
Abstract
Background
Plasmodium vivax malaria remains a major health problem in tropical and sub-tropical regions worldwide. Several rhoptry proteins which are important for interaction with and/or invasion of red blood cells, such as Pf RONs, Pf 92, Pf 38, Pf 12 and Pf 34, have been described during the last few years and are being considered as potential anti-malarial vaccine candidates. This study describes the identification and characterization of the P. vivax rhoptry neck protein 1 (Pv RON1) and examine its antigenicity in natural P. vivax infections.
Methods
The Pv RON1 encoding gene, which is homologous to that encoding the P. falciparum apical sushi protein (ASP) according to the plasmoDB database, was selected as our study target. The pvron1 gene transcription was evaluated by RT-PCR using RNA obtained from the P. vivax VCG-1 strain. Two peptides derived from the deduced P. vivax Sal-I Pv RON1 sequence were synthesized and inoculated in rabbits for obtaining anti-Pv RON1 antibodies which were used to confirm the protein expression in VCG-1 strain schizonts along with its association with detergent-resistant microdomains (DRMs) by Western blot, and its localization by immunofluorescence assays. The antigenicity of the Pv RON1 protein was assessed using human sera from individuals previously exposed to P. vivax malaria by ELISA.
Results
In the P. vivax VCG-1 strain, RON1 is a 764 amino acid-long protein. In silico analysis has revealed that Pv RON1 shares essential characteristics with different antigens involved in invasion, such as the presence of a secretory signal, a GPI-anchor sequence and a putative sushi domain. The Pv RON1 protein is expressed in parasite's schizont stage, localized in rhoptry necks and it is associated with DRMs. Recombinant protein recognition by human sera indicates that this antigen can trigger an immune response during a natural infection with P. vivax.
Conclusions
This study shows the identification and characterization of the P. vivax rhoptry neck protein 1 in the VCG-1 strain. Taking into account that Pv RON1 shares several important characteristics with other Plasmodium antigens that play a functional role during RBC invasion and, as shown here, it is antigenic, it could be considered as a good vaccine candidate. Further studies aimed at assessing its immunogenicity and protection-inducing ability in the Aotus monkey model are thus recommended.
Similar content being viewed by others

Background
Malaria remains one of the prevailing health problems worldwide. According to the World Health Organization (WHO) [1], nearly 225 million people are infected annually; about 785,000 of them die as a direct consequence of this disease, of which 85% are children aged less than five years. Although malaria in humans is caused by Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae and Plasmodium knowlesi, the first two species represent about 90% of the clinical cases reported [2]. P. falciparum is responsible for causing the disease's highest mortality rates while P. vivax represents significant morbidity having socioeconomic implications [3]. In spite of international control strategies and policies having been implemented during the last fifty years, mortality figures are still alarming; therefore, developing an efficient vaccine to combat this imminent threat has become an urgent need.
Invasion of red blood cells (RBC) by Plasmodium parasites involves highly coordinated events which are directed by a set of proteins secreted from the apical organelles (rhoptries and micronemes) [4]. It has been shown that several rhoptry proteins, such as Pf RON2, -4, and the Pf AMA-1 antigen (secreted by micronemes), are involved in tight junction formation between the parasite and its target cell [5–7]; it has also been found that some others (such as Pf RON1, Pf 92, Pf 38, Pf 12 and Pf 34) are associated with detergent-resistant membrane microdomains (DRM) through glycosylphosphatidylinositol (GPI)-anchor sequences [8], which are considered organizing centers for the assembly of molecules implicated in cell signaling [9, 10]. To date, several of these DRM proteins have been shown to play an active role in host cell interaction and to trigger antibody responses in the host [11–15].
Rhoptry neck protein 1 (RON1), initially described in Toxoplasma gondii (Tg RON1) [16], has been a particularly interesting protein. It is a highly-conserved antigen amongst Apicomplexa members. Different tgron1 homologous genes have also been found in members of the Plasmodium genus, such as P. falciparum[16, 17]. Pf RON1 is also known as the apical sushi protein (ASP), exhibiting a prominent transcriptional peak towards the end of the intraerythrocyte lifecycle [17]. This protein has 731 amino acids encoded by 4 exons and ~85.46 kDa molecular mass. It has been previously demonstrated that Pf ASP undergoes proteolytic processing, resulting in ~50 kDa and ~30 kDa polypeptides [18]. The protein also contains a signal peptide, a GPI-anchor sequence and a complement control protein (CCP) or sushi-like conserved domain [19]; this domain typically contains ~60 residues and represents a protein module involved in protein-protein interactions and/or cell adhesion, containing 4 cysteines, proline residues and highly conserved acidic amino acids (aspartic and glutamic acid). Furthermore, it has been recently found that Pf RON1 has 4 high activity binding peptide sequences (HABPs) to erythrocytes (manuscript in preparation), and could thus be important for parasite entry to target cells.
In the search for an effective anti-malarial vaccine several studies have been focused on identifying antigens expressed during the parasite's asexual stage due to the fact that some of these antigens are mainly responsible for invasion of RBC [20, 21]. Several proteins included in developing a promising vaccine against P. falciparum have been functionally characterized to date [21]. However, studies carried out with P. vivax have been limited due to the difficulty in maintaining an in vitro continuous parasite culture. Taking this into account and considering the degree of conservation observed between the genetic content of different Plasmodium species [23, 24], some bioinformatics tools have been used for homology searches of the complete P. vivax genome for antigens already described in P. falciparum. To date, a considerable number of surface [25–28] and rhoptry proteins [22, 29–35] have been experimentally identified and characterized in P. vivax using both its genome [21] and intraerythrocyte lifecycle transcriptome [36] as well as an Aotus-adapted strain [37]. These proteins belong to a list of antigens, which are being tested as vaccine candidates in the Aotus monkey model.
This study shows the identification and characterization of the Pv RON1 protein which is homologous to Pf ASP. Different aspects such as Pv RON1 transcription and expression at the end of the intraerythrocyte life cycle, the subcellular localization pattern and the ability to trigger an immune response in patients who have presented active episodes of P. vivax malarial infection have been determined by molecular biology and immunochemistry techniques.
Methods
Bioinformatics analysis
The P. vivax ron1 gene sequence (which is homologous to the Pf ASP encoding gene) was obtained from the plasmoDB database [38]; likewise, pfasp orthologous genes in other malarial species were searched. Identity and similarity values between P. falciparum - P. vivax and P. vivax - P. knowlesi hypothetical protein products were determined using ClustalW software [39]. The presence of a signal peptide, repeat sequences and a GPI-anchor sequence was determined by SignalP 3.0 [40], XSTREAM repeat [41] and GPI-anchor [42] bioinformatics tools, respectively. Putative domains were assessed using the Interpro database [43]. Antheprot software [44] was used for determining two peptides throughout the Pv RON1 hypothetical sequence (PlasmoDB accession number: PVX_000945) that could be good B-cell epitopes, taking the highest average hydrophilicity (>4.9), solvent accessibility (>162) and Parker's antigenicity (>37.8) values into account (calculated in a 20 residue sliding window), according to previously established parameters [45–47].
Animal handling
The experimental handling of animals used here was carried out in accordance with Colombian Law 84/1989 and resolution 504/1996. Aotus monkeys kept at FIDIC's primate station (Leticia, Amazon) and New Zealand rabbits provided by the Instituto Nacional de Salud (Bogotá, Colombia) were handled following the guidelines for the care and use of laboratory animals (National Institute of Health, USA) under the constant supervision of a veterinarian. Immunization and bleeding procedures for Aotus monkeys had been previously approved by our institute's ethics committee, and were carried out in agreement with the conditions stipulated by CorpoAmazonia (resolution 00066, September 13th2006). A monkey from the Aotus genus was experimentally infected with the VCG-1 strain (Vivax Colombia Guaviare 1) and subjected to daily monitoring to assess the progress of infection throughout the whole study (up to day 18) using acridine orange staining. The monkey was treated with pediatric doses of chloroquine (10 mg/kg on the first day and 7.5 mg/kg/day until the fifth day) and primaquine (0.25 mg/kg/day from the third to the fifth day) at the end of the study to guarantee total blood parasite clearance. Once experiments were over, CorpoAmazonia officers supervised the primate's return to its natural habitat in excellent health conditions.
Isolating the P. vivax parasite
The VCG-1 strain was obtained according to a previously described methodology [37]. Mature forms of the parasite, mainly schizonts, were purified from a blood sample (3 mL) infected with P. vivax using a Percoll discontinuous gradient (GE Healthcare, Uppsala, Sweden), according to a previously established protocol [48]. This schizont-enriched sample was then used for performing all of the following procedures: RNA, genomic DNA or parasite total protein extraction, as well as indirect immunofluorescence assays.
RNA extraction and cDNA synthesis
Total RNA from the schizont-enriched sample was extracted using the Trizol method and treated with RNase-free RQ1 DNase (Promega, Wisconsin, USA), according to the manufacturer's instructions. 5 μL RNA were used to synthesize cDNA by RT-PCR, using the SuperScript III enzyme (Invitrogen, California, USA). Complementary DNA synthesis was carried out in the following conditions: 65°C for 5 mins, 50°C for 1 hour and 70°C for 15 mins. After a 15-min incubation period with RNase (Promega, USA) at 37°C, the product was stored at -20°C until its use.
Cloning and sequencing
Three specific primer sets were designed to cover the whole gene sequence and produce two smaller-sized recombinant fragments (Forward 5'-ACAGAAGAGAAGAGAACAGA-3', Reverse 5'-GTTCACACATGCGGTCAC-3'; Forward 5'- ATGGCGAAGGAGCCCAAGTG -3', Reverse 5'- ATCCCTAGCAATGCTTCG -3'; Forward 5'-ATGCTGCTAGTGCCACCCG-3', Reverse 5'- CTGAACACCATCGAAATCG -3'). After PCR amplification using the Platinum Pfx DNA polymerase enzyme (Invitrogen, California, USA), each product was purified by Wizard PCR preps kit (Promega), ligated to the pEXP5 CT/TOPO vector (Invitrogen) and cloned in Escherichia coli TOP10 bacteria (Invitrogen), following the manufacturer's instructions. Recombinant DNA was purified using an UltraClean mini plasmid prep purification kit (MO BIO laboratories, California, USA) and each sequence's integrity was confirmed by sequencing two clones obtained from independent PCRs, using the automated genetic analyzer ABI PRISM 310 (PE Applied Biosystems, California, USA).
Peptide synthesis and polyclonal antibody production
Two 20-amino-acid-long peptides designed on the Pv RON1 deduced sequence (Sal-1 strain) (CG-KHKEGGAAKRHKKEPHEQRG-GC and CG-EDASVADKDGQPGERGQDGQ-GC) were synthesized according to a previously established methodology [49], polymerized, lyophilized and characterized by RP-HPLC and MALDI-TOF MS. Afterwards, a 150 μg dose of a mixture from both synthetic peptides, emulsified in Freund's complete adjuvant (FCA) (Sigma, Missouri, USA), was inoculated into New Zealand rabbits on day 0, while the same mixture emulsified in Freund's incomplete adjuvant (FIA) was inoculated on days 21 and 42. Sera were collected before the first immunization (pre-immune sera) and 20 days after the last booster dose (post III sera). Rabbit sera were absorbed with E. coli proteins coupled to a Sepharose column and then stored at -20°C until use, according to the manufacturer's instructions (Amersham Biosciences, Buckinghamshire, UK).
Parasite protein extraction
A parasite pellet was treated with 0.2% saponin, then washed six times with PBS and homogenized in lysis buffer (5% SDS, 10 mM PMSF, 10 mM iodoacetamide, 1 mM EDTA). The total lysate was quantified by using a micro BCA protein assay kit (Thermo scientific) and resolved in SDS-PAGE in reducing and non-reducing conditions. Immunoblot detection was carried out using sera from rabbits previously immunized with synthetic peptides.
Isolating detergent-resistant microdomains (DRMs)
Parasite mature forms extracted by the Percoll method were treated with 0.2% saponin in PBS for 5 minutes. After six washes in PBS, the pellet was suspended in a 200 μL TNET solution (1% Triton X-100, 25 mM Tris-HCl, 150 mM NaCl and 1 mM EDTA) which contained protease inhibitors (1 mM PMSF, 1 mM iodoacetamide, 1 mM EDTA and 1 mg/mL leupeptin). The sample was poured into two aliquots which were then treated in the following conditions: a 100 μL aliquot was incubated at 4°C for 30 minutes and subsequently centrifuged at 7,550 rpm for 10 minutes; the remaining sample was kept at 37°C for 30 minutes and then centrifuged in the same conditions described above. The supernatant was recovered in a new Eppendorf tube and the remaining pellet from each assay was suspended in lysis buffer (5% SDS, 10 mM PMSF, 10 mM iodoacetamide, 1 mM EDTA). Each fraction was quantified by Micro BCA protein assay kit (Thermo scientific) and resolved in 12% SDS-PAGE.
Expression and purification of recombinant protein fragments
Once the sequence from cloned inserts had been confirmed, these were transformed in the E. coli BL21-AI strain (Invitrogen), according to the manufacturer's recommendations. In brief, cells were grown overnight at 37°C and then inoculated in Terrific broth medium supplemented with 0.1 mg/mL ampicillin and 0.1% (w/v) D-glucose. Once the cells had reached their stationary phase (0.6 OD600), 0.2% L-arabinose (w/v) was added as inductor and these were incubated again in the same conditions described above for 4 hours. The culture was centrifuged at 13,000 rpm for 30 minutes and the pellet was suspended in extraction buffer (6 M Urea, 12 mM imidazole, 10 mM Tris-Cl, 100 mM NaH2PO4 and 10 mg/mL lysozyme) supplemented with protease inhibitors (1 mM PMSF, 1 mM iodoacetamide, 1 mM EDTA and 1 mg/mL leupeptin) and lysed by sonication. rPv RON1-a and rPv RON1-c recombinant fragment expression was assessed on 12% SDS-PAGE. pEXP5 CT/TOPO vector was used to facilitate purification and recognition by monoclonal anti-histidine antibodies. This vector adds a six-histidine tag towards the C-terminus of each fragment, which is used for purifying the recombinant protein by affinity chromatography using a Ni+2-NTA resin (Qiagen, California, USA). Briefly, the extraction buffer described above was used to elute the non-retained proteins and then the same buffer plus 500 mM imidazole was used to elute the recombinant protein. All individually collected fractions were analyzed by SDS-PAGE and Western blot. Pure fractions were dialyzed in PBS pH 7.0. The protein was ultrafiltered and concentrated using an Amicon filtration system (LabX scientific marketplace, Midland Canada) and quantified with a micro BCA protein assay kit (Thermo Fisher scientific, Pittsburgh, PA, USA).
SDS-PAGE and Western blot
5 μg of each recombinant and 50 μg of both parasite total lysate and the fractions obtained in DRMs were separated on SDS-PAGE gels and transferred to nitrocellulose membranes. These membranes were blocked with 5% skimmed milk in 0.05% PBS-Tween for one hour. After three washes with 0.05% PBS-Tween, each membrane was cut into strips which were individually analyzed as follows: strips with recombinant protein were incubated for 1 hour at room temperature in 5% skimmed milk in 0.05% PBS-Tween, including a 1:100 dilution of rabbit (pre-immune and post-III) or human sera. Strips with parasite total lysate and those containing DRM fractions were incubated for 1 hour at room temperature in 5% skimmed milk in 0.05% PBS-Tween, including a 1:100 dilution of anti-Pv RON1 antibodies. After 3 washes, strips were incubated for 1 hour with goat anti-rabbit IgG coupled to phosphatase as secondary antibody in a 1:4,500 dilution at room temperature, or goat anti-human IgG in a 1:5,000 dilution. Western blot for recombinant proteins was carried out as follows: a nitrocellulose strip was used as positive control; this strip was incubated with a peroxidase-coupled monoclonal anti-histidine antibody diluted 1:4,500 in skimmed milk plus 0.05% PBS-Tween. Blots were revealed with a substrate peroxidase VIP kit (Vector Laboratories, Burlingame, Canada) or BCIP/NBT color development substrate kits (Promega), according to the manufacturer's instructions.
Indirect immunofluorescence assay (IFA)
Blood smears from the Aotus monkey infected with P. vivax were fixed with 4% (v/v) formaldehyde. Slides were washed thrice with PBS and blocked at 37°C for 45 minutes using a 1% (v/v) bovine serum albumin (BSA) solution in PBS. After 3 washes, slides were incubated at 37°C for 1 hour with a mixture of anti-Pv RON1 rabbit sera in 1:30 dilution and anti-Pv RhopH3 mice sera (previously obtained in our institute [30]) in 1:40 dilution, suspended in PBS -1% BSA - 0.1% Triton X-100 solution. After three washes, plates were incubated with FITC-conjugated anti-rabbit IgG antibody (Sigma) and Red-conjugated anti-mouse IgG antibody (Millipore) in previously described conditions. Slides were stained with DAPI (2 μg/mL) for 20 minutes at room temperature and then observed under an Olympus BX51 fluorescence microscope, using 100× oil immersion objective.
Accession number: the nucleotide and amino acid sequences used here have been reported in the GenBank database, under accession number JN188400.
Results and Discussion
Pvron1 gene identification and localization in a syntenic chromosome region
The initial P. vivax ron1 gene sequence selected for this study was found in the plasmoDB database (Accession number PVX_000945). According to the information found there, the ron1 gene seemed to be ubiquitous within the Plasmodium genus, since orthologous genes were found in the Plasmodium falciparum, Plasmodium knowlesi and Plasmodium berghei genomes. Additionally, the database showed that open reading frame (ORF) orientation and exon-intron structure were highly conserved among the genes encoding the hypothetical protein products in these parasite species. Such genome organization was consistent with the identity (Id) and similarity (S) values obtained when P. falciparum - P. vivax and P. vivax - P. knowlesi contigs were analyzed (Figure 1). It is worth noting that the high degree of S observed between P. vivax - P. knowlesi was in agreement with the evolutionary proximity found in a previous phylogenetic analysis of conserved regions from the circumsporozoite protein (CSP) encoding gene [50] and in a comparative genomics study of P. vivax[23] which showed that these organisms share similar chromosomal regions. Interestingly, the Id value found between the alignment of ORFs encoded by pfasp (PFD0295c) and pvron1 (PVX_000945) genes was similar to the Id values found in antigens considered good candidates for designing an effective vaccine against P. vivax malaria [22, 28, 29], suggesting that Pv RON1 should be an interesting candidate for immunological trials.

Schematic scale organization showing the localization of pfasp (PFD0295c), pvron1 (PVX_000945) and pkron1 (PCH_030870) genes within P. falciparum, P. vivax and P. knowlesi chromosome fragments . Exon-intron organization, ORF orientation and gene IDs are shown in accordance with the plasmoDB database. Identity (Id) and similarity (S) values between each hypothetical protein product contained in P. falciparum - P. vivax and P. vivax - P. knowlesi upstream and downstream contigs are shown.
Pv RON1 characterization in silico
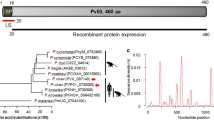
The pvron1 gene from SAL-1 strain has 2,801 bp; it encodes to a 772 amino acid-long protein (~84.5 kDa estimated molecular weight). This protein is 41 residues longer compared to Pf ASP (731 aa) [18]. Pv RON1 presents two hydrophobic regions in the N and C-terminus matching a secretory signal sequence composed by the first 21 amino acids and another located between amino acids 743 to 764 (Figure 2A). This latter feature plus the result of the GPI-anchor predictor, point out to the C-terminal anchoring of this protein by GPI. In addition, a sushi-like domain (also known as complement control protein (CCP) module) was predicted; this domain consists of 56 amino acids, four corresponding to cysteines (Cys) which intervene in the domain's structural conformation to mediate protein-protein binding and/or cell adhesion [19].

In silico characterization, alignment of a RON1 protein-fragment in different malarial species and pvron1 gene transcription. (A) pvron1 gene scheme, showing the signal peptide, GPI-anchor sequence, TR and sushi domain localizations. Cysteine residues are represented by lines. Peptides used for rabbit immunization are shown, as well as rPv RON1-a and rPv RON1-c fragments expressed as recombinants. (B) Alignment of a RON1 protein-fragment from P. falciparum, P. vivax, P. chabaudi and P. knowlesi species. This figure shows the region consisting of amino acids 120 to 210, which includes a repeat sequence which is exclusive for P. vivax species. Asterisks indicate identical amino acids. A dot refers to weakly similar amino acids and two dots indicate strongly similar amino acids. (C) pvron1 gene amplification by PCR. Line 1 shows the molecular weight marker. Lines 2 and 3 correspond to amplification of genomic and complementary DNA.
XSTREAM analysis revealed a tandem repeat (TR) between amino acids 148 to 171 (Figure 2A). According to the alignment performed with other homologous proteins in Plasmodium species (P. falciparum, P. knowlesi and P. chabaudi) this region has been shown to be exclusive for P. vivax (Figure 2B). The TR consists of four blocks of eight amino acids having the EKLPIGGG consensus sequence. Antheprot software prediction has shown that this could be a good B-cell lineal epitope. Another two blocks having a similar sequence to that of the TR were found between amino acids 133 to 147; these sequences showed substitutions or deletions of some residues, suggesting that this region might be under selective pressure. It has been previously described that the polymorphic repeat sequences found in some P. falciparum malarial antigens, such as the circumsporozoite protein (CSP), the ring-infected erythrocyte surface antigen (RESA) and the S-antigen, can affect antibody affinity maturation and mask critical epitopes involved in invasion from being recognized [51–54]; the present analysis has thus led us to suggesting that the TR regions found in Pv RON1 could be part of a mechanism used by the parasite to evade the host's humoral immune response. However, further experimental evidence is needed to confirm this hypothesis.
The pvron1 gene is transcribed during the blood stage
The presence of ron1 gene transcripts in the P. vivax VCG-1 strain was assessed by PCR, using cDNA as template. Figure 2C shows the amplification products obtained from RT-PCR and genomic DNA, indicating that the pvron1 gene is transcribed in schizont-enriched samples. These results agreed with previously reported studies showing a peak transcription from 35 to 40 hours of the P. vivax Sal-1 strain intraerythrocyte cycle [36] and between 35 and 48 hours in the P. falciparum 3D7 strain [17]. Once genomic and complementary DNA sequences were aligned, it was found that the pvron1 gene was encoded by four exons, similar to the homologous pfasp gene [18], the fourth of them containing a sushi domain. Two nucleotide substitutions and a thirty-two base pair deletion corresponding to a repeat block (EKLPIGGG) were observed when the pvron1 gene sequences from Sal-1 reference strain and VCG-1 Aotus- adapted strain were compared. These mutations were non-synonymous and produced changes in the following amino acids: glutamic acid for glycine in position 148 and isoleucine for threonine in position 740. Furthermore, 17 Cys residues were shown to be conserved between these two strains. When comparing these residues from the Pv RON1 sequence with its P. falciparum homologue (Pf ASP), these presented an identical localization, suggesting that there was probably a degree of conservation in the 3D structure of such molecules.
Pv RON1 expression and association with DRMs
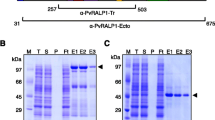
Two peptides derived from the deduced P. vivax Sal-I Pv RON1 sequence were synthesized and inoculated in rabbits for obtaining anti-Pv RON1 antibodies that could recognize the protein in parasite lysate. The antibodies' ability to recognize Pv RON1 was assessed by Western blot, using purified rPv RON1-a and rPv RON1-c fragments as antigens (Figure 3A). Immunodetection assays carried out here with polyclonal sera detected bands at ~100 kDa, corresponding to the protein without the signal peptide, and others at ~57 kDa and ~42 kDa, suggesting proteolytic products (Figure 3B; Line 2). Given that the expected weight of the complete protein was slightly lower (~81.35 kDa), it is more likely that the result corresponded to anomalous migration, similar to what occurs with other Apicomplexa antigens already identified in the rhoptries, such as Tg RON2 [7], Tg ROP1 [55] and Pv RON2 [30]. When the experiment was carried out in non-reducing conditions, the ~57 kDa and ~42 kDa bands were not detected, whilst the ~100 kDa band became intensified (Figure 3B; line 4) suggesting that, as occurs with its homologue [18], these two polypeptides were likely to remain bound by disulphide bridges formed between the cysteines contained in their sequences.

Detection of PvRON1 recombinant fragments and the native protein by polyclonal antibodies. (A) Evaluation of anti-Pv RON1 polyclonal antibody recognition by Western blot. Lines 1-3 and 8-10 correspond to recognition by pre-immune sera. Lines 4-6 and 11-13 correspond to detection with rabbits' post III sera . Lines 7 and 14 indicate the recognition of purified rPv RON1-a and rPv RON1-c fragments by anti-polyhistidine monoclonal antibody. (B) Detection of the protein in parasite total lysate and DRMs. Lines 1 and 2, parasite lysate in reducing conditions. Lines 3 and 4, parasite lysate in non-reducing conditions. Lines 5-12, DRM fractions treated at 4°C and 37°C. S and P letters represent the supernatant and pellet.
To evaluate Pv RON1 presence in DRMs, a parasite lysate was solubilized at different temperatures and used for rabbit sera recognition assays. Consistent with the results that recognized the protein in the parasite lysate (Figure 3B; Line 2), polyclonal antibodies detected bands at ~100 kDa, ~57 kDa and ~42 kDa in the pellet soluble fraction at 4°C, and in the treatment soluble fraction at 37°C (Figure 3B; lines 8 and 10); these bands disappeared when the pellet was treated with the non-ionic detergent at 37°C (Figure 3B; line 12). Interestingly, sera revealed bands in the soluble fraction at 4°C (Figure 3B; line 6), which can be attributed to the high protein expression levels during schizont stage. Together, these results led to confirming that PvR ON1 antigen was associated with DRMs, differing from what has been reported in the DRM proteome study [15].
Localization in rhoptries
An immunofluorescence assay was carried out on a blood smear of P. vivax- infected RBCs for assessing the protein's subcellular localization in the parasite. Figure 4A shows Pv RON1 distribution in mature schizonts as a punctuated fluorescence pattern, which is characteristic of apical organelle proteins (rhoptries and micronemes). Alternatively, double labeling was carried out using anti-Pv RhopH3 mice antibodies (rhoptry bulb marker) and anti-Pv RON1 rabbit antibodies (Figure 4B). Overlapping images showed partial localization of both proteins, supporting the fact that Pv RON1 antigen could be forming part of the rhoptry neck, as occurs with P. falciparum ASP protein [56].

Sub-cellular localization of the Pv RON1 protein in mature schizonts using fluorescence microscopy . (A) Apical localization of Pv RON1 protein. (1) Nuclei stained with DAPI; (2) Detection with anti-Pv RON1; (3) Overlapping of the two recognition patterns. (B) Double stained assay. (1) Nuclei stained with DAPI. (2) Recognition by anti-Pv RON1. (3) Detection of Pv RhopH3. (4) Overlapping of 1, 2 and 3 images.
Pv RON1 antigenicity
Two fragments located towards the N- and C-termini of the pvron1 gene were cloned, transformed and expressed as recombinants proteins. Each purified protein's antigenicity was evaluated by Western blot using sera from 20 patients living in P. vivax malarial endemic regions in Colombia and who had had active episodes of the disease. Sera from four individuals who had never suffered from malaria were used as negative controls. All individuals signed an informed consent after receiving detailed information regarding the study goals. Most sera, except for that from controls, presented reactivity towards rPv RON1-a and rPv RON1-c (Figure 5A and 5B) fragments, suggesting that Pv RON1 is expressed during natural malarial infection and that it can also trigger antibody responses in the host. It has been demonstrated that antibodies directed against antigenic proteins can inhibit parasite-host interaction [21] and, therefore, these have been considered as potential candidates in designing a vaccine against malaria. All the above supports the notion that Pv RON1 may be a promising candidate for further trials aimed at assessing its immunogenicity and protection-inducing ability in the Aotus monkey model.

Western blot analysis showing the recognition of Pv RON1 recombinant fragments by human sera . (A-B) rPv RON1-a and rPv RON1-c detection. Lines 1-21, detection with sera from P. vivax-infected patients; Lines 22-25, recognition by control patients' sera; Line 26, detection of the protein using anti-polyhistidine monoclonal antibody.
Conclusions
This study has described the identification and characterization of a DRM-associated rhoptry protein. Taking into account the multiple particularities of Pv RON1 identified here (i.e. the gene's transcription, the protein's expression towards the end of the intraerythrocyte lifecycle and its localization and the broad recognition presented by sera from individuals infected with P. vivax), it can be suggested that this protein is a good vaccine candidate. The presence of secretory and GPI-anchor signals and a domain probably involved in invasion also suggested that the Pv RON1 antigen could play a functional role during RBC invasion; further immunogenicity and protection-inducing ability studies are thus needed in the Aotus experimental model to confirm the value of this protein or its products, as components of a multi-epitope, subunit-based vaccine against P. vivax malaria.
References
WHO: World malaria report 2010. The WHO global malaria programme. 2010, Geneva
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI: The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005, 434: 214-217. 10.1038/nature03342.
Mendis K, Sina BJ, Marchesini P, Carter R: The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001, 64: 97-106.
Cowman AF, Crabb BS: Invasion of red blood cells by malaria parasites. Cell. 2006, 124: 755-766. 10.1016/j.cell.2006.02.006.
Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC: Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS pathogens. 2005, 1: e17-10.1371/journal.ppat.0010017.
Cao J, Kaneko O, Thongkukiatkul A, Tachibana M, Otsuki H, Gao Q, Tsuboi T, Torii M: Rhoptry neck protein RON2 forms a complex with microneme protein AMA1 in Plasmodium falciparum merozoites. Parasitol Int. 2009, 58: 29-35. 10.1016/j.parint.2008.09.005.
Alexander DL, Arastu-Kapur S, Dubremetz JF, Boothroyd JC: Plasmodium falciparum AMA1 binds a rhoptry neck protein homologous to TgRON4, a component of the moving junction in Toxoplasma gondii. Eukaryotic cell. 2006, 5: 1169-1173. 10.1128/EC.00040-06.
Gilson PR, Nebl T, Vukcevic D, Moritz RL, Sargeant T, Speed TP, Schofield L, Crabb BS: Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2006, 5: 1286-1299. 10.1074/mcp.M600035-MCP200.
Allen JA, Halverson-Tamboli RA, Rasenick MM: Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007, 8: 128-140. 10.1038/nrn2059.
Murphy SC, Hiller NL, Harrison T, Lomasney JW, Mohandas N, Haldar K: Lipid rafts and malaria parasite infection of erythrocytes. Mol Membr Biol. 2006, 23: 81-88. 10.1080/09687860500473440.
Schofield L, Vivas L, Hackett F, Gerold P, Schwarz RT, Tachado S: Neutralizing monoclonal antibodies to glycosylphosphatidylinositol, the dominant TNF-alpha-inducing toxin of Plasmodium falciparum: prospects for the immunotherapy of severe malaria. Ann Trop Med Parasitol. 1993, 87: 617-626.
Garcia J, Curtidor H, Pinzon CG, Vanegas M, Moreno A, Patarroyo ME: Identification of conserved erythrocyte binding regions in members of the Plasmodium falciparum Cys6 lipid raft-associated protein family. Vaccine. 2009, 27: 3953-3962. 10.1016/j.vaccine.2009.04.039.
Obando-Martinez AZ, Curtidor H, Arevalo-Pinzon G, Vanegas M, Vizcaino C, Patarroyo MA, Patarroyo ME: Conserved high activity binding peptides are involved in adhesion of two detergent-resistant membrane-associated merozoite proteins to red blood cells during invasion. J Med Chem. 2010, 53: 3907-3918. 10.1021/jm901474p.
Arevalo-Pinzon G, Curtidor H, Vanegas M, Vizcaino C, Patarroyo MA, Patarroyo ME: Conserved high activity binding peptides from the Plasmodium falciparum Pf34 rhoptry protein inhibit merozoites in vitro invasion of red blood cells. Peptides. 2010, 31: 1987-1994. 10.1016/j.peptides.2010.07.009.
Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ, McConville MJ, Schofield L, Hodder AN, Yates JR, Crabb BS: Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J Biol Chem. 2005, 280: 40169-40176. 10.1074/jbc.M509631200.
Bradley PJ, Ward C, Cheng SJ, Alexander DL, Coller S, Coombs GH, Dunn JD, Ferguson DJ, Sanderson SJ, Wastling JM, Boothroyd JC: Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in Toxoplasma gondii. J Biol Chem. 2005, 280: 34245-34258. 10.1074/jbc.M504158200.
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL: The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1: E5-
O'Keeffe AH, Green JL, Grainger M, Holder AA: A novel Sushi domain-containing protein of Plasmodium falciparum. Mol Biochem Parasitol. 2005, 140: 61-68. 10.1016/j.molbiopara.2004.12.003.
O'Leary JM, Bromek K, Black GM, Uhrinova S, Schmitz C, Wang X, Krych M, Atkinson JP, Uhrin D, Barlow PN: Backbone dynamics of complement control protein (CCP) modules reveals mobility in binding surfaces. Protein Science. 2004, 13: 1238-1250. 10.1110/ps.03582704.
Patarroyo ME, Patarroyo MA: Emerging rules for subunit-based, multiantigenic, multistage chemically synthesized vaccines. Accounts Chem Res. 2008, 41: 377-386. 10.1021/ar700120t.
Rodriguez LE, Curtidor H, Urquiza M, Cifuentes G, Reyes C, Patarroyo ME: Intimate molecular interactions of P. falciparum merozoite proteins involved in invasion of red blood cells and their implications for vaccine design. Chem Rev. 2008, 108: 3656-3705. 10.1021/cr068407v.
Moreno-Perez DA, Mongui A, Soler LN, Sanchez-Ladino M, Patarroyo MA: Identifying and characterizing a member of the RhopH1/Clag family in Plasmodium vivax. Gene. 2011, 481 (1): 17-23. 10.1016/j.gene.2011.03.007.
Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, Cheng Q, Coulson RM, Crabb BS, Del Portillo HA, Essien K, Feldblyum TV, Fernandez-Becerra C, Gilson PR, Gueye AH, Guo X, Kang'a S, Kooij TW, Korsinczky M, Meyer EV, Nene V, Paulsen I, White O, Ralph SA, Ren Q, Sargeant TJ, Salzberg SL, Stoeckert CJ, Sullivan SA, Yamamoto MM, Hoffman SL, Wortman JR, Gardner MJ, Galinski MR, Barnwell JW, Fraser-Liggett CM: Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008, 455: 757-763. 10.1038/nature07327.
Carlton J, Silva J, Hall N: The genome of model malaria parasites, and comparative genomics. Current issues in molecular biology. 2005, 7: 23-37.
Mongui A, Angel DI, Moreno-Perez DA, Villarreal-Gonzalez S, Almonacid H, Vanegas M, Patarroyo MA: Identification and characterization of the Plasmodium vivax thrombospondin-related apical merozoite protein. Malar J. 2010, 9: 283-10.1186/1475-2875-9-283.
Mongui A, Perez-Leal O, Soto SC, Cortes J, Patarroyo MA: Cloning, expression, and characterisation of a Plasmodium vivax MSP7 family merozoite surface protein. Biochem Biophys Res Comm. 2006, 351: 639-644. 10.1016/j.bbrc.2006.10.082.
Perez-Leal O, Sierra AY, Barrero CA, Moncada C, Martinez P, Cortes J, Lopez Y, Salazar LM, Hoebeke J, Patarroyo MA: Identifying and characterising the Plasmodium falciparum merozoite surface protein 10 Plasmodium vivax homologue. Biochem Biophys Res Comm. 2005, 331: 1178-1184. 10.1016/j.bbrc.2005.04.031.
Perez-Leal O, Sierra AY, Barrero CA, Moncada C, Martinez P, Cortes J, Lopez Y, Torres E, Salazar LM, Patarroyo MA: Plasmodium vivax merozoite surface protein 8 cloning, expression, and characterisation. Biochem Biophys Res Comm. 2004, 324: 1393-1399. 10.1016/j.bbrc.2004.09.202.
Mongui A, Angel DI, Gallego G, Reyes C, Martinez P, Guhl F, Patarroyo MA: Characterization and antigenicity of the promising vaccine candidate Plasmodium vivax 34kDa rhoptry antigen (Pv34). Vaccine. 2009, 28: 415-421. 10.1016/j.vaccine.2009.10.034.
Arevalo-Pinzon G, Curtidor H, Patino LC, Patarroyo MA: PvRON2, a new Plasmodium vivax rhoptry neck antigen. Malar J. 2011, 10: 60-10.1186/1475-2875-10-60.
Mongui A, Angel DI, Guzman C, Vanegas M, Patarroyo MA: Characterisation of the Plasmodium vivax Pv38 antigen. Biochem Biophys Res Comm. 2008, 376: 326-330. 10.1016/j.bbrc.2008.08.163.
Mongui A, Perez-Leal O, Rojas-Caraballo J, Angel DI, Cortes J, Patarroyo MA: Identifying and characterising the Plasmodium falciparum RhopH3 Plasmodium vivax homologue. Biochem Biophys Res Comm. 2007, 358: 861-866. 10.1016/j.bbrc.2007.05.015.
Angel DI, Mongui A, Ardila J, Vanegas M, Patarroyo MA: The Plasmodium vivax Pv41 surface protein: identification and characterization. Biochem Biophys Res Comm. 2008, 377: 1113-1117. 10.1016/j.bbrc.2008.10.129.
Patarroyo MA, Perez-Leal O, Lopez Y, Cortes J, Rojas-Caraballo J, Gomez A, Moncada C, Rosas J, Patarroyo ME: Identification and characterisation of the Plasmodium vivax rhoptry-associated protein 2. Biochem Biophys Res Comm. 2005, 337: 853-859. 10.1016/j.bbrc.2005.09.120.
Perez-Leal O, Mongui A, Cortes J, Yepes G, Leiton J, Patarroyo MA: The Plasmodium vivax rhoptry-associated protein 1. Biochem Biophys Res Comm. 2006, 341: 1053-1058. 10.1016/j.bbrc.2006.01.061.
Bozdech Z, Mok S, Hu G, Imwong M, Jaidee A, Russell B, Ginsburg H, Nosten F, Day NP, White NJ, Carlton JM, Preiser PR: The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci USA. 2008, 105: 16290-16295. 10.1073/pnas.0807404105.
Pico de Coana Y, Rodriguez J, Guerrero E, Barrero C, Rodriguez R, Mendoza M, Patarroyo MA: A highly infective Plasmodium vivax strain adapted to Aotus monkeys: quantitative haematological and molecular determinations useful for P. vivax malaria vaccine development. Vaccine. 2003, 21: 3930-3937. 10.1016/S0264-410X(03)00278-0.
PlasmoDB: Plasmodium genomics resource. [http://www.plasmodb.org]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340: 783-795. 10.1016/j.jmb.2004.05.028.
Newman AM, Cooper JB: XSTREAM: a practical algorithm for identification and architecture modeling of tandem repeats in protein sequences. BMC bioinformatics. 2007, 8: 382-10.1186/1471-2105-8-382.
Poisson G, Chauve C, Chen X, Bergeron A: FragAnchor: a large-scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genomics, proteomics & bioinformatics/Beijing Genomics Institute. 2007, 5: 121-130.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJ, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C: InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, 37: D211-215. 10.1093/nar/gkn785.
Deleage G, Combet C, Blanchet C, Geourjon C: ANTHEPROT: an integrated protein sequence analysis software with client/server capabilities. Computers in Biology and Medicine. 2001, 31: 259-267. 10.1016/S0010-4825(01)00008-7.
Janin J, Wodak S: Conformation of amino acid side-chains in proteins. J Mol Biol. 1978, 125: 357-386. 10.1016/0022-2836(78)90408-4.
Hopp TP, Woods KR: Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA. 1981, 78: 3824-3828. 10.1073/pnas.78.6.3824.
Welling GW, Weijer WJ, van der Zee R, Welling-Wester S: Prediction of sequential antigenic regions in proteins. FEBS Lett. 1985, 188: 215-218. 10.1016/0014-5793(85)80374-4.
Andrysiak PM, Collins WE, Campbell GH: Concentration of Plasmodium ovale- and Plasmodium vivax-infected erythrocytes from nonhuman primate blood using Percoll gradients. Am J Trop Med Hyg. 1986, 35: 251-254.
Houghten RA: General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci USA. 1985, 82: 5131-5135. 10.1073/pnas.82.15.5131.
Escalante AA, Barrio E, Ayala FJ: Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Molecular biology and evolution. 1995, 12: 616-626.
Burkot TR, Da ZW, Geysen HM, Wirtz RA, Saul A: Fine specificities of monoclonal antibodies against the Plasmodium falciparum circumsporozoite protein: recognition of both repetitive and non-repetitive regions. Parasite Immunol. 1991, 13: 161-170. 10.1111/j.1365-3024.1991.tb00272.x.
Ramasamy R: Molecular basis for evasion of host immunity and pathogenesis in malaria. Biochim Biophys Acta. 1998, 1406: 10-27.
Hisaeda H, Yasutomo K, Himeno K: Malaria: immune evasion by parasites. Int J Biochem Cell Biol. 2005, 37: 700-706. 10.1016/j.biocel.2004.10.009.
Anders RF: Multiple cross-reactivities amongst antigens of Plasmodium falciparum impair the development of protective immunity against malaria. Parasite Immunol. 1986, 8: 529-539. 10.1111/j.1365-3024.1986.tb00867.x.
Ossorio PN, Schwartzman JD, Boothroyd JC: A Toxoplasma gondii rhoptry protein associated with host cell penetration has unusual charge asymmetry. Mol Biochem Parasitol. 1992, 50: 1-15. 10.1016/0166-6851(92)90239-G.
Srivastava A, Singh S, Dhawan S, Mahmood Alam M, Mohmmed A, Chitnis CE: Localization of apical sushi protein in Plasmodium falciparum merozoites. Mol Biochem Parasitol. 2010, 174: 66-69. 10.1016/j.molbiopara.2010.06.003.
Acknowledgements
We would like to thank Rafael Areiza and Nohora Guayacan for their technical support and Jason Garry for translating and reviewing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DAMP designed experiments, analyzed data and wrote the initial manuscript. MM carried out molecular biology and immunochemical assays. MEP and MAP evaluated and coordinated the assays, and corrected the final manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Moreno-Perez, D.A., Montenegro, M., Patarroyo, M.E. et al. Identification, characterization and antigenicity of the Plasmodium vivax rhoptry neck protein 1 (Pv RON1). Malar J 10, 314 (2011). https://doi.org/10.1186/1475-2875-10-314
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-10-314