
Abstract
Background
Plasmodium vivax continues to be the most widely distributed malarial parasite species in tropical and sub-tropical areas, causing high morbidity indices around the world. Better understanding of the proteins used by the parasite during the invasion of red blood cells is required to obtain an effective vaccine against this disease. This study describes characterizing the P. vivax asparagine-rich protein (Pv ARP) and examines its antigenicity in natural infection.
Methods
The target gene in the study was selected according to a previous in silico analysis using profile hidden Markov models which identified P. vivax proteins that play a possible role in invasion. Transcription of the arp gene in the P. vivax VCG-1 strain was here evaluated by RT-PCR. Specific human antibodies against Pv ARP were used to confirm protein expression by Western blot as well as its subcellular localization by immunofluorescence. Recognition of recombinant Pv ARP by sera from P. vivax- infected individuals was evaluated by ELISA.
Results
VCG-1 strain Pv ARP is a 281-residue-long molecule, which is encoded by a single exon and has an N-terminal secretion signal, as well as a tandem repeat region. This protein is expressed in mature schizonts and is located on the surface of merozoites, having an apparent accumulation towards their apical pole. Sera from P. vivax-infected patients recognized the recombinant, thereby suggesting that this protein is targeted by the immune response during infection.
Conclusions
This study showed the characterization of Pv ARP and its antigenicity. Further assays orientated towards evaluating this antigen’s functional importance during parasite invasion are being carried out.
Similar content being viewed by others

Background
Malaria is a tropical disease that causes millions of deaths per year around the world. The World Health Organization’s (WHO) Malaria Report 2011 indicated that there were 216 million cases and 655,000 deaths, mainly in children aged less than five years [1]. In spite of the incidence of cases worldwide and mortality index having become substantially reduced by 17% and 25% between 2000 and 2010, respectively, the figures regarding cases of malaria continue to be alarming. This is due to two main aspects impeding the total eradication of the disease: a gradual increase of parasite strains which are resistant to anti-malarial drugs [2] and populations of the mosquito vector which are insecticide-resistant [3].
Plasmodium vivax stands out as the most widespread parasite species causing malaria in humans; it is found throughout tropical and subtropical areas of the world and causes the disease’s highest morbidity indices on the Asian and American continents [4]. Even though it has been thought that P. vivax was a benign species, recent studies have shown that infection caused by this parasite could cause severe clinical symptoms [5, 6], similar to those found in Plasmodium falciparum infection, thereby making it a potential menace.
Synthetic vaccines have been considered a good choice among control strategies when combating infectious diseases. Regarding malarial blood stages, vaccine development has been focused on the recombinant expression of parasite antigens (MSP-1 [7–9] and AMA-1 [10, 11] having been the most studied) or on using synthetic peptides [12, 13]; however, no fully effective vaccine against any species has been reported to date.
Recent work has established that the key to achieving an effective vaccine lies in blocking the interaction of parasite ligands which facilitate adhesion to target cell receptors [14]; this means that molecules localized on parasite surface and apical organelles (rhoptries and micronemes) must be identified. Unfortunately, data regarding the P. vivax proteins involved in invasion of reticulocytes that have been functionally characterized to date lag behind that available for their P. falciparum counterparts [15]. The foregoing has been due to the difficulty of standardizing an in vitro culture given poor reticulocyte recovery from adult human total blood [16]. Such experimental limitation has led to several study alternatives having been suggested; probabilistic techniques have been most useful when predicting possible vaccine candidates. A recent study involving hidden Markov models for analyzing the transcriptome of the P. vivax Sal-1 strain’s intra-erythrocyte life-cycle has led to the identification of 45 proteins that play a potential role in invasion; the role in cell adhesion for 13 of them (localized in merozoite rhoptries or on their surface) had previously been determined [17]. It was particularly interesting that an asparagine-rich protein (ARP) was found, this being conserved throughout the Plasmodium genus [17]. Only its P. falciparum orthologue has been described to date, called the apical asparagine-rich protein (Pf AARP) [18]. The Pf AARP-encoding gene has a prominent expression pattern towards the last intra-erythrocyte parasite development stage (48 hours post-invasion), which has been shown by real-time PCR and Northern blot. Antigenicity assays have shown that the N-terminal protein’s region (Pf AARP-N) obtained as a recombinant is recognized by antibodies from patients who have been naturally infected by P. falciparum. Rabbit antibodies directed against Pf AARP-N have been able to significantly inhibit parasite invasion of RBC in vitro. The foregoing, together with an RBC binding assay involving the expression of the complete protein on COS cell surface, has highlighted this antigen’s functional role in parasite binding to and invasion of target cells [18].
The present study was thus aimed at characterizing the asparagine-rich protein orthologue for Pf AARP in P. vivax. Molecular biology assays and immunochemistry techniques were used to demonstrate Pvarp gene transcription, protein expression and localization, as well as the ability to induce an antigenic response in patients who had suffered episodes of P. vivax malaria.
Methods
Selecting the gene and designing the primers and synthetic peptides
Pv ARP was selected, bearing in mind the in silico study by Restrepo-Montoya et al.[17] of P. vivax proteins playing a potential role in invasion. The PlasmoDB [19] database was then scanned to obtain the Pvarp gene sequence from the Salvador 1 (Sal-1) reference strain and to analyze adjacent genes’ synteny in different Plasmodium species. Specific primers were designed manually using Gene Runner software (version 3.05). B-cell lineal epitopes were predicted with AntheProt software [20] using the deduced amino-acid (aa) sequence. A tBlastn analysis of the predicted B-cell epitopes was then carried out to select peptide sequences exclusive for the P. vivax ARP.
Animal handling
The experimental animals used were handled in accordance with Colombian Law 84/1989 and resolution 504/1996. Aotus monkeys kept at FIDIC’s primate station (Leticia, Amazon) were handled following established guidelines for the care and use of laboratory animals (National Institute of Health, USA) under the constant supervision of a primatologist. All experimental procedures involving Aotus monkeys had been previously approved by the Fundación Instituto de Inmunología's ethics committee and were carried out in agreement with the conditions stipulated by CorpoAmazonia (resolution 00066, 13 September, 2006). An Aotus monkey was experimentally infected with the Vivax Colombia Guaviare 1 (VCG-1) strain and monitored daily to assess infection progress throughout the entire study (up to day 18) using Acridine Orange staining. The monkey was treated with paediatric doses of chloroquine (10 mg/kg on the first day and 7.5 mg/kg/day until the fifth day) and primaquine (0.25 mg/kg/day from the third to the fifth day) at the end of the study to guarantee parasite clearance from total blood. Once experiments were over, CorpoAmazonia officers supervised the primate’s return to its natural habitat in excellent health.
Isolating the Plasmodium vivax parasite
VCG-1 strain parasites were maintained in vivo according to previously described methodology [21]. A P. vivax-infected blood sample (3 mL) was passed through a discontinuous Percoll gradient (GE Healthcare, Uppsala, Sweden) according to an already established protocol [22] for obtaining schizont-stage enriched parasite. The sample was then used as RNA, genomic DNA (gDNA) and total protein source.
Extracting RNA and cDNA synthesis
Total RNA was extracted from the schizont-enriched sample using the Trizol method and treated with RQ1 (RNA-qualified) RNase-free DNase (Promega, Wisconsin, USA) according to the manufacturer’s recommendations. Complementary DNA (cDNA) was synthesized using a SuperScript III enzyme (RT+) (Invitrogen, California, USA) in the following conditions: 65°C for 5 min, 50°C for 1 hour and 70°C for 15 min. An additional reaction without the SuperScript III enzyme (RT-) was made for use as control. Following 15 min’ incubation at 37°C with RNase (Promega, USA) the product was stored at −70°C until its later use.
Cloning, sequencing and bioinformatics analysis
The cDNA RT + and RT- samples, as well as the gDNA obtained using a DNA Wizard Genomic purification kit (Promega), were used as template in 10 μL PCR reactions containing 0.5 U/μL Accuzyme DNA polymerase (Bioline), 1x AccuBuffer, 2 mM MgCl2, 0.5 mM dNTP, 0.5 μM primers and DNAse-free water for completing the reaction volume. Specific primers were designed for amplifying a region containing the entire Pvarp gene (direct 5′- CATTTGATCAGAGACGAC -3′ and reverse 5′- TTGGCACTTTTGTCACGA -3′), or the encoding sequence without the signal peptide (direct 5′- atgTGCAACACAAATGGGAAAA -3′ and reverse 5′- CACGCCAAACAGCTTCA -3′); the protein expression start codon was included in the direct primer’s 5′ end. A set of primers which had been previously designed for amplifying the Pvron1-a region (direct 5′- atgGCGAAGGAGCCCAAGTG-3′ and reverse 5′- ATCCCTAGCAATGCTTCG -3′) [23] was used as control for cDNA contamination with gDNA. The PCR for the Pvarp gene began with a denaturing step at 95°C for 5 min, followed by 35 cycles at 95°C for 30 sec, 52°C for 10 sec and 72°C for 1 min. Pvron1-a PCR began with a denaturing step at 95°C for 5 min, followed by 35 cycles at 95°C for 30 sec, 56°C for 10 sec and 72°C for 1.5 min. A Wizard PCR preps kit (Promega) was used for purifying Pvarp gene amplicons obtained from independent PCRs done with the RT + sample, once quality had been evaluated by 1% agarose gel. Pure products were then ligated to the pEXP5 CT/TOPO expression vector and transformed in TOP10 Escherichia coli cells (Invitrogen). Various clones were grown to purify the plasmid, using an UltraClean mini plasmid prep purification kit (MO BIO laboratories, California, USA); insert integrity and its correct orientation were confirmed by sequencing using an ABI PRISM 310 genetic analyzer (PE Applied Biosystems, California, USA). VCG-1 strain Pv ARP was characterized in silico using SignalP 3.0 [24], FragAnchor [25], XSTREAM [26], tools and the Interpro database [27] to search for secretion signal or GPI-anchor sequences, tandem repeats and putative domains, respectively. Clustal W software was used for aligning genes and pertinent encoding sequences [28].
Recombinant protein expression and purification
The pEXP5-Pv ARP recombinant plasmid which encodes the entire Pv ARP sequence without the signal peptide (confirmed by sequencing) was transformed in E. coli BL21-AI (Invitrogen), according to the manufacturer’s recommendations. A protocol described by Sivashanmugam and his group [29] with some modifications, was used for improving expression yield. Briefly, the cells were grown overnight at 37°C in 10 mL Luria Bertani (LB) medium containing 100 μg/mL ampicillin and 0.1% (w/v) D-glucose. The initial inoculum was then seeded in 100 mL LB volume with the same amount of the aforementioned ampicillin and D-glucose and left to grow at 37°C using ~300 rpm until reaching 0.5 OD600; 0.2% L-arabinose (w/v) was used for five hours to induce expression. The culture was spun at 13,000 rpm for 30 min and lysed in extraction buffer (EB) (6 M urea, 12 mM imidazole, 10 mM Tris-Cl, 100 mM NaH2PO4 and 10 mg/mL lysozyme) supplemented with protease inhibitors (1 mM PMSF, 1 mM iodoacetamide, 1 mM EDTA and 1 mg/mL leupeptin). Pv ARP recombinant expression (rPv ARP) was verified by Western blot and the protein was then purified by solid-phase affinity chromatography using Ni+2-NTA resin (Qiagen, California, USA) following the manufacturer’s recommendations. Briefly, total lysate was incubated with the resin pre-equilibrated with EB overnight at 4°C. The rPv ARP mixture coupled to the resin was placed on a column and then washed several times with EB to eliminate weakly bound proteins. The recombinant protein was eluted with EB containing imidazole at differing concentrations (20, 100, 250 and 500 mM) in 3 mL fractions, which were analyzed by Coomassie blue staining to verify the presence of a single band and then dialyzed in PBS, pH 7.0. A micro BCA protein assay kit (Thermo Scientific) was used for quantifying every fraction so obtained; a bovine serum albumin (BSA) curve was used as reference.
Peptide synthesis and obtaining polyclonal antibodies
A 20 aa-long peptide (predicted to be a good B-cell epitope), located at the N-terminus of Pv ARP (CG- LDNLKAKESPSSNDDGVYAKG-GC), was synthesized according to a previously-established methodology [30], polymerized, lyophilized and characterized by RP-HPLC and MALDI-TOF MS. Five mg of peptide (called 38582 herein) were immobilized on a CNBr-activated Sepharose 4B column, according to the manufacturer’s recommendations. A pool of fifteen sera taken from patients who had suffered previous P. vivax malarial episodes (stored in FIDIC’s serum-bank, see the ‘Sample source’ section) was incubated with the peptide coupled to a Sepharose 4B column overnight at 4°C with constant shaking to purify specific antibodies against peptide 38582 (anti-Pv ARP38582). The retained antibodies were eluted with gradients of increasing salt concentration (50 mM-0.3 M NaCl); they were then dialyzed in PBS, pH 7.8, and stored at −20°C until use.
SDS-PAGE and Western blot
Five μg rPv ARP and 50 μg total parasite proteins were separated on 12% SDS-PAGE and then transferred to nitrocellulose membranes. After having been blocked with 5% skimmed milk in PBS-0.05% Tween for one hour, each membrane was cut into strips and individually analyzed as follows: strips with the recombinant protein were incubated for two hours at room temperature (RT) with anti-Pv ARP38582 serum fractions (1:100 dilution) in a solution of 5% skimmed milk in PBS-0.05% Tween to assess which of them contained anti-Pv ARP specific antibodies; one strip was incubated with an anti-histidine monoclonal antibody coupled to peroxidase (1:4,500) as positive control for Western blot. Serum fractions recognizing the recombinant protein were then used to detect Pv ARP in total parasite lysate in the aforementioned conditions. Once antibody reactivity had been eliminated by incubating anti-Pv ARP38582 serum with peptide 38582 for one hour at 37°C, then this solution was used as control. Following three washes with PBS-0.05% Tween (5 min per wash), the strips were incubated for one hour with phosphatase-conjugated goat anti-human IgG as secondary antibody (1:5,000) at RT. The blots were revealed with a VIP peroxidase (Vector Laboratories, Burlingame, Canada) or BCIP/NBT colour development substrate kits (Promega), according to the manufacturers’ indications.
Indirect immunofluorescence assay (IFA)
Plasmodium vivax-parasitized reticulocytes were washed thrice with PBS and then diluted in this solution until obtaining five to seven schizonts per field evaluated by staining with Acridine orange. Twenty μL of the sample were fed per well on eight-well multitest glass slides (Biomedicals, Inc) and the supernatant was removed 10 min later. Once the samples were dry, they were fixed with 4% formaldehyde for 5 min at RT. Following five washes with PBS, the sample was incubated with 1% Triton X-100 for 5 min in the previously described conditions. After 10-min blocking at RT with 1% (v/v) skimmed milk in PBS, each sample was incubated for one hour at RT with anti-Pv ARP38582 antibodies (20 μL). The samples were then incubated with FITC-conjugated anti-human IgG antibody (Sigma) at 1:30 dilution for 45 min in the dark. The DNA was stained with DAPI (0.5 μg/mL) for 10 min at RT and the excess was removed by washing several times with PBS-0.05% Tween. Once the slides had been examined under an Olympus BX51 florescence microscope (using 100× oil immersion objective), Volocity software (version 5.3.2) was used for superimposing the images.
Enzyme-linked immunosorbent assay (ELISA)
Pv ARP antigenicity was evaluated in triplicate using serum from patients who had been living in malaria-endemic areas in Colombia and had presented episodes of such infection. Sera taken from healthy individuals who had never suffered the disease were used as negative controls. Briefly, 96-well polysorb plates were covered with 1 μg/mL rPv ARP overnight at 4°C and then incubated at 37°C for one hour. The dishes were blocked with 200 μL 5% skimmed milk - PBS-0.05% Tween for one hour at 37°C. Antibody reactivity against the recombinant protein was evaluated by incubating the plates with a 1:100 dilution of each human serum in 5% skimmed milk -PBS-0.05% Tween for one hour at 37°C. Following incubation of the dishes with peroxidase-coupled anti-human IgG secondary antibody (1:10,000) diluted in 5% skimmed milk - PBS-0.05% Tween for one hour at 37°C, a peroxidase substrate solution (KPL Laboratories, WA, USA) was added to reveal the reaction, according to the manufacturer’s recommendations. Optical density (OD) was detected at 620 nm with an MJ ELISA multiskan reader and then calculated by subtracting the OD value obtained from the control well (no antigen). A 0.11 cut-off value for evaluating the positivity threshold was determined by taking the average of the OD plus twice the standard deviation (2 ± SD) of healthy individuals’ sera reactivity.
Statistical analysis
Differences in average OD for rPv ARP recognition by P. vivax-infected patients’ sera and in the control group were evaluated using the Kruskal-Wallis rank-sum test. A 0.05 significance level was used for testing a stated hypothesis.
Sample source
Sera were obtained from 38 patients who were living in malaria-endemic areas of Colombia and who had suffered previous episodes of P. vivax malaria (but not P. falciparum), as well as from 15 healthy individuals who had never been affected by the disease. All individuals signed an informed consent form after receiving detailed information regarding the study’s goals.
Accession number
The nucleotide and aa sequences used here have been reported in the GenBank database, under accession number KC514070.
Results and discussion
Analyzing the arp gene in Plasmodium species
The P. vivax proteins identified as playing a potential role in invasion by profile hidden Markov models [17] led to Pv ARP being selected. According to the information provided by the PlasmoDB database, the Pvarp gene (access number: PVX_090210) was found to be located between base pairs 1,230,371 and 1,231,228 in chromosome 5 of the Sal-1 strain. Similar genes were also found in the genome of other Plasmodium species known to be causing malaria in humans (P. falciparum and Plasmodium knowlesi), apes (Plasmodium cynomolgi) and rodents (Plasmodium berghei, Plasmodium yoelii and Plasmodium chabaudi). When analyzing alignment, the Pvarp gene codified product was 61.19%, identical to its orthologue in P. knowlesi (PKH_052690), 53.15% to its orthologue in P. cynomolgi (PCYB_053680) and 33.68% to its orthologue in P. falciparum (PF3D7_0423400), while identity ranged from 23.61% to 22.22% regarding orthologues in P. chabaudi (PCHAS_052400), P. yoelii (PY06454) and P. berghei (PBANKA_052380). Such genes were located in a syntenic region, as corroborated by their open reading frame orientation and exon-intron structure. The foregoing supported the idea that the Pvarp gene has been derived from a common ancestor; however, experimental evidence concerning the functional role that the encoded protein might have in different parasite species remains to be determined.
The Pvarp gene is transcribed in schizonts
The presence of Pvarp gene transcripts in the P. vivax VCG-1 strain was confirmed by PCR using the cDNA from a parasite sample as template. Figure 1 shows the Pvarp gene amplification products (excluding the signal peptide-encoding region) (lanes 2–4) and the Pvron1 gene’s a region (lanes 5–8) from cDNA and gDNA. A ~810 bp band (Figure 1; lane 2) obtained from cDNA amplification (RT+) showed that the Pvarp gene was transcribed in the schizont-enriched sample, similar to that reported in the transcriptional profile for the Sal-1 strain showing a maximum transcription level after 35 hours of intra-erythrocyte life cycle [31]. It was also confirmed that the Pvarp gene was encoded by a single exon once the sequences obtained from cDNA and gDNA products (Figure 1; lanes 2 and 4) had been aligned. The presence of a single ~1,053 bp band in Pvron1-a PCR (Figure 1; lane 6) indicated that the cDNA had not been contaminated by gDNA given that the expected product for the latter would have been ~1,559 bp (Figure 1; lane 8). No amplification was observed in the negative controls for each PCR (Figure 1; lanes 3 and 7 (RT-), and 5 (DNA-free water)).

Pvarp gene transcription during blood stage. Lane 1 indicates the molecular weight marker. Lanes 2–4 show Pvarp gene amplification using cDNA RT+, RT- and gDNA, respectively. Lanes 6–8 show the amplification of the Pvron1-a region using cDNA RT+, RT- and gDNA as template, respectively. Lane 5 shows the negative control for amplifying the Pvarp gene.
Comparing Aotus monkey-adapted VCG-1 strain Pvarp gene sequences to those from the Sal-1 reference strain led to identifying four synonymous mutations, two non-synonymous ones producing aa changes (i e, methionine (M) for asparagine (N) and glycine (G) for N in aa position 217 and 219, respectively) and a 12-base pair deletion related to an asparagine-methionine-asparagine-glycine (NMNG) repeat block (Table 1). It has been found that parasite proteins have both highly polymorphic and conserved regions; the former are the target for an immune response while conserved sequences implicated in interaction with cell receptors are usually not antigenic [32]. Considering that the latter regions might be suitable targets for blocking parasite entry to host cells, further studies aimed at evaluating Pvarp gene polymorphism in different isolates are required to determine which sequences could be used as components of a vaccine against malaria caused by P. vivax.
Characterizing Pv ARP in silico
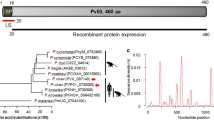
The VCG-1 strain Pvarp gene encoded a 281 aa long protein having ~30 kDa molecular mass, this being 64 residues longer when compared to its homologue Pf AARP (217 aa) [18]. Pv ARP consists of 20% asparagine residues and has a signal peptide with a cleavage site between aa TNG-KS (Figure 2). A post-translational modification false positive consisting of a C-terminal glycosylphosphatidylinositol (GPI) anchor sequence has been predicted [17], differing to its P. falciparum homologue which has a true positive one. Asparagine- and proline-rich regions were found towards the C-terminal extreme of the protein sequence; the first of these covered residues 212 to 235, while the another one was found downstream between aa 242 and 259 (Figure 2). Additionally, a tandem repeat region (TR), a feature shared with other vaccine candidates described to date, was also found using XSTREAM software [26] (Figure 2); this region consisted of 11 repeat blocks from the (D/N/S)(V/M)NG consensus sequence found in aa 168 to 211. The sequence was seen to be exclusive for P. vivax and had mutations (two substitutions and four deletions), thereby suggesting that it was under pressure from the immune system. TR have been common in several P. vivax antigens described to date, which are mainly located on the surface or in apical organelles; these would include the circumsporozoite protein (CSP) [33], merozoite surface protein 9 (MSP-9) [34], Pv 34 [35] and rhoptry neck proteins 1 and 2 [23, 36]. Even though several studies have shown that the tandem repeats of Pv CSP trigger an immune response when inoculated in primates and humans [33, 37, 38], the response so produced did not completely inhibit infection caused by the parasite. It has been shown in other Plasmodium species that TR could act as a smokescreen against the immune system, thereby diverting strong reactions towards functionally-relevant regions [39]; however, their exact role in P. vivax antigens remains unknown.

In silico characterization of Pv ARP, showing signal peptide localization, tandem repeats (TR), asparagine (ARR) and proline (PRR) amino acid repeat regions and the peptide selected for the antibody purification assay (shown in the box).
Pv ARP expression in schizonts and subcellular localization
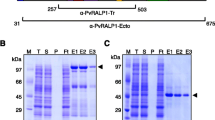
Specific human antibodies against an N-terminal Pv ARP synthetic peptide (Figure 2) were used for checking protein expression and localization in the schizont-enriched sample. Pv ARP was recombinantly expressed excluding the signal peptide and then purified (Figure 3A). Once human anti-Pv ARP38582 antibody ability to detect the recombinant protein in Western blot assays had been checked (Figure 3B), they were then used for detecting the protein on a blot containing parasite total lysate (Figure 3C). Both the parasite and recombinant Pv ARP proteins were detected above the expected weight (~40 and ~49 kDa, respectively), probably due to the presence of acidic aa (aspartic acid and glutamic acid) thereby causing anomalous migration on SDS-PAGE gel. The antibodies had specific reactivity to a ~40 kDa band; such reactivity was eliminated by using serum which had been pre-incubated with peptide 38582 (Figure 3C; lane 2).

Detecting recombinant and parasite protein by human antibodies. (A) Recombinant protein expression and purification. Lanes 2–3 show non-induced and induced cell lysate, respectively (Coomassie staining). Lanes 4–5 show purified rPv ARP stained with Coomassie or analyzed by Western blot using anti-polyhistidine antibodies, respectively. (B and C) Antibody ability to recognize recombinant and parasite Pv ARP by Western blot, respectively. Lane 2 shows the absence of human serum reactivity after being pre-incubated with peptide 38582. Lane 3 indicates Pv ARP recognition. Lane 4 shows detection of recombinant protein (positive control). MW kDa indicates molecular weight marker in kDa.
A strong fluorescence signal, having an apparent concentration towards the apical pole, was found on free merozoites’ surface and in mature schizonts when using the serum as primary antibody in the parasitized reticulocyte sample (Figure 4). The results led to the suggestion that Pv ARP could be expressed in apical organelles and then become relocated to the surface. However, other confocal or electron microscope assays are needed to determine the protein’s exact localization pattern.

Pv ARP sub-cellular localization in mature schizonts. (A) Shows the detection of the protein on free merozoite surface. (B) Pv ARP labelling on mature schizonts. The nuclei are labelled with DAPI (blue). An amplified image of a merozoite (indicated by an arrow) is shown in small boxes.
Antigenicity in humans
Pv ARP antigenic ability was evaluated by ELISA, using the sera from 38 patients who had suffered P. vivax malaria and 15 serum samples from people who had never suffered from the disease. The statistical test revealed a statistically significant difference between the medians (m) of the groups (Wilcoxon rank-sum test. Z = 5.1, p = 0.000); it gave m = 0.5 for the group of infected patients and m = 0.1 for the control group (Figure 5), thereby corroborating the fact that the protein was able to trigger an antibody response in the host during natural P. vivax malaria infection, most sera being able to recognize native and recombinant protein, as demonstrated by IFA and Western blot, respectively. The results supported the idea of analyzing this protein’s potential as a candidate for an anti-P. vivax vaccine.

r Pv ARP antigenicity. The box diagram shows OD distribution (Y axis) for detecting rPv ARP by sera from non-infected and infected individuals (X axis). *: Infected individuals (n = 38; X̄±S = 0.5 ± 0.2; 95%CI = 0.16-1.1) and control (n = 15; X̄±S = 0.1 ± 0.07; 95%IC = 0.03-0.24). p value = 0.000.
Conclusions
This study has described how the P. vivax asparagine-rich protein was characterized. As demonstrated, Pv ARP was conserved among different species belonging to the Plasmodium genus and shared some features of well-characterized surface and/or apical proteins being studied as candidates for a vaccine, such as prominent transcription and expression towards the end of the intra-erythrocyte life cycle and broad recognition by sera from patients infected with P. vivax malaria. The results supported the notion that this antigen could be a promising candidate for inclusion when developing an anti-malarial vaccine. Further immunogenicity assays and studies of the ability to induce protection in the experimental Aotus model are required.
References
WHO: World malaria report. 2011, Geneva: World Health Organization
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ: Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009, 361: 455-467. 10.1056/NEJMoa0808859.
Hunt RH, Fuseini G, Knowles S, Stiles-Ocran J, Verster R, Kaiser ML, Choi KS, Koekemoer LL, Coetzee M: Insecticide resistance in malaria vector mosquitoes at four localities in Ghana, West Africa. Parasit Vectors. 2011, 4: 107-10.1186/1756-3305-4-107.
Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, Kabaria CW, Tatem AJ, Manh BH, Elyazar IR, Baird JK, Snow RW, Hay SI: The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010, 4: e774-10.1371/journal.pntd.0000774.
Genton B, D’Acremont V, Rare L, Baea K, Reeder JC, Alpers MP, Muller I: Plasmodium vivax and mixed infections are associated with severe malaria in children: a prospective cohort study from Papua New Guinea. PLoS Med. 2008, 5: e127-10.1371/journal.pmed.0050127.
Tjitra E, Anstey NM, Sugiarto P, Warikar N, Kenangalem E, Karyana M, Lampah DA, Price RN: Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 2008, 5: e128-10.1371/journal.pmed.0050128.
Barrero CA, Delgado G, Sierra AY, Silva Y, Parra-Lopez C, Patarroyo MA: Gamma interferon levels and antibody production induced by two PvMSP-1 recombinant polypeptides are associated with protective immunity against P. vivax in Aotus monkeys. Vaccine. 2005, 23: 4048-4053. 10.1016/j.vaccine.2005.02.012.
Sierra AY, Barrero CA, Rodriguez R, Silva Y, Moncada C, Vanegas M, Patarroyo MA: Splenectomised and spleen intact Aotus monkeys’ immune response to Plasmodium vivax MSP-1 protein fragments and their high activity binding peptides. Vaccine. 2003, 21: 4133-4144. 10.1016/S0264-410X(03)00455-9.
Xue X, Ding F, Zhang Q, Pan X, Qu L, Pan W: Stability and potency of the Plasmodium falciparum MSP1-19/AMA-1(III) chimeric vaccine candidate with Montanide ISA720 adjuvant. Vaccine. 2010, 28: 3152-3158. 10.1016/j.vaccine.2010.02.054.
Gentil F, Bargieri DY, Leite JA, Francoso KS, Patricio MB, Espindola NM, Vaz AJ, Palatnik-de-Sousa CB, Rodrigues MM, Costa FT, Soares IS: A recombinant vaccine based on domain II of Plasmodium vivax Apical Membrane Antigen 1 induces high antibody titres in mice. Vaccine. 2010, 28: 6183-6190. 10.1016/j.vaccine.2010.07.017.
Dicko A, Diemert DJ, Sagara I, Sogoba M, Niambele MB, Assadou MH, Guindo O, Kamate B, Baby M, Sissoko M, Malkin EM, Fay MP, Thera MA, Miura K, Dolo A, Diallo DA, Mullen GE, Long CA, Saul A, Doumbo O, Miller LH: Impact of a Plasmodium falciparum AMA1 vaccine on antibody responses in adult Malians. PLoS One. 2007, 2: e1045-10.1371/journal.pone.0001045.
Herrera S, Bonelo A, Perlaza BL, Valencia AZ, Cifuentes C, Hurtado S, Quintero G, Lopez JA, Corradin G, Arevalo-Herrera M: Use of long synthetic peptides to study the antigenicity and immunogenicity of the Plasmodium vivax circumsporozoite protein. Int J Parasitol. 2004, 34: 1535-1546. 10.1016/j.ijpara.2004.10.009.
Patarroyo ME, Patarroyo MA: Emerging rules for subunit-based, multiantigenic, multistage chemically synthesized vaccines. Acc Chem Res. 2008, 41: 377-386. 10.1021/ar700120t.
Patarroyo ME, Bermudez A, Patarroyo MA: Structural and immunological principles leading to chemically synthesized, multiantigenic, multistage, minimal subunit-based vaccine development. Chem Rev. 2011, 111: 3459-3507. 10.1021/cr100223m.
Patarroyo MA, Calderon D, Moreno-Perez DA: Vaccines against Plasmodium vivax: a research challenge. Expert Rev Vaccines. 2012, 11: 1249-1260. 10.1586/erv.12.91.
Udomsangpetch R, Kaneko O, Chotivanich K, Sattabongkot J: Cultivation of Plasmodium vivax. Trends Parasitol. 2008, 24: 85-88. 10.1016/j.pt.2007.09.010.
Restrepo-Montoya D, Becerra D, Carvajal-Patino JG, Mongui A, Nino LF, Patarroyo ME, Patarroyo MA: Identification of Plasmodium vivax proteins with potential role in invasion using sequence redundancy reduction and profile hidden Markov models. PLoS One. 2011, 6: e25189-10.1371/journal.pone.0025189.
Wickramarachchi T, Devi YS, Mohmmed A, Chauhan VS: Identification and characterization of a novel Plasmodium falciparum merozoite apical protein involved in erythrocyte binding and invasion. PLoS One. 2008, 3: e1732-10.1371/journal.pone.0001732.
PlasmoDB: plasmodium genomics resource.http://www.plasmodb.org,
Deleage G, Combet C, Blanchet C, Geourjon C: ANTHEPROT: an integrated protein sequence analysis software with client/server capabilities. Comput Biol Med. 2001, 31: 259-267. 10.1016/S0010-4825(01)00008-7.
Pico de Coana Y, Rodriguez J, Guerrero E, Barrero C, Rodriguez R, Mendoza M, Patarroyo MA: A highly infective Plasmodium vivax strain adapted to Aotus monkeys: quantitative haematological and molecular determinations useful for P. vivax malaria vaccine development. Vaccine. 2003, 21: 3930-3937. 10.1016/S0264-410X(03)00278-0.
Andrysiak PM, Collins WE, Campbell GH: Concentration of Plasmodium ovale- and Plasmodium vivax-infected erythrocytes from nonhuman primate blood using Percoll gradients. Am J Trop Med Hyg. 1986, 35: 251-254.
Moreno-Perez DA, Montenegro M, Patarroyo ME, Patarroyo MA: Identification, characterization and antigenicity of the Plasmodium vivax rhoptry neck protein 1 (PvRON1). Malar J. 2011, 10: 314-10.1186/1475-2875-10-314.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340: 783-795. 10.1016/j.jmb.2004.05.028.
Poisson G, Chauve C, Chen X, Bergeron A: FragAnchor: a large-scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genomics Proteomics Bioinformatics. 2007, 5: 121-130. 10.1016/S1672-0229(07)60022-9.
Newman AM, Cooper JB: XSTREAM: a practical algorithm for identification and architecture modeling of tandem repeats in protein sequences. BMC Bioinformatics. 2007, 8: 382-10.1186/1471-2105-8-382.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJ, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C: InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, 37: D211-D215. 10.1093/nar/gkn785.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Sivashanmugam A, Murray V, Cui C, Zhang Y, Wang J, Li Q: Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 2009, 18: 936-948. 10.1002/pro.102.
Houghten RA: General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci U S A. 1985, 82: 5131-5135. 10.1073/pnas.82.15.5131.
Bozdech Z, Mok S, Hu G, Imwong M, Jaidee A, Russell B, Ginsburg H, Nosten F, Day NP, White NJ, Carlton JM, Preiser PR: The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci U S A. 2008, 105: 16290-16295. 10.1073/pnas.0807404105.
Rodriguez LE, Curtidor H, Urquiza M, Cifuentes G, Reyes C, Patarroyo ME: Intimate molecular interactions of P. falciparum merozoite proteins involved in invasion of red blood cells and their implications for vaccine design. Chem Rev. 2008, 108: 3656-3705. 10.1021/cr068407v.
Yang C, Collins WE, Xiao L, Saekhou AM, Reed RC, Nelson CO, Hunter RL, Jue DL, Fang S, Wohlhueter RM, Udhayakumar V, Lal AA: Induction of protective antibodies in Saimiri monkeys by immunization with a multiple antigen construct (MAC) containing the Plasmodium vivax circumsporozoite protein repeat region and a universal T helper epitope of tetanus toxin. Vaccine. 1997, 15: 377-386. 10.1016/S0264-410X(97)00200-4.
Oliveira-Ferreira J, Vargas-Serrato E, Barnwell JW, Moreno A, Galinski MR: Immunogenicity of Plasmodium vivax merozoite surface protein-9 recombinant proteins expressed in E. coli. Vaccine. 2004, 22: 2023-2030. 10.1016/j.vaccine.2003.07.021.
Mongui A, Angel DI, Gallego G, Reyes C, Martinez P, Guhl F, Patarroyo MA: Characterization and antigenicity of the promising vaccine candidate Plasmodium vivax 34 kDa rhoptry antigen (Pv34). Vaccine. 2009, 28: 415-421. 10.1016/j.vaccine.2009.10.034.
Arevalo-Pinzon G, Curtidor H, Patino LC, Patarroyo MA: PvRON2, a new Plasmodium vivax rhoptry neck antigen. Malar J. 2011, 10: 60-10.1186/1475-2875-10-60.
Herrera S, De Plata C, Gonzalez M, Perlaza BL, Bettens F, Corradin G, Arevalo-Herrera M: Antigenicity and immunogenicity of multiple antigen peptides (MAP) containing P. vivax CS epitopes in Aotus monkeys. Parasite Immunol. 1997, 19: 161-170. 10.1046/j.1365-3024.1997.d01-193.x.
Herrera S, Bonelo A, Perlaza BL, Fernandez OL, Victoria L, Lenis AM, Soto L, Hurtado H, Acuna LM, Velez JD, Palacios R, Chen-Mok M, Corradin G, Arevalo-Herrera M: Safety and elicitation of humoral and cellular responses in colombian malaria-naive volunteers by a Plasmodium vivax circumsporozoite protein-derived synthetic vaccine. Am J Trop Med Hyg. 2005, 73: 3-9.
Hisaeda H, Yasutomo K, Himeno K: Malaria: immune evasion by parasites. Int J Biochem Cell Biol. 2005, 37: 700-706. 10.1016/j.biocel.2004.10.009.
Acknowledgements
We would like to thank Daniela Prieto Borja for her technical support, Jason Garry for translating and reviewing this manuscript and especially Prof Manuel Elkin Patarroyo for his invaluable comments and suggestions. This research was supported by the “Instituto Colombiano para el Desarrollo de la Ciencia ‘Francisco José de Caldas” (COLCIENCIAS) contract RC#309-2013.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DAMP designed experiments, analyzed data and wrote the initial manuscript. AS carried out molecular biology and immunochemical assays. MAP designed, evaluated and coordinated the assays and corrected the final manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Moreno-Pérez, D.A., Saldarriaga, A. & Patarroyo, M.A. Characterizing Pv ARP, a novel Plasmodium vivax antigen. Malar J 12, 165 (2013). https://doi.org/10.1186/1475-2875-12-165
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-12-165