMP-1 involved in binding to host chondroitin sulphate A (CSA), ICAM-1, CD36 and uninfected erythrocytes, thus mediating parasite sequestration to the placenta and vascular endothelium as well as erythrocyte rosetting in infected individuals [171–174]. A concomitant advantage of surface expression has been to map out conserved epitopes recognized by inhibitory monoclonal antibodies and human sera (e.g. [175–177]). To facilitate the surface binding experiments, parasite protein domains are modified by N- and C-terminal fusion to mammalian or viral secretory signal sequences and transmembrane domains/GPI-anchoring motifs, respectively, and tagged with an epitope tag and/or GFP to monitor expression. The COS-7 green monkey kidney cell-line has generally been used for these (and other plasmodial expression) experiments, but the well-known CHO-K1 and HeLa cell lines have also been employed. Interestingly, with few exceptions (e.g. [174, 177]), the plasmodial sequences were rarely altered to rectify A+T-codon bias and remove potential N-glycosylation sites.
The second most numerous application of mammalian cell expression in malaria research is to support the development of DNA plasmid-based vaccines. Transfection of mammalian cells with the vaccine plasmids is used to verify expression of the relevant plasmodial sequences prior to proceeding with animal vaccination. In addition, mammalian cell expression may also be used to assess the generation of the desired specific antibodies subsequent to vaccination (e.g. [178–181]). Again, codon optimization is rarely reported. This raises the possibility that mammalian cells may be more accommodating for the expression of the A+T-biased plasmodial sequences.
Despite the advantages offered by the extensive characterization of mammalian cell biology, the cells appear to be rarely used for functional characterization of plasmodial proteins, beyond the ligand binding studies described above. One likely reason is the comprehensive difficulty of preparing functionally compromized mutants, which hampers the complementation studies that have been extensively done in S. cerevisiae (and potentially D. discoideum). A prospective (unreported) alternative may be to use post-transcriptional silencing (e.g. RNAi-mediated) to generate deficient cells for complementation assays. In addition, provided the functional readout is sufficiently sensitive and robust, overexpression of plasmodial proteins above the endogenous background may yield useful results. A further advantage for functional characterization is the high transient transfection efficiencies that can be achieved with current transfection reagents and cell types (e.g. COS cells), which may increase the turnaround time from transfection to analysis. Examples have included the cytoplasmic expression of CDP-diacylglycerol synthase (Pf CDS), mannosyl transferase (Pf PIG-M), Pf VPS4 and histidine rich protein II HRPII, which has shed light on the potential roles of these parasite proteins in phospholipid synthesis, GPI-anchor synthesis, endocytic trafficking and pH-sensing, respectively [158, 182–184].
Both Plasmodium membrane transporters implicated in CQ resistance, Pgh1 and Pf CRT, have been functionally expressed in mammalian cells. Although the intracellular location of Pgh1 expressed in CHO cells could not be determined accurately, it increased CQ uptake and sensitivity in these cells, whilst Pgh1 incorporating CQ resistance-associated mutations did not [185]. After synthetically introducing a yeast codon bias into the Pf CRT cDNA sequence, the protein was expressed in HEK 293 cells and faithfully targeted to lysosomal membranes [186]. Surprisingly, in contrast to the results obtained in D. discoideum ([150]; see earlier discussion), Pf CRT did not alter CQ accumulation in the host cells but increased acidification of lysosomes, an effect which was enhanced when the CQ resistance-conferring mutation K76T was included in the protein. The latter contradiction highlights the possibility that conclusions drawn from characterization in heterologous cell types may not always translate to accurate functional prediction in the parasite and does not substitute for characterization in the parasite itself.
Xenopus laevis
Once the technical and logistical challenges of Xenopus oocyte micro-manipulation and cRNA injection have been overcome, the cells may be extremely useful for the functional expression and characterization of membrane proteins, notably transport proteins. It has become the method of choice for characterizing Plasmodium transporters, particularly since the oocytes appear to be more tolerant than other eukaryotic cells to the peculiar codon preferences of Plasmodium genes [187, 188]. The expression system has been used most extensively to characterize Pf HT1, the principal hexose transporter of P. falciparum [188], in order to define its substrate specificity, probe structure-function relationships by mutational analysis with a view to therapeutic inhibitor design and confirm it as a potential drug target [189–191]. The substrate specificity of Pf ENT1, one of the four equilibrative nucleoside transporters encoded by the P. falciparum genome, was also determined using Xenopus oocyte expression and compared favourably with results obtained from transport experiments using isolated parasites, thus validating the oocyte system as a more convenient surrogate for studying this transporter [192–195]. A similar comparison of properties between isolated parasites and heterologous expression in Xenopus oocytes was obtained for Na+- and membrane potential-dependent transport of inorganic monovalent phosphate by Pf PiT [196]. Additional transporters characterized by expression in Xenopus oocytes coupled with oocyte swelling assays include the aquaglyceroporins of P. falciparum and P. berghei (Pf AQP and Pb AQP) [197, 198].
A significant contribution of the Xenopus oocyte expression system to malaria drug discovery was the identification and characterization of the foremost target of the artemisinins, Pf ATP6, a SERCA-type Ca2+ ATPase [199]. The study followed on the characterization of Pf ATP4, a P. falciparum P-type membrane Ca2+-ATPase, vis-à-vis its susceptibility to inhibitors and Ca2+ concentration-dependent activation [200]. Of further relevance to drug discovery was the use of Xenopus oocyte expression to investigate the role of Pf CRT in conferring CQ resistance. The findings led the authors to propose that Pf CRT affects CQ susceptibility in parasites by a modulation of food vacuole transporters, rather than by directly affecting vacuole pH or transporting CQ [201].
It should be noted that, although the Xenopus oocyte expression system has been used almost exclusively to characterize malaria membrane transporters, it is a functional cell and can be a useful system for studying other cellular functions as well, for example, cell signalling. After expression in oocytes, the parasite leucine-rich repeat antigen (Pf LRR1) was found to bind to the endogenous Xenopus protein phosphatase PP1 and disrupt the oocyte G2/M cell cycle arrest, observed as germinal vesicle breakdown. By inference, Pf LRR1 probably regulates protein phosphatase activity and cell cycle progression in malaria parasites [202]. In addition, measurement of cAMP levels in oocytes was used to characterize the properties of a X. laevis codon-optimized version of the parasite adenylyl cyclase (Pf ACα) and confirm results obtained by complementation assays in Dictyostelium [151].
Plants
The use of plants as expression hosts for foreign proteins has received increasing attention. Several heterologous proteins have been expressed in plants due to their ability to successfully express, fold and post-translationally modify foreign proteins. The real potential lies in the possibility of transgenic plants as bioreactors for the production of vaccines since the feasibility and low cost have been validated in several human and animal disease vaccines. It is therefore attractive to speculate that plants may be a host for 1) the transient production of functional plasmodial proteins or 2) the transgenic production of e.g. malaria vaccine targets. The latter has been under investigation with reports by Ghosh et al. (2002) in which they successfully obtained constitutive expression of a 19 kDa proteolytic product of P. falciparum MSP1 in tobacco plants resulting in the same immunogenic properties as an E. coli expressed form of the protein [203]. More recently, the concept of edible vaccines derived from antigen production in plants was supported by evidence from Hepatitis-B trials in humans and mice and has now been extended to the P. yoelii MSP4/5 expressed in transgenic tobacco [204]. This protein was codon optimized for production in plants and resulted in the high level expression of the antigen that could elicit protective antibodies in mice fed orally. These reports therefore indicate the feasibility of exploiting plants for the production of malaria proteins.
Cell-free protein expression
Cell-free transcription-translation systems have been exploited with varying degrees of success for numerous proteins including membrane proteins and cytotoxic proteins (reviewed in [205, 206]). Advantages include production of proteins that undergo proteolysis or that usually accumulate in inclusion bodies, as well as allowing selective labelling. Of recent interest is the wheat germ cell-free platform [206], which allows the high-level expression of eukaryotic proteins that can not be expressed in E. coli cells/cell-free systems. Optimized wheat germ systems have been created by removal of ribosomal protein synthesis inhibitors (tritin) from the wheat embryo to allow the robust production of proteins. This technology has been adopted by structural genomics consortia (e.g. Centre for Eukaryotic Structural Genomics, Madison, WI). The difficulties experienced over many years in obtaining significant soluble expression of Pf DHFR-TS are well-documented [207]. In a recent study it was shown that a wheat germ cell-free expression system yielded bioactive protein as an obligate dimer without the associated toxicity observed in cell-based systems [207]. Moreover, several malaria vaccine targets (Pfs 25, Pf CSP and Pf AMA-1) were successfully expressed in both native and codon-optimized forms which did not seem to influence the efficiency of expression, at least of Pfs 25 [208]. Yields between 50 and 200 μg/ml of soluble protein were obtained which was subsequently shown to be immunogenically active in raising specific antibodies in mice. Ambitiously, this study was expanded to include 124 additional P. falciparum blood stage proteins that were expressed in their native forms. Of these, 75% of the proteins were produced with an average yield of 1.9 μg/150 μl reaction mixture and 65% solubility. Inverse correlations were observed between soluble protein production and protein size, frequency of low-complexity regions and pI of the proteins.
Conclusion and future perspectives
Heterologous expression of plasmodial proteins as described above is principally carried out for preparative purposes, for example, crystallization and structural studies, preparation of recombinant vaccines, drug screening, antigen preparation, enzymatic and other in vitro functional assays, etc. However, in light of our poor understanding of parasite cell biology and the fact that the majority of malaria proteins predicted by the genome have unknown or uncertain functions, an important concomitant activity is the elucidation of Plasmodium in vivo protein function by heterologous expression in experimentally tractable, well-characterized eukaryotic model organisms and cell types. Model eukaryotes are especially favoured for the expression and characterization of integral membrane proteins, for which prokaryotes are generally ill-suited, provided that spurious, incorrect post-translational modifications (notably glycosylation) in the secretory pathway of the host cell can be avoided or tolerated. In addition to in situ functional characterization of malaria proteins, eukaryotic cells have also been explored for preparative expression in an attempt to circumvent problems experienced in prokaryotes, notably incorrect protein folding, insolubility and codon incompatibility, as described in the preceding sections.
From the above, it is evident that each protein has to be assessed on an individual basis since there is no universal procedure for the heterologous expression of malaria proteins or proteins of other organisms. However, a protocol is suggested worthy of consideration when attempting to express a plasmodial protein (Figure 1). Bioinformatics analyses of the gene and its encoded protein is clearly of paramount importance to guide the selection of the appropriate gene construct, the host/vector combination and the expression and isolation procedures. Expression of proteins from E. coli systems is clearly the first choice due to its ease of genetic manipulation and the availability of a variety of different vectors and E. coli strains. Fortuitously, new strains, vectors and methods are continuously being developed to address the multitude of challenging impediments revealed by structural genomics consortia in their endeavours to determine the three-dimensional structures of proteins encoded by the genomes of Eubacteria, Archea and Eukarya.
Alternative hosts including yeasts, baculovirus and cell-free systems, with or without codon adaptation strategies, are additional options to be explored if expression from E. coli systems is problematic. For preparative expression of soluble proteins, Pichia pastoris, baculovirus and cell free systems may be the most successful whereas alternatives such as Saccharomyces, Dictyostelium and mammalian systems may be more beneficial for functional analyses with the first two additionally having the major advantage of complementation studies thereby circumventing soluble protein isolation. Attempts to express transmembrane proteins for functional analyses should be focussed on systems including Xenopus and mammalian systems with a potential application of Dictyostelium as host.
The introduction and ongoing development of reverse genetics methodology for P. falciparum [e.g[209]], coupled with sporadic reports of successful post-transcriptional silencing by antisense RNA, RNAi and ribozymes [e.g. [210, 211]], provides the primary tools for elucidating protein function in the parasite. Nonetheless, transgene expression in more tractable heterologous model organisms (notably through mutant complementation in yeast and surface expression in Xenopus oocytes and mammalian cells) offers a powerful parallel approach to aid functional characterization. These transgene expression strategies are especially useful for determining substrate or ligand specificity, structure-function relationships through mutational analysis, and inhibitor sensitivity/resistance, and, under optimal circumstances, may even replace the requirement for preparative expression of purified soluble protein.
It is clear that numerous options are available, and will continue to be developed, for the successful expression of plasmodial proteins. This facet of research, however, still represents a major challenge to scientists in the malaria field, but it is a vital first step in the post-genomic functional and structural characterization of these proteins. This knowledge is crucial to identify new proteins that may serve as potential drug or vaccine targets, which will enhance our chances of producing new therapies to combat malaria in the future.
References
Birkholtz LM, Bastien O, Wells G, Grando D, Joubert F, Kasam V, Zimmermann M, Ortet P, Jacq N, Saidani N, Roy S, Hofmann-Apitius M, Breton V, Louw AI, Marechal E: Integration and mining of malaria molecular, functional and phamacological data: how far are we from a chemogenomic knowledge space?. Malaria J. 2006, 17: 110-10.1186/1475-2875-5-110.
THE SOUTHEAST COLLABORATORY FOR STRUCTURAL GENOMICS (SECSG). [http://www.secsg.org]
Flick K, Ahuja S, Chene A, Bejarano MT, Chen Q: Optimized expression of Plasmodium falciparum erythrocyte membrane protein I domains in Escherichia coli. Malaria J. 2004, 3: 1-8. 10.1186/1475-2875-3-50.
Carrio MM, Villaverde A: Construction and deconstruction of bacterial inclusion bodies. J Biotechnol. 2002, 96: 3-12. 10.1016/S0168-1656(02)00032-9.
Mehlin C, Boni E, Buckner FS, Engel L, Feist T, Gelb MH, Haji L, Kim D, Liu C, Mueller N, Myler PJ, Reddy JT, Sampson JN, Subramanian E, Van Voorhis WC, Worthey E, Zucker F, Hol WGJ: Heterologous expression of proteins from Plasmodium falciparum: Results from 1000 genes. Mol Biochem Parasitol. 2006, 148: 144-160. 10.1016/j.molbiopara.2006.03.011.
Aguiar JC, LaBaer J, Blair PL: High-throughput generation of P. falciparum functional molecules by recombinational cloning. Genome Res. 2004, 2004: 2076-2082. 10.1101/gr.2416604.
Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong A, Wasney GA, Gao M, Hills T, Brokx S, Qiu W, Sharma S, Diassiti A, Alam Z, Melone M, Mulichak A, Wernimont A, Bray J, Loppnau P, Plotnikova O, Newberry K, Sundararajan E, Houston S, Walker J, Tempel W, Bockharev A, Kozieradzki I, Edwards A, Arrowsmith C, Roos D, Kain K, Hui R: Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol. 2007, 151: 100-110. 10.1016/j.molbiopara.2006.10.011.
Sorensen HP, Mortensen KK: Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005, 115: 113-128. 10.1016/j.jbiotec.2004.08.004.
Sahdev S, Khattar SK, Saini KS: Production of active eukaryotic proteins through bacterial expression systems: a review of the existing biotechnology strategies. Mol Cell Biochem. 2008, 307: 249-264. 10.1007/s11010-007-9603-6.
Esposito D, Chatterjee DK: Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006, 17: 353-358. 10.1016/j.copbio.2006.06.003.
Tonkin CJ, van Dooren GG, Spurck TP, Struck NS, Good RT, Handman E, Cowman AF, McFadden GI: Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol Biochem Parasitol. 2004, 137: 13-21. 10.1016/j.molbiopara.2004.05.009.
Structural Genomics Consortium, China Structural Genomics Consortium, Northeast Structural Genomics Consortium, Gräslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, dhe-Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schütz A, Heinemann U, Yokoyama S, Büssow K, Gunsalus KC: Protein production and purification. Nature Methods. 2008, 5: 135-146. 10.1038/nmeth.f.202.
Peti W, Page R: Strategies to maximize heterologous protein expression in Eschericihia coli with minimal cost. Protein Expr Purif. 2007, 51: 1-10. 10.1016/j.pep.2006.06.024.
Suzuki M, Roy R, Zheng H, Woychik N, Inouye M: Bacterial Bioreactors for High yield production of recombinant protein. J Biol Chem. 2006, 281: 37559-37565. 10.1074/jbc.M608806200.
Smith HE: The transcriptional response of Escherichia coli to recombinant protein insolubility. J Struct Funct Genomics. 2007, 8: 27-35. 10.1007/s10969-007-9030-7.
Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA: Regulon and promotor analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes Dev. 2006, 20: 1776-1789. 10.1101/gad.1428206.
Yazdani SS, Shakri AR, Pattnaik P, Rizvi MMA, Chitnis CE: Improvement in yield and purity of a recombinant malaria vaccine candidate based on the receptor-binding domain of Plasmodium vivax Duffy binding protein by codon optimization. Biotechnol Lett. 2006, 28: 1109-1114. 10.1007/s10529-006-9061-3.
Zhou Z, Schnake P, Xiao L, Lal AA: Enhanced expression of a recombinant malaria candidate vaccine in Escherichia coli by codon optimization. Protein Expr Purif. 2004, 34: 87-94. 10.1016/j.pep.2003.11.006.
Coppel RL, Black CG: Malaria Parasite DNA. Malaria parasite biology, pathogenesis and protection. Edited by: Sherman IW. 1998, Washington DC: ASM Press
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan M-S, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Feng Z-P, Zhang X, Han P, Arora N, Anders RF, Norton RS: Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006, 150: 256-267. 10.1016/j.molbiopara.2006.08.011.
DePristo MA, Zilversmit MM, Hartl DL: On the abundance, amino acid composition, and evolutionary dynamics of low-complexity regions in proteins. Gene. 2006, 378: 19-30. 10.1016/j.gene.2006.03.023.
Baca AM, Hol WGJ: Overcoming codon bias: A method for high-level over expression of Plasmodium and other AT-rich parasite genes in Escherichia coli. Int J Parasitol. 2000, 30: 113-118. 10.1016/S0020-7519(00)00019-9.
Radivojac P, Lakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK: Intrinsic Disorder and Functional Proteomics. Biophys J. 2007, 92: 1439-1456. 10.1529/biophysj.106.094045.
Casta LJ, Buguliskis JS, Matsumoto Y, Taraschi TF: Expression and biochemical characterization of the Plasmodium falciparum DNA repair enzyme, flap endonuclease-1 (PfFEN-1). Mol Biochem Parasitol. 2008, 157: 1-12. 10.1016/j.molbiopara.2007.08.008.
Bandaru V, Cooper W, Wallace SS, Doublie S: Overproduction, crystallization and preliminary crystallographic analysis of a novel human DNA-repair enzyme that recognizes oxidative DNA damage. Acta Crystallogr D Biol Crystallogr. 2004, 60: 1142-1144. 10.1107/S0907444904007929.
Gräslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, Hammarström M, Johansson I, Kotenyova T, Nilsson M, Nordlund P, Weigelt J: The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif. 2008, 58: 210-221. 10.1016/j.pep.2007.11.008.
Kalthoff C: A novel strategy for the purification of recombinantly expressed unstructured protein domains. J Chromatogr B Analyt Technol Biomed Life Sci. 2003, 786: 247-254. 10.1016/S1570-0232(02)00908-X.
Zhang X, Perugini MA, Yao S, Adda CG, Murphy VJ, Low A, Anders RF, Norton RS: Solution Conformation, Backbone Dynamics and Lipid Interactions of the Intrinsically Unstructured Malaria Surface Protein MSP2. J Mol Biol. 2008, 379 (1): 105-121. 10.1016/j.jmb.2008.03.039.
Romier C, Ben Jelloul M, Albeck S, Buchwald G, Busso D, Celie PH, Christodoulou E, De Marco V, van Gerwen S, Knipscheer P, Lebbink JH, Notenboom V, Poterszman A, Rochel N, Cohen SX, Unger T, Sussman JL, Moras D, Sixma TK, Perrakis A: Co-expression of protein complexes in prokaryotic and eukaryotic hosts: experimental procedures, database tracking and case studies. Acta Crystallogr D Biol Crystallogr. 2006, 62: 1232-1242. 10.1107/S0907444906031003.
Scheich C, Kummel D, Soumailakakis D, Udo Heinemann U, Büssow K: Vectors for co-expression of an unrestricted number of proteins. Nucleic Acids Res. 2007, 35: 1-7. 10.1093/nar/gkm067.
Tolia NJ, Joshua-Tor L: Strategies for protein coexpression in Escherichia coli. Nature Methods. 2006, 3: 55-64. 10.1038/nmeth0106-55.
Ahuja S, Ahuja S, Chen Q, Wahlgren M: Prediction of solubility on recombinant expression of Plasmodium falciparum erythrocyte membrane protein 1 domains in Escherichia coli. Malaria J. 2006, 5: 52-10.1186/1475-2875-5-52.
Sirawaraporn W, Prapunwattana P, Sirawaraporn R, Yuthavong Y, Santi DV: The dihydrofolate reductase domain of Plasmodium falciparum thymidylate synthase-dihydrofolate reductase. J Biol Chem. 1993, 268: 21637-21644.
Wrenger C, Lüersen K, Krause T, Müller S, Walter RD: The Plasmodium falciparum bifunctional ornithine decarboxylase, S-adenosylmethionine decaroxylase enables a well-balanced polyamine synthesis without domain-domain interaction. J Biol Chem. 2001, 276: 29651-29656. 10.1074/jbc.M100578200.
WHO TDR Targets database. [http://www.tdrtargets.org]
Prapunwattana P, Sirawaraporn R, Yuthavong Y, Santi DV: Chemical synthesis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Mol Biochem Parasitol. 1996, 83: 93-106. 10.1016/S0166-6851(96)02756-9.
Angov E, Aufiero BM, Turgeon AM, Van Handenhove M, Ockenhouse CF, Kester KE, Walsh DS, McBride JS, Dubois MC, Cohen J, Haynes JD, Eckels KH, Heppner DG, Ballou WR, Diggs CL, Lyon JA: Development and pre-clinical analysis of a Plasmodium falciparum Merozoite Surface Protein-1(42) malaria vaccine. Mol Biochem Parasitol. 2003, 128: 195-204. 10.1016/S0166-6851(03)00077-X.
Angov E, Hillier CJ, Kincaid RL, Lyon JA: Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE. 2008, 3: e2189-10.1371/journal.pone.0002189.
Narum DL, Kumar S, Rogers WO, Fuhrmann SR, Liang H, Oakley M, Taye A, Sim BKL, Hoffman SL: Codon optimization of gene fragments encoding Plasmodium falciparum merozoite proteins enhances DNA vaccine protein expression and immunogenicity. Infection and Immunity. 2001, 69: 7250-7253. 10.1128/IAI.69.12.7250-7253.2001.
Yadava A, Ockenhouse CF: Effect of codon optimization on expression levels of a functionally folded malaria vaccine candidate in Prokaryotic and Eukaryotic expression systems. Infect Immun. 2003, 71: 4961-4969. 10.1128/IAI.71.9.4961-4969.2003.
Coetzee L: Plasmodium falciparum dihydrofolate synthase-folylpolyglutamate synthase (DHFS-FPGS): Gene synthesis and recombinant expression. 2003, Pretoria: University of Pretoria
Kincaid RL, Angov E, Lyon JA: Protein expression by codon harmonization and translational attenuation. United States., C12P 21/02, C12N 1/21, C07H 21/04, C12N 15/74, edn. 2004, San Francisco, CA, US: Veritas, 37-
Thanaraj TA, Argos P: Ribosome-mediated translational pause and protein domain organization. Protein Science. 1996, 5: 1594-1612.
Hillier CJ, Ware LA, Barbosa A, Angov E, Lyon JA, Heppner DG, Lanar DE: Process development and analysis of liver-stage antigen 1, a preerythrocyte-stage protein-based vaccine for Plasmodium falciparum. Infect Immun. 2005, 73: 2109-2115. 10.1128/IAI.73.4.2109-2115.2005.
Longshaw VM, Nicoll WS, Botha M, Ludewig MH, Shonhai A, Stephens LL, Blatch GL: Getting practical with molecular chaperones. Biotechnology International. 2006, 18: 24-27.
Mayer MP, Bukau B: Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005, 62: 670-684. 10.1007/s00018-004-4464-6.
de Marco A, De Marco V: Bacteria co-transformed with recombinant proteins and chaperones cloned in independent plasmids are suitable for expression tuning. J Biotechnol. 2004, 109: 45-52. 10.1016/j.jbiotec.2003.10.025.
Sargeant TJ, Marti M, Caler E, Charlton Jm, Simpson K, Speed TP, Cowman AF: Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 2006, 7: R12-10.1186/gb-2006-7-2-r12.
Shonhai A, Boshoff A, Blatch GL: The structural and functional diversity of Hsp70 proteins from Plasmodium falciparum. Prot Sci. 2007, 16: 1803-1818. 10.1110/ps.072918107.
Matambo TS, Odunuga OO, Boshoff A, Blatch GL: Overproduction, purification and characterisation of the Plasmodium falciparum heat shock protein 70. Protein Expr Purif. 2004, 33: 214-222. 10.1016/j.pep.2003.09.010.
Shonhai A, Boshoff A, Blatch GL: Plasmodium falciparum heat shock protein 70 is able to suppress the thermosensitivity of an Escherichia coli DnaK mutant strain. Mol Genet Genomics. 2005, 274: 70-78. 10.1007/s00438-005-1150-9.
Gragerov A, Zeng L, Zhao X, Burkholder W, Gottesman ME: Specificity of DnaK-peptide binding. J Mol Biol. 1994, 235: 848-854. 10.1006/jmbi.1994.1043.
Ramya TNC, Surolia NN, Surolia A: 15-Deoxyspergualin modulates Plasmodium falciparum heat shock protein function. Biochem Biophys Res Commun. 2006, 348: 585-592. 10.1016/j.bbrc.2006.07.082.
LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE: A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005, 438: 103-107. 10.1038/nature04104.
Botha M, Pesce E-R, Blatch GL: The Hsp40 proteins of Plasmodium falciparum and other apicomplexa: Regulating chaperone power in the parasite and the host. Int J Biochem Cell Biol. 2007, 39 (10): 1781-1803. 10.1016/j.biocel.2007.02.011.
Foth BJ, Ralph SA, Tonkin CJ, Struck NS, Fraunholz M, Roos DS, Cowman AF, McFadden GI: Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science. 2003, 299: 705-708. 10.1126/science.1078599.
Nyalwidhe J, Lingelbach K: Proteases and chaperones are the most abundant proteins in the parsitophorous vacuole of Plasmodium falciparum-infected erythrocytes. Proteomics. 2006, 6: 1-11. 10.1002/pmic.200500379.
Lanzer M, Wickert H, Krohne G, Vincensini L, Breton CB: Maurer's clefts: A novel multi-functional organelle in the cytoplasm of Plasmodium falciparum-infected erythrocytes. Int J Parasitol. 2006, 36: 23-36. 10.1016/j.ijpara.2005.10.001.
Kumar N, Koski G, Harada M, Aikawa M, Zheng H: Induction and localization of Plasmodium falciparum stress proteins related to the heat shock protein 70 family. Mol Biochem Parasitol. 1991, 48: 47-58. 10.1016/0166-6851(91)90163-Z.
Carrio MM, Villaverde A: Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 2001, 489: 29-33. 10.1016/S0014-5793(01)02073-7.
Villaverde A, Carrio MM: Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol Lett. 2003, 25: 1385-1395. 10.1023/A:1025024104862.
Singh SM, Panda AK: Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng. 2005, 99: 303-310. 10.1263/jbb.99.303.
Vallejo LF, Rinas U: Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins. Microb Cell Fact. 2004, 3: 11-22. 10.1186/1475-2859-3-11.
Jungbauer A, Kaar W: Current status of technical protein refolding. J Biotechnol. 2007, 128: 587-596. 10.1016/j.jbiotec.2006.12.004.
Kumar A, Kumar K, Korde R, Puri SK, Malhotra P, Cauhan VS: Falcipain-1, a Plasmodium falciparum Cysteine protease with vaccine potential. Infect Immun. 2007, 75: 2026-2034. 10.1128/IAI.01533-06.
Sijwali PS, Brinen LS, Rosenthal PJ: Systematic optimization of expression and refolding of the Plasmodium falciparum cysteine protease Falcipain-2. Protein Expr Purif. 2001, 22: 128-134. 10.1006/prep.2001.1416.
Leung WH, Meng ZQ, Hui G, Ho WKK: Expression and immunologically reactive merozoite surface protein (MSP-142) in E. coli. Biochim Biophys Acta. 2004, 1675: 62-70.
Shimp RL, Martin LB, Zhang Y, Henderson BS, Duggan P, MacDonald NJ, Lebowitz J, Saul A, Narum DL: Production and characterization fo clinical grade Escherichia coli derived Plasmodium falciparum 42 kDa merozoite surface protein 1 (MSP142) in the absence of an affinity tag. Protein Expr Purif. 2006, 50: 58-67. 10.1016/j.pep.2006.06.018.
Dominquez A, Ferminan E, Sanchez M, Gonzalez J, Perez-Campo FM, Garcia S, Herrero AB, San Vincente A, Cabello J, Prado M, Iglesias FJ, Choupina A, Burguillo FJ, Fernandez-Lago L, Lopez MC: Non-conventional yeasts as hosts for heterologous protein production. Int Microbiol. 1998, 1: 131-142.
Bathurst IC: Protein expression in yeast as an approach to production of recombinant malaria antigens. Am J Trop Med Hyg. 1994, 50: 20-26.
Jeffress M, Fields S: Identification of putative Plasmodium falciparum mefloquine resistance genes. Mol Biochem Parasitol. 2005, 139: 133-139. 10.1016/j.molbiopara.2004.10.005.
Brake AJ: Secretion of heterologous proteins directed by the yeast alpha-factor leader. Biotechnology. 1989, 13: 269-280.
Vallari RC, Cook WJ, Audino DC, Morgan MJ, Jensen DE, Laudano AP, Denis CL: Glucose repression of the yeast ADH2 gene occurs through multiple mechanisms, including control of the protein synthesis of its transcriptional activator, ARD1. Mol Cell Biol. 1992, 12: 1663-1673.
Miles AP, Zhang Y, Saul A, Stowers AW: Large-scale purification and characterization of malaria vaccine candidate antigen Pvs25H for use in clinical trials. Prot Express Purif. 2002, 25: 87-96. 10.1006/prep.2001.1613.
Stowers AW, Zhang Y, Shimp RL, Kaslow DC: Structural conformers produced during malaria vaccine production in yeast. Yeast. 2001, 18: 137-150. 10.1002/1097-0061(20010130)18:2<137::AID-YEA657>3.0.CO;2-X.
Milek RLB, Stunnenberg HG, Konings RNH: Assembly and expression of a synthetic gene encoding the antigen Pfs48/45 of the human malaria parasite Plasmodium falciparum in yeast. Vaccine. 2000, 18: 1402-1411. 10.1016/S0264-410X(99)00392-8.
Blackman MJ, Holder AA: Use of a recombinant baculovirus product to measure naturally acquired human antibodies to disulphide-constrained epitopes on the P. falciparum merozoite surface protein-1 (MSP1). FEMS Immunol Med Microbiol. 1993, 6: 307-315. 10.1111/j.1574-695X.1993.tb00343.x.
Tanner W, Lehle L: Protein glycosylation in yeast. Biochim Biophys Acta. 1987, 906: 81-99.
Chang VT, Crispin M, Aricescu AR, Harvey DJ, Nettleship JE, Fennelly JA, Yu C, Boles KS, Evans EJ, Stuart DI, Dwek RA, Jones EY, Owens RJ, Davis SJ: Glycoprotein structural genomics: solving the glycosylation problem. Structure. 2007, 15: 267-273. 10.1016/j.str.2007.01.011.
Mattanovich D, Gasser B, Hohenblum H, Sauer M: Stress in recombinant protein producing yeasts. J Biotechnol. 2004, 113: 121-135. 10.1016/j.jbiotec.2004.04.035.
Cereghino GPL, Cregg JM: Applications of yeasts in biotechnology: protein production and genetic analysis. Curr Opin Biotechnol. 1999, 10: 422-427. 10.1016/S0958-1669(99)00004-X.
Romanos MA, Scorer CA, Clare JF: Foreign gene expression in yeast: a review. Yeast. 1992, 8: 423-488. 10.1002/yea.320080602.
Sibley CH, Macreadie I: Novel approaches to tackling malarial drug resistance using yeast. IUBMB Life. 2001, 52: 285-289. 10.1080/152165401317291138.
Kaslow DC, Bathurst IC, Lensen T, Ponnudurai T, Barr PJ, Keister DB: Saccharomyces cerevisiae recombinant Pfs25 adsorbed to alum elicits antibodies that block transmission of Plasmodium falciparum. Infect Immun. 1994, 62: 5576-5580.
Malkin EM, Durbin AP, Diemert DJ, Sattabongkot J, Wu Y, Miura K, Long CA, Lambert L, Miles AP, Wang J, Stowers A, Miller LH, Saul A: Phase 1 vaccine trial of Pvs25H: a transmission blocking vaccine for Plasmodium vivax malaria. Vaccine. 2005, 23: 3131-3138. 10.1016/j.vaccine.2004.12.019.
Moelans II, Cohen J, Marchand M, Molitor C, de Wilde P, van Pelt JF, Hollingdale MR, Roeffene WFG, Elinge WMC, Atkinsonf CT, Aikawaf M, Schoenmakersa JGG, Koningsa RNH: Induction of Plasmodium falciparum sporozoite-neutralizing antibodies upon vaccination with recombinant Pfs16 vaccinia virus and/or recombinant Pfs16 protein produced in yeast. Mol Biochem Parasitol. 1995, 72: 179-192. 10.1016/0166-6851(95)00072-9.
Hirunpetcharat C, Tian JH, Kaslow DC, van Rooijen N, Kumar S, Berzofsky JA, Miller LH, Good MF: Complete protective immunity induced in mice by immunization with the 19-kilodalton carboxyl-terminal fragment of the merozoite surface protein-1 (MSP1[19]) of Plasmodium yoelii expressed in Saccharomyces cerevisiae. J Immunol. 1997, 159: 3400-3411.
Kumar S, Yadava A, Keister DB, Tian JH, Ohl M, Perdue-Greenfield KA, Miller LH, Kaslow DC: Immunogenicity and in vivo efficacy of recombinant Plasmodium falciparum merozoite surface protein-1 in Aotus monkeys. Mol Med. 1995, 1: 325-332.
EUROSCARF. [http://web.uni-frankfurt.de/fb15/mikro/euroscarf/]
Saccharomyces Genome Deletion Project. [http://www-sequence.stanford.edu/group/yeast_deletion_project/]
ATCC. [http://www.atcc.org/]
Volkman SK, Cowman AF, Wirth DF: Functional complementation of the ste6 gene of Saccharomyces cerevisiae with the pfmdr1 gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1995, 92: 8921-8925. 10.1073/pnas.92.19.8921.
Yatsushiro S, Taniguchi S, Mitamura T, Omoto H, Moriyama Y: Proteolipid of vacuolar H+-ATPase of Plasmodium falciparum : cDNA cloning, gene organization and complementation of a yeast null mutant. Biochim Biophys Acta. 2005, 1717: 89-96. 10.1016/j.bbamem.2005.08.011.
Zeuthen T, Wu B, Pavlovic-Djuranovic S, Holm LM, Uzcategui NL, Duszenko M, Kun JF, Schultz JE, Beitz E: Ammonia permeability of the aquaglyceroporins from Plasmodium falciparum, Toxoplasma gondii and Trypanosoma brucei. Mol Microbiol. 2006, 61: 1598-1608. 10.1111/j.1365-2958.2006.05325.x.
Santiago TC, Zufferey R, Mehra RS, Coleman RA, Ben Mamoun C: The Plasmodium falciparum PfGatp is an endoplasmic reticulum membrane protein important for the initial step of malarial glycerolipid synthesis. J Biol Chem. 2004, 279: 9222-9232. 10.1074/jbc.M310502200.
Wengelnik K, Vial HJ: Characterization of the phosphatidylinositol synthase gene of Plasmodium species. Res Microbiol. 2007, 158: 51-59. 10.1016/j.resmic.2006.11.005.
Pessi G, Choi JY, Reynolds JM, Voelker DR, Mamoun CB: In vivo evidence for the specificity of Plasmodium falciparum phosphoethanolamine methyltransferase and its coupling to the Kennedy pathway. J Biol Chem. 2005, 280: 12461-12466. 10.1074/jbc.M414626200.
Shams-Eldin H, Azzouz N, Kedees MH, Orlean P, Kinoshita T, Schwarz RT: The GPI1 homologue from Plasmodium falciparum complements a Saccharomyces cerevisiae GPI1 anchoring mutant. Mol Biochem Parasitol. 2002, 120: 73-81. 10.1016/S0166-6851(01)00434-0.
Fan Q, An L, Cui L: Plasmodium falciparum histone acetyltransferase, a yeast GCN5 homologue involved in chromatin remodelling. Eukar Cell. 2004, 3: 264-276. 10.1128/EC.3.2.264-276.2004.
Bhattacharyya MK, Hong Z, Kongkasuriyachai D, Kumar N: Plasmodium falciparum protein phosphatase 1 functionally complements a glc7 mutant in Saccharomyces cerevisiae. Int J Parasitol. 2002, 32: 739-747. 10.1016/S0020-7519(02)00007-3.
Vinkenoog R, Aparecida Sperança M, Ramesar J, Thomas AW, del Portillo HA, Janse CJ, Waters AP: Characterisation of the Cdc2-related kinase 2 gene from Plasmodium knowlesi and P. berghei. Mol Biochem Parasitol. 1998, 95: 229-240. 10.1016/S0166-6851(98)00104-2.
Aruna K, Chakraborty T, Rao PN, Santos C, Ballesta JPG, Sharma S: Functional complementation of yeast ribosomal P0 protein with Plasmodium falciparum P0. Gene. 2005, 357: 9-17. 10.1016/j.gene.2005.04.007.
Wrenger C, Eschbach ML, Muller IB, Warnecke D, Walter RD: Analysis of the vitamin B6 biosynthesis pathway in the human malaria parasite Plasmodium falciparum. J Biol Chem. 2005, 280: 5242-5248. 10.1074/jbc.M412475200.
Salcedo E, Cortese JF, Plowe CV, Sims PFG, Hyde JE: A bifunctional dihydrofolate synthetase-folylpolyglutamate synthetase in Plasmodium falciparum identified by functional complementation in yeast and bacteria. Mol Biochem Parasitol. 2001, 112: 239-252. 10.1016/S0166-6851(00)00370-4.
Hankins EG, Warhurst DC, Sibley CH: Novel alleles of the Plasmodium falciparum dhfr highly resistant to pyrimethamine and chlorcycloguanil, but not WR99210. Mol Biochem Parasitol. 2001, 117: 91-102. 10.1016/S0166-6851(01)00335-8.
Hastings MD, Maguire JD, Bangs MJ, Zimmerman PA, Reeder JC, Baird JK, Sibley CH: Novel Plasmodium vivax dhfr alleles from the Indonesian archipelago and Papua New Guinea: association with pyrimethamine resistance determined by a Saccharomyces cerevisiae expression system. Antimicrob Agents Chemother. 2005, 49: 733-740. 10.1128/AAC.49.2.733-740.2005.
Ferlan JT, Mookherjee S, Okezie IN, Fulgence L, Sibley CH: Mutagenesis of dihydrofolate reductase from Plasmodium falciparum : analysis in Saccharomyces cerevisiae of triple mutant alleles resistant to pyrimethamine or WR99210. Mol Biochem Parasitol. 2001, 113: 139-150. 10.1016/S0166-6851(01)00207-9.
Lau H, Ferlan JT, Brophy VH, Rosowsky A, Sibley CH: Efficacies of lipophilic inhibitors of dihydrofolate reductase against parasitic protozoa. Antimicrob Agents Chemother. 2001, 45: 187-195. 10.1128/AAC.45.1.187-195.2001.
Djapa LY, Basco LK, Zelikson L, Rosowsky A, Djaman JA, Yonkeu JN, Bolotin-Fukuhara M, Mazabraud A: Antifolate screening using yeast expressing Plasmodium vivax dihydrofolate reductase and in vitro drug susceptibility assay for Plasmodium falciparum. Mol Biochem Parasitol. 2007, 156: 89-92. 10.1016/j.molbiopara.2007.07.009.
Houard S, Heinderyckx M, Bollen A: Engineering of non-conventional yeasts for efficient synthesis of macromolecules: the methyltrophic genera. Biochimie. 2002, 84: 1089-1093. 10.1016/S0300-9084(02)00011-1.
Giersing B, Miura K, Shimp R, Wang J, Zhou H, Orcutt A, Stowers A, Saul A, Miller LH, Long C, Singh S: Posttranslational modification of recombinant Plasmodium falciparum apical membrane antigen 1: impact on functional immune responses to a malaria vaccine candidate. Infect Immun. 2005, 73: 3963-3970. 10.1128/IAI.73.7.3963-3970.2005.
Zou L, Miles AP, Wang J, Stowers AW: Expression of malaria transmission-blocking vaccine antigen Pfs25 in Pichia pastoris for use in human clinical trials. Vaccine. 2003, 21: 1650-1657. 10.1016/S0264-410X(02)00701-6.
Kennedy MC, Wang J, Zhang Y, Miles AP, Chitsaz F, Saul A, Long CA, Miller LH, Stowers AW: In vitro studies with recombinant Plasmodium falciparum apical membrane antigen 1 (AMA 1): production and activity of an AMA1 vaccine and generation of a multiallelic response. Infect Immun. 2002, 70: 6948-6960. 10.1128/IAI.70.12.6948-6960.2002.
Kocken CH, Dubbeld MA, Wel van der A, Pronck JT, Waters AP, Langermans JAM, Thomas AW: High-level expression of Plasmodium vivax apical membrane antigen 1 (AMA-1) in Pichia pastoris: strong immunogenicity in Macaca mulatta immunized with P. vivax AMA-1 and adjuvant SBAS2. Infect Immun. 1999, 67: 43-49.
Brady CP, Shimp RL, Miles AP, Whitmore M, Stowers AW: High-level production and purification of P30P2MSP119, an important vaccine antigen for malaria, epressed in the methyltropic yeast Pichia pastoris. Protein Expr Purif. 2001, 23: 468-475. 10.1006/prep.2001.1526.
Pan W, Huang D, Zhang Q, Qu L, Zhang D, Zhang X, Xue X, Qian F: Fusion of two malaria vaccine candidate antigens enhances product yield, immunogenicity, and antibody-mediated inhibition of parasite growth in vitro. J Immunol. 2004, 172: 6167-6174.
Faber BW, Remarque EJ, Morgan WD, Kocken CH, Holder AA, Thomas AW: Malaria vaccine-related benefits of a single protein comprising Plasmodium falciparum apical membrane 1 domains I and II fused to a modified form of the 19-kilodalton C-terminal fragment of merozoite surface protein 1. Infect Immun. 2007, 75: 5947-5955. 10.1128/IAI.01804-06.
Zhang D, Weiqing P: Evaluation of three Pichia pastoris-expressed Plasmodium falciparum merozoite proteins as a combination vaccine against infection with blood-stage parasites. Infect Immun. 2005, 73: 6530-6536. 10.1128/IAI.73.10.6530-6536.2005.
Zhang D, Xue X, Qu L, Pan W: Construction and evaluation of a multistage combination vaccine against malaria. Vaccine. 2007, 25: 2112-2119. 10.1016/j.vaccine.2006.11.015.
Tsai CW, Duggan PF, Shimp RL, Miller LH, Narum DL: Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J Biotechnol. 2006, 121: 458-470. 10.1016/j.jbiotec.2005.08.025.
Zhang Q, Ding F, Xue X, Xu X, Weiqing P: Changing the N-terminal sequence protects recombinant Plasmodium falciparum circumsporozoite protein from degradation in Pichia pastoris. Appl Microbiol Biotechnol. 2008, 78: 139-145. 10.1007/s00253-007-1294-0.
Stowers AW, Kennedy MC, Keegan BP, Saul A, Long CA, Miller LH: Vaccination of monkeys with recombinant Plasmodium falciparum apical membrane antigen 1 confers protection against blood-stage malaria. Infect Immun. 2002, 70: 6961-6967. 10.1128/IAI.70.12.6961-6967.2002.
Malkin EM, Diemert DJ, McArthur JH, Perrault JR, Miles AP, Giersing BK, Mullen GE, Orcutt A, Muratova O, Awkal M, Zhou H, Wang J, Stowers A, Long CA, Mahanty S, Miller LH, Saul A, Durbin AP: Phase 1 clinical trial of apical membrane antigen 1: an asexual blood-stage vaccine for Plasmodium falciparum malaria. Infect Immun. 2005, 73: 3677-3685. 10.1128/IAI.73.6.3677-3685.2005.
Arakawa T, Komesu A, Otsuki H, Sattabongkot J, Udomsangpetch R, Matsumoto Y, Tsuji N, Wu Y, Torii M, Tsuboi T: Nasal immunization with a malaria transmission-blocking vaccine candidate, Pfs25, induces complete protective immunity in mice against field isolates of Plasmodium falciparum. Infect Immun. 2005, 73: 7375-7380. 10.1128/IAI.73.11.7375-7380.2005.
Zhang ZG, Yu WG, Qiu WS, Zhao HM: Immunogenicity of C-terminus of Plasmodium falciparum merozoite surface protein 1 expressed as a non-glycosylated peptide in yeast. Acta Biochim Biophys Sin. 2006, 38: 403-409. 10.1111/j.1745-7270.2006.00178.x.
Wang J, Nguyen V, Glen J, Henderson B, Saul A, Miller LH: Improved yield of recombinant merozoite surface protein 3 (MSP3) from Pichia pastoris using chemically defined media. Biotechnol Bioeng. 2005, 90: 838-847. 10.1002/bit.20491.
Zhang H, Paguio M, Roepe PD: The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochem. 2004, 43: 8290-8296. 10.1021/bi049137i.
Zhang H, Howard EM, Roepe PD: Analysis of the antimalarial drug resistance protein Pfcrt expressed in yeast. J Biol Chem. 2002, 277: 49767-49775. 10.1074/jbc.M204005200.
Amoah LE, Lekostaj JK, Roepe PD: Heterologous expression and ATPase activity of mutant versus wild type PfMDR1 protein. Biochemistry. 2007, 46: 6060-6073. 10.1021/bi7002026.
Tan W, Gou DM, Tai E, Zhao YZ, Chow LMC: Functional reconstitution of purified chloroquine resistance membrane transporter expressed in yeast. Arch Biochem Biophys. 2006, 452: 119-128. 10.1016/j.abb.2006.06.017.
Kost TA, Condrey JP, Jarvis DL: Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnol. 2005, 23: 567-575. 10.1038/nbt1095.
Chang SP, Gibson HL, Lee-Ng CT, Barr PJ, Hui GS: A carboxyl-terminal fragment of Plasmodium falciparum gp195 expressed by a recombinant baculovirus induces antibodies that completely inhibit parasite growth. J Immunol. 1992, 149: 548-555.
Pizarro JC, Chitarra V, Verger D, Holm I, Pêtres S, Dartevelle S, Nato F, Longacre S, Bentley GA: Crystal structure of a Fab complex formed with PfMSP1-19, the C-terminal fragment of merozoite surface protein 1 from Plasmodium falciparum : a malaria vaccine candidate. J Mol Biol. 2003, 328: 1091-1103. 10.1016/S0022-2836(03)00376-0.
Murphy VF, Rowan WC, Page MJ, Holder AA: Expression of hybrid malaria antigens in insect cells and their engineering for correct folding and secretion. Parasitology. 1990, 100: 177-183.
Jacobs P, Massaer M, Heinderyckx M, Milican F, Gilles P, van Opstal O, Voet P, Gheysen D, Bollen A: Plasmodium falciparum : recombinant baculoviruses direct the expression of circumsporozoite proteins in Spodoptera frugiperda cell cultures. Mol Biol Rep. 1991, 15: 73-79. 10.1007/BF00364842.
Barfod L, Bernasconi NL, Dahlbäck M, Jarrossay D, Andersen PH, Salanti A, Ofori MF, Turner L, Resende M, Nielsen MA, Theander TG, Sallusto F, Lanzavecchia A, Hviid L: Human pregnancy-associated malaria-specific B cells target polymorphic, conformational epitopes in VAR2CSA. Mol Microbiol. 2007, 63: 335-347. 10.1111/j.1365-2958.2006.05503.x.
Daugherty JR, Murphy CI, Doros-Richert LA, Barbosa A, Kashala LO, Ballou WR, Snellings NJ, Ockenhouse CF, Lanar DE: Baculovirus-mediated expression of Plasmodium falciparum erythrocyte binding antigen 175 polypeptides and their recognition by human antibodies. Infect Immun. 1997, 65: 3631-3637.
Liang H, Narum DL, Fuhrmann SR, Luu T, Sim BK: A recombinant baculovirus-expressed Plasmodium falciparum receptor-binding domain of erythrocyte binding protein EBA-175 biologically mimics native protein. Infect Immun. 2000, 68: 3564-3568. 10.1128/IAI.68.6.3564-3568.2000.
Narum DL, Welling GW, Thomas AW: Ion-exchange-immunoaffinity purification of a recombinant baculovirus Plasmodium falciparum apical membrane antigen, PF83/AMA-1. J Chromatogr. 1993, 657: 357-363. 10.1016/0021-9673(93)80291-F.
Li J, Matsuoka H, Mitamura T, Horii T: Characterisation of proteases involved in the processing of Plasmodium falciparum serine repeat antigen (SERA). Mol Biochem Parasitol. 2002, 120: 177-186. 10.1016/S0166-6851(01)00452-2.
Teuscher F, Lowther J, Skinner-Adams TS, Spielmann T, Dixon MW, Stack CM, Donnelly S, Mucha A, Kafarski P, Vassiliou S, Gardiner DL, Dalton JP, Trenholme KR: The M18 aspartyl aminopeptidase of the human malaria parasite Plasmodium falciparum. J Biol Chem. 2007, 282: 30817-30826. 10.1074/jbc.M704938200.
Stack CM, Lowther J, Cunningham E, Donnelly S, Gardiner DL, Trenholme KR, Skinner-Adams TS, Teuscher F, Grembecka J, Mucha A, Kafarski P, Lua L, Bell A, Dalton JP: Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase. A protease involved in amino acid regulation with potential for antimalarial drug development. J Biol Chem. 2007, 282: 2069-2080. 10.1074/jbc.M609251200.
Jean L, Hackett F, Martin SR, Blackman MJ: Functional characterization of the propeptide of Plasmodium falciparum subtilisin-like protease-1. J Biol Chem. 2003, 278: 28572-28579. 10.1074/jbc.M303827200.
Yuda M, Yano K, Tsuboi T, Torii M, Chinzei Y: von Willebrand Factor A domain-related protein, a novel microneme protein of the malaria ookinete highly conserved throughout Plasmodium parasites. Mol Biochem Parasitol. 2001, 116: 65-72. 10.1016/S0166-6851(01)00304-8.
Kedees MH, Azzouz N, Gerold P, Shams-Eldin H, Iqbal J, Eckert V, Schwarz RT: Plasmodium falciparum : glycosylation status of Plasmodium falciparum circumsporozoite protein expressed in the baculovirus system. Exp Parasitol. 2002, 101: 64-68. 10.1016/S0014-4894(02)00030-9.
Gowda DC, Davidson EA: Protein glycosylation in the malaria parasite. Parasitol Today. 1999, 15: 147-152. 10.1016/S0169-4758(99)01412-X.
Pang ALY, Hashimoto CN, Tam LQ, Meng ZQ, Hui GSN, HO WKK: In Vivo Expression and immunological studies of the 42-Kilodalton carboxyl-terminal processing fragment of Plasmodium falciparum merozoite surface protein 1 in the Baculovirus-Silkworm system. Infect Immun. 2002, 70: 2772-2779. 10.1128/IAI.70.6.2772-2779.2002.
An Online Informatics Resource for Dictyostelium. [http://www.dictybase.org]
Naudé B, Brzostowski JA, Kimmel AR, Wellems TE: Dictyostelium discoideum expresses a malaria chloroquine resistance mechanism upon transfection with mutant, but not wild-type, Plasmodium falciparum transporter PfCRT. J Biol Chem. 2005, 280: 25596-25603. 10.1074/jbc.M503227200.
Muhia DK, Swales CA, Eckstein-Ludwig U, Saran S, Polley SD, Kelly JM, Schaap P, Krishna S, Baker DA: Multiple splice variants encode a novel adenylyl cyclase of possible plastid origin expressed in the sexual stage of the malaria parasite Plasmodium falciparum. J Biol Chem. 2003, 278: 22014-22022. 10.1074/jbc.M301639200.
Raymond CD, Beghdadi-Rais C, Roggero M, Duarte EA, Desponds C, Bernard M, Groux D, Matile H, Bron C, Corradin G, Fasel NJ: Anchoring of an immunogenic Plasmodium falciparum circumsporozoite protein to the surface of Dictyostelium discoideum. J Biol Chem. 1995, 270: 12941-12947. 10.1074/jbc.270.21.12941.
Szafranski K, Lehmann R, Parra G, Guigo R, Glöckner G: Gene organization features in A/T-rich organisms. J Mol Evol. 2005, 60: 90-98. 10.1007/s00239-004-0201-2.
Fasel N, Begdadi-Rais C, Bernard M, Bron C, Corradin G, Reymond CD: Dictyostelium discoideum as an expression host for the circumsporozoite protein of Plasmodium falciparum. Gene. 1992, 111: 157-163. 10.1016/0378-1119(92)90683-G.
Van Bemmelen MX, Beghdadi-Rais C, Desponds C, Vargas E, Herrera S, Reymond CD, Fasel N: Expression and one-step purification of Plasmodium proteins in Dictyostelium. Mol Biochem Parasitol. 2000, 111: 377-390. 10.1016/S0166-6851(00)00330-3.
Kim K, Weiss LM: Toxoplasma gondii : the model apicomplexan. Int J Parasitol. 2004, 34: 423-432. 10.1016/j.ijpara.2003.12.009.
Pinder JC, Fowler RE, Bannister LH, Dluzewski AR, Mitchell GH: Motile systems in malaria merozoites: how is the red blood cell invaded?. Parasitol Today. 2000, 16: 240-245. 10.1016/S0169-4758(00)01664-1.
Yang M, Coppens I, Wormsley S, Baevova P, Hoppe HC, Joiner KA: The Plasmodium falciparum Vps4 homolog mediates multivesicular body formation. J Cell Sci. 2004, 117: 3831-3838. 10.1242/jcs.01237.
Di Cristina M, Ghouze F, Kocken CH, Naitza S, Cellini P, Soldati D, Thomas AW, Crisanti A: Transformed Toxoplasma gondii tachyzoites expressing the circumsporozoite protein of Plasmodium knowlesi elicit a specific immune response in Rhesus monkeys. Infect Immun. 1999, 67: 1677-1682.
Charest H, Sedegah M, Yap GS, Gazzinelli RT, Caspar P, Hoffman SL, Sher A: Recombinant attenuated Toxoplasma gondii circumsporozoite protein provides highly effective priming for CD8+ T cell-dependent protective immunity against malaria. J Immunol. 2000, 165: 2084-2092.
Diaz CA, Allocco J, Powles MA, Yeung L, Donald RGK, Anderson JW, Liberator PA: Characterization of Plasmodium falciparum cGMP-dependent protein kinase (PfPKG): Antiparasitic activity of a PKG inhibitor. Mol Biochem Parasitol. 2006, 146: 78-88. 10.1016/j.molbiopara.2005.10.020.
Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH: Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science. 1994, 264: 1941-1944. 10.1126/science.8009226.
Tolia NH, Enemark EJ, Sim BKL, Joshua-Tor L: Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005, 122: 183-193. 10.1016/j.cell.2005.05.033.
Kato K, Mayer DCG, Singh S, Reid M, Miller LH: Domain III of Plasmodium falciparum apical membrane antigen 1 binds to the erythrocyte membrane protein Kx. Proc Natl Acad Sci USA. 2005, 102: 5552-5557. 10.1073/pnas.0501594102.
Fraser TS, Kappe SHI, Narum DL, VanBuskirk KM, Adams JH: Erythrocyte-binding activity of Plasmodium yoelii apical membrane antigen-1 expressed on the surface of transfected COS-7 cells. Mol Biochem Parasitol. 2001, 117: 49-59. 10.1016/S0166-6851(01)00326-7.
VanBuskirk KM, Sevova E, Adams JH: Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc Natl Acad Sci USA. 2004, 101: 15754-15759. 10.1073/pnas.0405421101.
Han HJ, Park SG, Kim SH, Hwang SY, Han J, Traicoff J, Kho WG, Chung JY: Epidermal growth factor-like motifs 1 and 2 of Plasmodium vivax merozoite surface protein 1 are critical domains in erythrocyte invasion. Biochem Biophys Res Comm. 2004, 320: 563-570. 10.1016/j.bbrc.2004.06.008.
Mayer DCG, Mu JB, Feng X, Su X-z, Miller LH: Polymorphism in a Plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J Exp Med. 2002, 196: 1523-1528. 10.1084/jem.20020750.
Mayer DCG, Mu JB, Kanedo O, Duan J, Su X-z, Miller LH: Polymorphism in the Plasmodium falciparum erythrocyte-binding ligand JESEBL/EBA-181 alters its receptor specificity. Proc natl Acad Sci USA. 2004, 101: 2518-2523. 10.1073/pnas.0307318101.
Wickramarachchi T, Devi YS, Mohmmed A, Chauhan VS: Identification and characterization of a novel Plasmodium falciparum merozoite apical protein involved in erythrocyte binding and invasion. PLoS ONE. 2008, 3: e1732-10.1371/journal.pone.0001732.
Gamain B, Smith JD, Avril M, Baruch DI, Scherf A, Gysin J, Miller LH: Identification of a 67-amino acid region of the Plasmodium falciparum variant surface antigen that binds chondroitin sulphate A and elicits antibodies reactive with the surface of placental isolates. Mol Microbiol. 2004, 53: 445-455. 10.1111/j.1365-2958.2004.04145.x.
Smith JD, Kyes S, Craig AG, Fagan T, Hudson-Taylor D, Miller LH, Baruch DI, Newbold CI: Analysis of adhesive domains from the A4VAR Plasmodium falciparum erythrocyte membrane proten-1 identifies a CD36 binding domain. Mol Biochem Parasitol. 1998, 97: 133-148. 10.1016/S0166-6851(98)00145-5.
Smith JD, Craig AG, Kriek N, Hudson-Taylor D, Kyes S, Fagen T, Pinches R, Baruch DI, Newbold CI, Miller LH: Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: a parasite adhesion trait implicated in cerebral malaria. Proc Natl Acad Sci USA. 2000, 97: 1766-1771. 10.1073/pnas.040545897.
Russell C, Mercereau-Puijalon O, Le Scanf C, Steward M, Arnot DE: Further definition of PfEMP-1 DBL-1α domains mediating rosetting adhesion of Plasmodium falciparum. Mol Biochem Parasitol. 2005, 144: 109-113. 10.1016/j.molbiopara.2005.06.009.
Gamain B, Miller LH, Baruch DI: The surface variant antigens of Plasmodium falciparum contain cross-reactive epitopes. Proc Natl Acad Sci USA. 2001, 98: 2664-2669. 10.1073/pnas.041602598.
Ohas EA, Adams JH, Waitumbi JN, Orago ASS, Barbosa A, Lanar DE, Stoute JA: Measurement of antibody levels against region II of the erythrocyte-binding antigen 175 of Plasmodium falciparum in an area of malaria holoendemicity in western Kenya. Infect Immun. 2004, 72: 735-741. 10.1128/IAI.72.2.735-741.2004.
Collins CR, Withers-Martinez C, Bentley GA, Batchelor AH, Thomas AW, Blackman MJ: Fine mapping of an epitope recognized by an invasion-inhibitory monoclonal antibody on the malaria vaccine candidate apical membrane antigen 1. J Biol Chem. 2007, 282: 7431-7441. 10.1074/jbc.M610562200.
Rainczuk A, Scorza T, Spithill TW, Smooker PM: A bicistronic DNA vaccine containing apical membrane antigen 1 and merozoite surface protein 4/5 can prime humoral and cellular immune responses and partially protect mice against virulent Plasmodium chabaudi adami DS malaria. Infect Immun. 2004, 72: 5565-5573. 10.1128/IAI.72.10.5565-5573.2004.
Qazi KR, Wikman M, Vasconcelos NM, Berzins K, Ståhl S, Fernández C: Enhancement of DNA vaccine potency by linkage of Plasmodium falciparum malarial antigen gene fused with a fragment of HSP70 gene. Vaccine. 23: 1114-1125. 10.1016/j.vaccine.2004.08.033.
Fanning SL, Czesny B, Sedegah M, Carucci DJ, van Gemert GJ, Eling W, Williamson KC: A glycosylphosphatidylinositol anchor signal sequence enhances the immunogenicity of a DNA vaccine encoding Plasmodium falciparum sexual-stage antigen, Pfs230. Vaccine. 2003, 21: 3228-3235. 10.1016/S0264-410X(03)00265-2.
Sim BKL, Narum DL, Liang H, Fuhrman SR, Obaldia N, Gramzinski R, Aguiar J, Haynes JD, Moch JK, Hoffman SL: Induction of biologically active antibodies in mice, rabbits, and monkeys by Plasmodium falciparum EBA-175 region II DNA vaccine. Mol Med. 2001, 7: 247-254.
Martin D, Gannoun-Zaki L, Bonnefoy S, Eldin P, Wengelnik K, Vial H: Characterization of Plasmodium falciparum CDP-diacylglycerol synthase, a proteolytically cleaved enzyme. Mol Biochem Parasitol. 2000, 110: 93-105. 10.1016/S0166-6851(00)00260-7.
Kim YU, Hong Y: Functional analysis of the first mannosyltransferase (PIG-M) involved in glycosylphosphatidylinositol synthesis in Plasmodium falciparum. Mol Cells. 2007, 24: 294-300.
Benedetti CE, Kobarg J, Pertinhez TA, Gatti RM, de Souza OM, Spisni A, Meneghini R: Plasmodium falciparum histidine-rich protein II binds to actin, phosphatidylinositol 4,5-bisphosphate and erythrocyte ghosts in a pH-dependent manner and undergoes coil-to-helix transitions in anionic micelles. Mol Biochem Parasitol. 2003, 128: 157-166. 10.1016/S0166-6851(03)00057-4.
Van Es HHG, Karcz S, Chu F, Cowman AF, Vidal S, Gros P, Schurr E: Expression of the plasmodial pfmdr1 gene in mammalian cells is associated with increased susceptibility to chloroquine. Mol Cell Biol. 1994, 14: 2419-2428.
Reeves DC, Liebelt DA, Lakshmanan V, Roepe PD, Fidock DA, Akabas MH: Chloroquine-resistant isoforms of the Plasmodium falciparum chloroquine resistance transporter acidify lysosomal pH in HEK293 cells more than chloroquine-sensitive isoforms. Mol Biochem Parasitol. 2006, 150: 288-299. 10.1016/j.molbiopara.2006.09.001.
Penny JI, Hall ST, Woodrow CJ, Cowan GM, Gero AM, Krishna S: Expression of substrate-specific transporters encoded by Plasmodium falciparum in Xenopus laevis oocytes. Mol Biochem Parasitol. 1998, 93: 81-89. 10.1016/S0166-6851(98)00024-3.
Woodrow CJ, Penny JI, Krishna S: Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem. 1999, 274: 7272-7277. 10.1074/jbc.274.11.7272.
Manning SK, Woodrow C, Zuniga FA, Iserovich P, Fischbarg J, Louw AI, Krishna S: Mutational analysis of the hexose transporter of Plasmodium falciparum and development of a three-dimensional model. J Biol Chem. 2002, 277: 30942-30949. 10.1074/jbc.M204337200.
Joët T, Holterman L, Stedman TT, Kocken CHM, Wel van der A, Thomas AW, Krishna S: Comparative characterization of hexose transporters of Plasmodium knowlesi, Plasmodium yoelii and Toxoplasma gondii highlights functional differences within the apicomplexan family. Biochem J. 2002, 368: 923-929. 10.1042/BJ20021189.
Fayolle M, Ionita M, Krishna S, Morin C, Patel AP: Probing structure/affinity relationships for the Plasmodium falciparum hexose transporter with glucose derivatives. Bioorg Med Chem Lett. 2006, 16: 1267-1271. 10.1016/j.bmcl.2005.11.068.
Downie MJ, Saliba KJ, Bröer S, Howitt SM, Kirk K: Purine nucleobase transport in the intraerythrocytic malaria parasite. Int J Parasitol. 2008, 38: 203-209. 10.1016/j.ijpara.2007.07.005.
Carter NS, Ben Mamoun C, Liu W, Silva EO, Landfear SM, Goldberg DE, Ullman B: Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J Biol Chem. 2000, 275: 10683-10691. 10.1074/jbc.275.14.10683.
Parker MD, Hyde RJ, McRobert L, Cass CE, Young JD, McConkey GA, Baldwin SA: Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem J. 2000, 349: 67-75. 10.1042/0264-6021:3490067.
Downie MJ, Saliba KJ, Howitt SM, Bröer S, Kirk K: Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1. Mol Microbiol. 2006, 60: 738-748. 10.1111/j.1365-2958.2006.05125.x.
Saliba KJ, Martin RE, Bröer A, Henry RI, McCarthy S, Downie MJ, Allen RJW, Mullin KA, McFadden GI, Bröer S, Kirk K: Sodium-dependent uptake of inorganic phosphate by the intracellular malaria parasite. Nature. 2006, 443: 582-585.
Hansen M, Kun JFJ, Schultz JE, Beitz E: A single, bi-functional aquaglyceroporin in blood-stage Plasmodium falciparum malaria parasites. J Biol Chem. 2002, 277: 4874-4882. 10.1074/jbc.M110683200.
Promeneur D, Liu Y, Maciel J, Agre P, King LS, Kumar N: Aquaglyceroporin PbAQP during intraerythrocytic development of the malaria parasite Plasmodium berghei. Proc Natl Acad Sci USA. 2007, 104: 2211-2216. 10.1073/pnas.0610843104.
Eckstein-Ludwig U, Webb RJ, van Goethem DA, East JM, Lee AG, Kimura M, O'Neill PM, Bray PG, Ward SA, Krishna S: Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003, 424: 957-961. 10.1038/nature01813.
Krishna S, Woodrow C, Webb R, Penny J, Takeyasu K, Kimura M, East JM: Expression and functional characterization of a Plasmodium falciparum Ca2+-ATPase (PfATP4) belonging to a subclass unique to apicomplexan organisms. J Biol Chem. 2001, 276: 10782-10787. 10.1074/jbc.M010554200.
Nessler S, Friedrich O, Bakouh N, Fink RHA, Sanchez CP, Planelles G, Lanzer M: Evidence for activation of endogenous transporters in Xenopus laevis oocytes expressing the Plasmodium falciparum chloroquine resistance transporter, PfCRT. J Biol Chem. 2004, 279: 39438-39446. 10.1074/jbc.M404671200.
Daher W, Browaeys E, Pierrot C, Jouin H, Dive D, Meurice E, Dissous C, Capron M, Tomavo S, Doerig C, Cailliau K, Khalife J: Regulation of protein phosphatase type 1 and cell cycle progression by PfLRR1, a novel leucine-rich repeat protein of the human malaria parasite Plasmodium falciparum. Mol Microbiol. 2006, 60: 578-590. 10.1111/j.1365-2958.2006.05119.x.
Ghosh S, Malhotra P, Lalitha PV, Guha-Mukherjee S, Chauhan VS: Expression of Plasmodium falciparum C-terminal region of merozoite surface protein (PfMSP119), a potential malaria vaccine candidate in tobacco. Plant Science. 2002, 162: 335-343. 10.1016/S0168-9452(01)00555-6.
Wang L, Richie TL, Stowers A, Hanh Nhan D, Coppel RL: Naturally acquired antibody responses to Plasmodium falciparum merozoite surface protein 4 in a population living in an area of endemicity in Vietnam. Infect Immun. 2001, 69: 4390-4397. 10.1128/IAI.69.7.4390-4397.2001.
Shimizu Y, Kumura Y, Ying BW, Umekage S, Ueda T: Cell-free translation systems for protein engineering. FEBS J. 2006, 273: 4133-4140. 10.1111/j.1742-4658.2006.05431.x.
Vinarov DA, Loushin CL, Newman L, Markley JL: Wheat germ cell-free platform for eukaryotic protein production. FEBS J. 2006, 273: 4160-4169. 10.1111/j.1742-4658.2006.05434.x.
Mudeppa DG, Pang CKT, Tsuboi T, Endo E, Buckner FS, Varani G, PK R: Cell-free production of functional Plasmodium falciparum dihydrofolate reductase-thymidylate synthase. Mol Biochem Parasitol. 2007, 151: 216-219. 10.1016/j.molbiopara.2006.10.016.
Tsuboi T, Takeo S, Iriko H, Jin L, Tsuchimochi M, Matsuda s, Han ET, Otsuki H, Kaneko O, Sattabongkot J, Udomsangpetch R, Sawasaki T, Torii M, Endo Y: Wheat germ cell-free system based production of malaria proteins for discovery of Novel Vaccine candidates. Infect Immun. 2008, 76: 1702-1708. 10.1128/IAI.01539-07.
Bhalu B, Adams JH: Advancements in transfection technologies for Plasmodium. Int J Parasitol. 2007, 37: 1-10. 10.1016/j.ijpara.2006.10.001.
Siau A, Touré FS, Ouwe-Missi-Oukem-Boyer O, Cicéron L, Mahmoudi N, Vaquero C, Froissard P, Bisvigou U, Bisser S, Coppée JY, Bischoff E, David PH, Mazier D: Whole-transcriptome analysis of Plasmodium falciparum field isolates: identification of new pathogenicity factors. J Infect Dis. 2007, 196: 1603-1612. 10.1086/522012.
Agop-Nersesian C, Pfahler J, Lanzer M, Meissner M: Functional expression of ribozymes in Apicomplexa: towards exogenous control of gene expression by inducible RNA-cleavage. Int J Parasitol. 2008, 38: 673-681. 10.1016/j.ijpara.2007.10.015.
Bracchi-Ricard V, Moe D, Chakrabarti D: Two Plasmodium falciparum ribonucleotide reductase small subunits, PfR2 and PfR4, interact with each other and are components of the in vivo enzyme complex. J Mol Biol. 2005, 347: 749-759. 10.1016/j.jmb.2005.01.051.
Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA: Malarial dihydroorotate dehydrogenase substrate inhibitor and specificity. J Biol Chem. 2002, 277: 41827-41834. 10.1074/jbc.M206854200.
Barik S, Taylor RE, Chakrabarti D: Identification, cloning, and mutational analysis of the casein kinase 1 cDNA of the malaria parasite, Plasmodium falciparum. J Biol Chem. 1997, 272: 26132-26138. 10.1074/jbc.272.42.26132.
Faber PM, Arscott LD, Williams CH, Becker K, Schirmer RH: Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett. 1998, 422: 311-314. 10.1016/S0014-5793(98)00031-3.
Kanzok SM, Schirmer RH, Turbachova I, Iozef R, Becker K: The thioredoxin system of the malaria parasite Plasmodium falciparum. J Biol Chem. 2000, 275: 40180-40186. 10.1074/jbc.M007633200.
Holton S, Merckx A, Burgess D, Doerig C, Noble M, Endicott J: Structures of P. falciparum PfPK5 test the CKD regulation paradigm and suggest mechnisms of small molecule inhibition. Structure. 2003, 11: 1329-1337. 10.1016/j.str.2003.09.020.
Droucheau E, Primot A, Thomas V, Mattei D, Knockaert M, Richardson C, Salllicandro P, Alano P, Jafarshad A, Baratte B, Kunick C, Parzy D, Pearl L, Doerig C, Meijer L: Plasmodium falciparum glycogen synthase kinase-3: molecular model, expression, intracellular localisation and selective inhibitors. Biochim Biophys Acta. 2004, 1697: 181-196.
Kumar A, Vaid A, Syin C, Sharma P: PfPKB, a Novel Protein Kinase B-like Enzyme from Plasmodium falciparum. J Biol Chem. 2004, 279: 24255-24264. 10.1074/jbc.M312855200.
Li CM, Tyler PC, Furneaux RH, Kicska G, Xu Y, Grubmeyer C, Girvin ME, Schramm VL: Transition-state analogs as inhibitors of human and malarial hypoxanthine-guanine phosphoribosyltransferases. Nat Struct Biol. 1999, 6 (6): 582-587. 10.1038/9367.
Ridley RG, White JH, McAleese SM, Goman M, Alano P, de Vries E, Kilbey BJ: DNA polymerase δ: gene sequences from Plasmodium falciparum indicate that this enzyme is more highly conserved than DNA polymerase α. Nucleic Acids Res. 1991, 24: 6731-6736. 10.1093/nar/19.24.6731.
Muller S, Da'dara A, Luersen K, Wrenger C, Das Gupta R, Madhubala R, Walter RD: In the human parasite Plasmodium falciparum, polyamines are synthesized by a bifunctional ornithine decarboxylase, S-adenosylmethionine decarboxylase. J Biol Chem. 2000, 275: 8097-8102. 10.1074/jbc.275.11.8097.
Shoemark DK, Cliff MJ, Sessions RB, Clarke AR: Enzymatic properties of the lactate dehydrogenase enzyme from Plasmodium falciparum. FEBS J. 2007, 274: 2738-2748. 10.1111/j.1742-4658.2007.05808.x.
Berwal R, Gopalan N, Chandel K, Prasad GBKS, Prakash S: Plasmodium falciparum : Enhanced soluble expression, purification and biochemical characterization of lactate dehydrogenase. Exp Parasitol. 2008
Jayalakshmi R, Sumathy K, Balaram H: Purification and Characterization of Recombinant Plasmodium falciparum Adenylosuccinate Synthetase Expressed in Escherichia coli. Protein Expr Purif. 2002, 25: 65-72. 10.1006/prep.2001.1610.
Chiang PH, Chamberlin ME, Nicholson D, Soubes S, Su XZ, Subramanian G, Lanar DE, Prigge ST, Scovill JP, Miller LH, Chou JY: Molecular characterization of Plasmodium falciparum S-adenosylmethionine synthetase. Biochem J. 1999, 344: 571-576. 10.1042/0264-6021:3440571.
Prade L, Jones AF, Boss C, Richard-Bilstein S, Meyer S, Binkert C, Bur D: X-ray structure of Plasmepsin II complexed with a potent achiral inhibitor. J Biol Chem. 2005, 280: 23837-23843. 10.1074/jbc.M501519200.
Gunaratne RS, Sajid M, Ling IT, Tripathi R, Pachebat JA, Holder AA: Characterization of N-myristoyltransferase from Plasmodium falciparum. Biochem J. 2000, 348: 459-463. 10.1042/0264-6021:3480459.
Kumar R, Musiyenko A, Barik S: The heat shock protein 90 of Plasmodium falciparum and antimalarial activity of its inhibitor, geldanamycin. Malaria J. 2003, 2: 1-11. 10.1186/1475-2875-2-30.
Wagner JT, Ludemann H, Färber PM, Lottspeich F, Krauth-Siegel RL: Glutamate dehydrogenase, the marker protein of Plasmodium falciparum. Eur J Biochem. 1998, 258: 813-819. 10.1046/j.1432-1327.1998.2580813.x.
Werner C, Stubbs MT, Krauth-Siegel RL, Klebe G: The crystal structure of Plasmodium falciparum glutamate dehydrogenase, a putative target for novel antimalarial drugs. J Mol Biol. 2005, 349: 587-607. 10.1016/j.jmb.2005.03.077.
Ranie J, Kumar VP, Balaram H: Cloning of the triosephosphate isomerase gene of Plasmodium falciparum and expression in Escherichia coli. Mol Biochem Parasitol. 1993, 61: 159-169. 10.1016/0166-6851(93)90062-3.
Creedon KA, Pradipsinh KR, Wellems TE: Plasmodium falciparum S-Adenosylhomocysteine hydrolase. cDNA identification, predicted protein sequence and expression in Escherichia coli. J Biol Chem. 1994, 269: 16364-16370.
Tanaka N, Nakanishi M, Kusakabe Y, Shiraiwa K, Yabe S, Ito Y, Kitade Y, Nakamura KT: Crystal Structure of S-Adenosyl-l-Homocysteine Hydrolase from the Human Malaria Parasite Plasmodium falciparum. J Mol Biol. 2004, 343: 1007-1017. 10.1016/j.jmb.2004.08.104.
Muench SP, Rafferty JB, McLeod R, Rice DW, Prigge ST: Expression, purification and crystallization of the Plasmodium falciparum enoyl reductase. Acta Crystallogr D Biol Crystallogr. 2003, 59: 1246-1248. 10.1107/S0907444903008813.
Joshi MB, Lin DT, Chiang PH, Goldman ND, Fujioka H, Aikawa M, Syin C: Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol Biochem Parasitol. 1999, 99: 11-19. 10.1016/S0166-6851(98)00177-7.
Pal B, Pybus B, Muccio DD, Chattopadhyay : Biochemical characterization and crystallization of recombinant 3-phosphoglycerate kinase of Plasmodium falciparum. Biochim Biophys Acta. 2004, 1699: 277-280.
Acknowledgements
All authors are members of the South African Malaria Initiative and are listed alphabetically except for the corresponding author (SAMI: http://www.acgt.co.za/SAMI). The authors wish to acknowledge the Department of Science and Technology of South Africa for funding the SAMI programme. In addition, we acknowledge Colin Kenyon and Jaco de Ridder for expert opinions on heterologous plasmodial protein expressions and codon harmonization, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The South African Malaria Initiative http://www.acgt.co.za/SAMI was initiated with the purpose of integrating malaria research expertise in South Africa to accelerate novel drug and drug target discovery. All authors are members of the Functional Expression of malaria proteins Core Expertise Group of SAMI. AIL conceptualized the manuscript; all authors were involved in drafting the manuscript, particularly LB, EH who were also responsible for the codon harmonization, protein refolding, plant and cell free sections; GB, AS and LS were responsible for the chaperone section; TLC for the baculovirus section; HH for the Dictyostelium, Toxoplasma, mammalian cell and Xenopus sections; LO contributed to the yeast section; ZN coordinates the SAMI research effort; EJM chairs the SAMI steering committee.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Birkholtz, LM., Blatch, G., Coetzer, T.L. et al. Heterologous expression of plasmodial proteins for structural studies and functional annotation. Malar J 7, 197 (2008). https://doi.org/10.1186/1475-2875-7-197
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-7-197