
Abstract
Background
Some strains of Bacillus licheniformis have been improved by target-directed screening as well as by classical genetic manipulation and used in commercial thermostable α-amylase and alkaline protease production for over 40 years. Further improvements in production of these enzymes are desirable.
Results
A new strain of B. licheniformis CBBD302 carrying a recombinant plasmid pHY-amyL for Bacillus licheniformis α-amylase (BLA) production was constructed. The combination of target-directed screening and genetic recombination led to an approximately 26-fold improvement of BLA production and export in B. licheniformis. Furthermore, a low-cost fermentation medium containing soybean meal and cottonseed meal for BLA production in shake-flasks and in a 15 liter bioreactor was developed and a BLA concentration of up to 17.6 mg per ml growth medium was attained.
Conclusion
This production level of BLA by B. licheniformis CBBD302(pHY-amyL) is amongst the highest levels in Gram-positive bacteria reported so far.
Similar content being viewed by others

Background
In any industrial biotechnological process, achieving high productivity is an essential factor for commercial success. The maximum specific productivities of a production strain are usually independent of process parameters and determined by the genetic and physiological properties of the organism. Bacterial extracellular enzymes are an important class of industrial enzymes constituting approximately 20% of the enzyme market [1, 2]. To obtain a high yield in bacterial extracellular enzyme production, the following genetic and physiological properties of the strain are important: a) the metabolic flux for amino acids synthesis and ATP regeneration, b) the cell growth rate and cell density in an inexpensive medium, c) mainly vegetative growth by spore-forming strains, d) secretion capacity for extracellular enzymes, e) long-term preservation in an active form in broth, and f) a high expression level of the specific gene encoding a bacterial extracellular enzyme [3].
Genetic improvement of bacterial extracellular enzyme production is achieved by applying a range of strategies based on molecular cloning tools. These include: 1) enhancement of expression level through amplification of gene copy number [4], codon usage optimization [5], or strong promoters being used to boost gene transcription [6]; 2) enhancement of secretion by modulation of signal peptides [7, 8], fusion to heterologous signal peptides for efficient targeting to the translocase [9], increasing the copy number of signal peptidase genes [10, 11], or deregulation and/or co-expression of chaperon encoding genes to make efficient protein folding [12]; 3) improvement of productivity through re-designing the capacity of the secretion machinery by targeted deletion of genes encoding non-beneficial extracellular enzymes [13], or genome reduction [14]; and 4) improvement of the productivity by preventing degradation of extracellular enzymes using protease deficient strains [15].
In general, a specific strain should possess a definite maximum capacity for synthesis and secretion of extracellular enzymes [4]. Overproduction of secreted proteins sometimes severely affects the secretory system of an organism [16] and eventually results in a secretion stress response that may limit secretion [17, 18]. Moreover, it is almost impossible at the present stage to enhance the maximum synthesis and secretion capacity by site-directed gene disruption and expression in a specific strain. Alternatively, data from genome shuffling and genome size reduction studies [14] strongly suggest that a natural strain should exist with a maximum capacity for synthesis and secretion of extracellular enzymes as a result of accumulation of mutations and shuffling of the genomes.
In present work, the combination of target-directed screening and genetic recombination yielded a novel B. licheniformis strain that produced up to 17.6 mg B. licheniformis α-amylase per ml growth medium.
Results and discussion
Rationale for the selection approach of the B. licheniformis strain and property identification
Enzyme synthesis and export is an energy-dependent event [3]. In order to select a suitable host cell for BLA overproduction, following genetic and physiological criteria were applied: 1) the ability of the strain to sporulate should be poor in order to extend the duration of BLA production; 2) the strain should form little or no lichenysin in order to reduce consumption of ATP and the amino acid pool; 3) the strain should not clump during cultivation to maintain efficiency in a production bioreactor; 4) the strain should grow well on either low-cost fermentation medium containing a high substrate concentration appropriate for an industrial process; 5) the strain should contain no native plasmids but be sensitive to kanamycin or tetracyclin to facilitate further genetic manipulation and 6) the strain should be amenable to transformation to enable genetic modification.
B. licheniformis strain selection and biological property identification
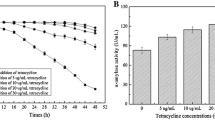
Following a selection procedure described above, based on these criteria, a candidate strain designated as CBB0302 was selected out of a total of 526 B. licheniformis isolates (Figure 1). The strain produced catalase, amylase and protease, utilized citrate, propionate and nitrate, grew in 7% NaCl, and at 50°C but not at 60°C, typical of B. licheniformis strains. Notably, fewer than 0.5% of the cells in culture formed spores and no cell clumping was found after 72 h cultivation. Furthermore the strain did not produce lichenysin-like pigments and harboured no native plasmids. The cell shape of the strain CBB0302 was identical to that of B. licheniformis CICIM B30306, an industrial BLA-producing strain, when both strains were cultivated in LB medium for 10-12 h at 45°C and 220 rpm but the average cell volume was 30-40% less (Figure 2).

B. licheniformis strain selection strategy and selection. A: selection routine; B: selection results.

The scanning electron micrograph of B. licheniformis. 108 cells were inoculated in a 250 ml flask with 30 ml of LB medium and cultivated for 10-12 h at 45°C and 220 rpm. A drop of culture was collected and analyzed by scanning electron microscope. A: B. licheniformis CICIM B30306; B: B. licheniformis CBB0302.
Increased transformation efficiency by genetic modification
The transformation efficiency of the natural B. licheniformis cells is poor and strains routinely need long periods with difficulty to finally obtain a desired transformant [[19]; our own unpublished results]. This is mainly due to the existence of two type I restriction modification systems (RMS) in B. licheniformis [20, 21]. Single as well as double knock-outs of the RMS resulted in strains being readily transformable with plasmids isolated from Bacilli. Introduction of shuttle plasmids isolated from Escherichia coli is routinely possible when the double mutant B. licheniformis MW3 (ΔhsdR1, ΔhsdR2) was used in transformation experiments [19].
A strain CBBD302 was developed by deletion of a type I RMS locus in strain CBB0302 by using homolog-mediated recombination according to the method described by Waschkau et al [19]. The growth and secretion of extracellular enzymes by this strain were unaffected by the deletion (data not shown). The transformation efficiency with a shuttle plasmid pHY-300PLK isolated from E. coli was significantly improved and 42 cfu/μg DNA in strain CBBD302 but only 5 cfu/μg DNA in strain CBB0302 were attained.
Increased recombinant BLA production and secretion by using B. licheniformis CBBD302 as host
In order to test BLA production in B. licheniformis CBBD302, a recombinant plasmid pHY-amyL was constructed. A 1.6 kb fragment containing B. licheniformis B0204 amyL coding for the mature BLA peptide was recovered from pET28a-amyLNEW by PCR, inserted into the Eco RI and Sma I sites of pHY-WZX and functionally tested in E. coli (Figure 3), yielded hybrid plasmid pHY-amyL. Subsequently, pHY-amyL was transferred into B. licheniformis CBBD302, yielding B. licheniformis CBBD302 (pHY-amyL). The BLA production was carried out in LB supplemented with 40 g/l lactose in shake-flasks (Figure 4). B. licheniformis CBBD302, B. licheniformis B30306, B. licheniformis B0204 and B. licheniformis CBBD302 (pHY-amyL) produced 0.1, 0.7, 0.9 and 2.6 mg BLA per ml growth medium, respectively. Strain CBBD302 carrying pHY-amyL produced a 26-fold improvement in BLA production compared to the parent strain CBBD302 and about three times compared to the B. licheniformis B0204. Although the growth rate was slower than its parent CBBD302, strain CBBD302 (pHY-amyL) grew significantly faster than industrial strains B0204 and B30306 did (Figure 4).

Development and identification of pHY-amyL. A: The physical map of pHY-amyL. B: α-Amylase was expressed by E. coli carrying pHY-amyL, a: E. coli(pHY-WZX); b: E. coli(pHY-amyL).

BLA production by B. licheniformis CBBD302(pHY-amyL). B. licheniformis CBBD302 (circles), B. licheniformis CBBD302(pHY-amyL) (triangles), B. licheniformis B30306 (squares) and B. licheniformis B0204 (diamonds) were cultivated in LB medium supplemented with 40 g/l lactose for up to 120 h at 42°C and 220 rpm. Open symbols for cell growth and closed symbols for BLA production.
Bacterial extracellular enzyme production is a complex process, in which the efficiencies of transcription and translation of the enzyme-encoding genes as well as protein translocation define the enzyme concentration in the growth medium and are under control of the bacterial host. A B. licheniformis strain has been found to have a specific capacity maximum for protein synthesis and secretion by the introduction of different copy numbers of amyL in B. licheniformis B0204 [3]. Evidently the capacity could be improved since B. licheniformis CBB0302 with physiological properties of less than 0.5% sporulation, no production of lichenysin-like pigments, reduced nutrient requirement for growth, smaller cell volume, identical cell shape and no cell clumping during propagation gave an increased BLA yield.
Optimization of fermentation medium
α-Amylase production is subjected to catabolite repression by glucose and other sugars, similar to most inducible enzymes. Therefore, the use of glucose in the production of α-amylase in certain cases is problematic [22, 23]. A number of other substrates such as lactose [24] have also been used for the production of α-amylase. The effect of the addition of different carbon sources to the fermentation medium on BLA production was investigated and the results are summarized in Figure 5. BLA secretion by strain CBBD302 carrying pHY-amyL varied between carbon sources and the lactose gave the highest BLA concentration and glucose, starch and corn cob hydrolysate supported a 70~80% BLA production level compared to lactose (Figure 5). These results indicate that many various carbon sources can be used for BLA production and BLA synthesis in CBBD302 is not subject to catabolite repression.

The time-course for BLA production in B. licheniformis CBBD302 (pHY-amyL) on different carbon sources. Symbols: triangle, lactose; square, glucose; diamond, starch; circle, corn cob hydrolysate. The highest activity of BLA on lactose is designated as 100% activity.
Yeast extract alone or in conjunction with other nitrogen sources such as bactopeptone and ammonium sulfate has been used for the production of α-amylase from Bacillus sp. [25]. In this study, the effect of various nitrogen sources in the fermentation medium on BLA production was investigated. When soybean meal, fish meal or (NH4)2SO4 was added as nitrogen source for BLA production to the fermentation medium, lower respective BLA production levels of 95.8, 28.6 and 25.3% were obtained relative to cottonseed meal. Much higher BLA production levels were obtained with cottonseed meal and/or soybean meal supplemented 0.01 mol/l ammonium sulphate as nitrogen source (respectively 132 and 140% of cottonseed level).
The maximum production of BLA by B. licheniformis CBBD302(pHY-amyL) of 17.6 mg/ml was obtained when the strain was cultivated in the fermentation medium consisting of 40 g/l lactose, 25 g/l soybean meal, 20 g/l cottonseed meal, 30 g/l corn-steep liquor and 0.01 mol/l ammonium sulfate in a pH-controlled 15 l bioreactor fermentations (Figure 6). The BLA production was boosted by the 40 g/l lactose when fed at the 54th hour.

A time-course of BLA production with B. licheniformis CBBD302(pHY-amyL) in 15 l fermentor. Arrow indicates one more 40 g/L lactose was fed at that point during the fermentation.
Conclusion
The combination of target-directed screening and genetic recombination led to an overall 26-fold improvement of BLA production and export in B. licheniformis. In a low-cost fermentation medium containing 40 g/l lactose, 25 g/l soybean meal, 20 g/l cottonseed meal, 30 g/l corn-steep liquor and 0.01 mol/l ammonium sulphate and a 15 l bioreactor up to 17.6 mg BLA per ml growth medium was produced. This production level of BLA by B. licheniformis CBBD302(pHY-amyL) is amongst the highest levels in Gram-positive bacteria reported so far.
Methods
Plasmids and strains
Plasmid pHY300PLK [26] was used to determine transformation efficiency and plasmid pHY-WZX [27] was used as an expression vector in B. licheniformis. E. coli XL1 was used as a host cell for functional identification of the recombinant plasmid. Molecular biology methods were described previously [28]. A 1.6 kb fragment containing amyL coding for the mature peptide as well as its 180 bp downstream sequence was amplified with primers F2-Eco RI (5'-CGGAATTC CTTAATGGGACGCTGATGC-3') and R1-Sma I (5'-TACCCGGG TACATCAGATAACGTTGCC-3') using pET28a-amyLNEW [29] as template. The amplified product was purified and digested with Eco RI and Sma I and subsequently cloned into the same sites of pHY-WZX to yield recombinant pHY-amyL. B. licheniformis isolates as well as B0204 and B030306 were purchased from CICIM-CU http://cicim-cu.jiangnan.edu.cn. CBBD302 was developed from a native B. licheniformis isolate CBB0302, in which the restriction modification system locus was deleted according to the method described by Waschkau et al [19]. B. licheniformis CBBD302(pHY-amyL) was CBBD302 harboring pHY-amyL by using electroporation [30].
Strain screening and biological property identification
B. licheniformis strains were recovered from a culture collection held at -70°C and single colonies were picked onto LB plate and cultivated at 45°C for up to 72 h. Colonies with no or reduced lichenysin formation were picked onto LB plate supplemented with 5 μg/ml kanamycin or 10 μg/ml tetracycline and cultivated for 48 h. Colonies that failed to grow were examined for the existence of the native plasmids by plasmid extraction and agarose electrophoresis as described previously [28]. Strains carrying no native plasmids and sensitive to kanamycin and tetracycline were cultivated in LB containing 2% glucose at 45°C for up to 72 h and their degree of sporulation and cells dispersion were checked by light microscopy. Strains with sporaulation rate less than 5% after 72 h and with no cell clumping were inoculated onto LB supplemented 30% glucose or 30% starch and cultivated at 45°C for 24 h. The rapidly growing strains were selected and cultivated at 45°C and 24 h on a nutrient limited medium consisting of 0.02% peptone, 0.01% yeast extract, 1% NaCl and 0.01% glucose. Those strains that grew well on the nutrient limited medium were examined for their biological properties including the major secreted enzymes, genetic transformation efficiency as well as plasmid stability as described by Zhuge & Wang [31]. The cells were examined with a Quanta-200 scanning electron microscope (FEI, Netherland).
Cultivation
E. coli XL1 was cultivated at 37°C in LB medium. As required, 100 μg/ml ampicillin and/or 25 μg/ml kanamycin were added to the medium. B. licheniformis B0204, B030306 and CBBD302 were cultivated at 42°C in LB medium. For the shake-flask fermentation evaluation, B. licheniformis strains were grown in 500 ml Erlenmeyer flasks containing 50 ml LB supplemented with 40 g/l lactose at 42°C and 220 rpm. For optimization studies the fermentation medium consisted of 30 g/l corn-steep liquor, 30 g/l nitrogen source (cottonseed meal, soybean meal and fish meal, and former two combination with 0.01 mol/l (NH4)2SO4 and 40 g/l) carbon source (lactose, glucose, starch or corn cob hydrolysate) at pH 6.0 was used. For bioreactor cultivation studies, a Biostat (B. Braun, Melsungen, Germany) with a 15 l working volume was used. The bioreactor was inoculated with 5% (v/v) broth and cultivated at 42°C with a controlled pH of 6.0.
Analytical procedures
For shake flask cultivation, samples for cell density and BLA activity were taken at regular intervals. The optical density (OD600 nm) was measured in triplicate with an Ultrospec 3100 pro spectrophotometer (Amersham Pharmacia, UK). SDS-PAGE was performed using a Mini Protean 3 apparatus (Bio-Rad, USA). Proteins were stained by Coomassie Brilliant Blue G250. Directly after sampling, BLA activity was measured spectrophotometrically (Ultrospec 3100 pro, Amersham Pharmacia, UK) as described previously [32]. One unit was defined as the amount of enzyme that hydrolyzes 1 mg water soluble corn starch per minute at 70°C and pH 6.0. The amount of BLA protein (g/ml) in the medium was calculated using BLA specific activity parameter of 1 mg BLA is equal to 996 U.
References
Rao MB, Tanksale AM, Gathe MS, Deshpande VV: Molecular and biotechnological aspects of microbial proteases. Micrbiol Mol Biol Rev. 1998, 62: 597-635.
Gupta R, Paresh G, Mohapatra H, Goswami VK, Chauhan B: Microbial α-amylases: a biotechnological perspective. Process Biochem. 2003, 38: 1599-1616. 10.1016/S0032-9592(03)00053-0.
Zhuge J, Li H, Wang ZX: Genetic Improvement on Microorganisms. 2009, Beijing, Chemical Industry Press
Niu D, Shi GY, Wang ZX: Genetic improvement of α-amylase producing Bacillus licheniformis by homolog-mediated α-amylase gene amplification. Chin J Biotech. 2009, 25: 375-80.
Micheelsen PO, Ostergaard PR, Lange L, Skjøt M: High-level expression of the native barley alpha-amylase/subtilisin inhibitor in Pichia pastoris. J Biotechnol. 2008, 133: 424-32. 10.1016/j.jbiotec.2007.11.012.
Wang PZ, Doi RH: Overlapping promoters transcribed by Bacillus subtilis 55 and 37 RNA polymerase holoenzymes during growth and stationary phases. J Biol Chem. 1984, 259: 8619-25.
Hazebrouck S, Pothelune L, Azevedo V, Corthier G, Wal J-M, Langella P: Efficient production and secretion of bovine β-lactoglobulin by Lactobacillus casei. Microbial Cell Factories. 2007, 6: 12- 10.1186/1475-2859-6-12.
Yang Y, Biedendieck R, Wang W, Gamer M, Malten M, Jahn D, Deckwer WD: High yield recombinant penicillin G amidase production and export into the growth medium using Bacillus megaterium. Microbial Cell Factories. 2006, 5: 36- 10.1186/1475-2859-5-36.
Bolhuis A, Matzen A, Hyyryläinen HL, Kontinen VP, Meima R, Chapuis J, Venema G, Bron S, Freudl R, van Dijl JM: Signal peptide peptidase- and ClpP-like proteins of Bacillus subtilis required for efficient translocation and processing of secretory proteins. J Biol Chem. 1999, 274: 24585-92. 10.1074/jbc.274.35.24585.
Pummi T, Leskelä S, Wahlström E, Gerth U, Tjalsma H, Hecker M, Sarvas M, Kontinen VP: ClpXP protease regulates the signal peptide cleavage of secretory preproteins in Bacillus subtilis with a mechanism distinct from that of the Ecs ABC transporter. J Bacteriol. 2002, 184: 1010-8. 10.1128/jb.184.4.1010-1018.2002.
Nahrstedt H, Wittchen K, Rachman MA, Meinhardt F: Identification and functional characterization of a type I signal peptidase gene of Bacillus megaterium DSM319. Appl Microbiol Biotechnol. 2004, 64: 243-9. 10.1007/s00253-003-1469-2.
Balbás P: Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Mol Biotechnol. 2001, 19: 251-67. 10.1385/MB:19:3:251.
Tjalsma H, Koetje EJ, Kiewiet R, Kuipers OP, Kolkman M, Laan van der J, Daskin R, Ferrari E, Bron S: Engineering of quorum-sensing systems for improved production of alkaline protease by Bacillus subtilis. J Appl Microbiol. 2004, 96: 569-78. 10.1111/j.1365-2672.2004.02179.x.
Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N: Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res. 2008, 15: 73-81. 10.1093/dnares/dsn002.
Wittchen KD, Meinhardt F: Inactivation of the major extracellular protease from Bacillus megaterium DSM319 by gene replacement. Appl Microbiol Biotechnol. 1995, 42: 871-7. 10.1007/BF00191184.
Wong SL: Advances in the use of Bacillus subtilis for the expression and secretion of heterologous proteins. Curr Opin Biotechnol. 1995, 6: 517-22. 10.1016/0958-1669(95)80085-9.
Tjalsma H, Stover AG, Driks A, Venema G, Bron S, van Dijl JM: Conserved serine and histidine residues are critical for activity of the ER-type signal peptidase SipW of Bacillus subtilis. J Biol Chem. 2000, 275: 25102-8. 10.1074/jbc.M002676200.
Antelmann H, Darmon E, Noone D, Veening JW, Westers H, Bron S, Kuipers OP, Devine KM, Hecker M, van Dijl JM: The extracellular proteome of Bacillus subtilis under secretion stress conditions. Mol Microbiol. 2003, 49: 143-56. 10.1046/j.1365-2958.2003.03565.x.
Waschkau B, Waldeck J, Wieland S, Eichstädt R, Meinhardt F: Generation of readily transformable Bacillus licheniformis mutants. Appl Microbiol Biotechnol. 2008, 78: 181-8. 10.1007/s00253-007-1278-0.
Veith B, Herzberg C, Steckel S, Feesche J, Maurer KH, Ehrenreich P, Baumer S, Henne A, Liesegang H, Merkl R, Ehrenreich A, Gottschalk G: The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol. 2004, 7: 204-11. 10.1159/000079829.
Rey MW, Ramaiya P, Nelson BA, Brody-Karpin SD, Zaretsky EJ, Tang M, Lopez de Leon A, Xiang H, Gusti V, Clausen IG, Olsen PB, Rasmussen MD, Andersen JT, Jorgensen PL, Larsen TS, Sorokin A, Bolotin A, Lapidus A, Galleron N, Ehrlich SD, Berka RM: Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 2004, 5: R77- 10.1186/gb-2004-5-10-r77.
Morkeberg R, Carlsen M, Neilsen J: Induction and repression of α-amylase production in batch and continuous cultures of Aspergillus oryzae. Microbiol. 1995, 141: 2449-54. 10.1099/13500872-141-10-2449. 10.1099/13500872-141-10-2449.
Bhella RS, Altosaar I: Purification and some properties of the extracellular α-amylase from Aspergillus awamori. Can J Microbiol. 1985, 31: 149-
Kelly CT, Bolton DJ, Fogarty WM: Biphasic production of α-amylase of Bacillus flavothermus in batch fermentation. Biotechnol Lett. 1997, 19: 75-7. 10.1023/A:1018347017004. 10.1023/A:1018347017004.
Hamilton LM, Kelly CT, Fogarty WM: Production and properties of the raw starch-digesting α-amylase of Bacillus sp. IMD 435. Process Biochem. 1999, 35: 27-31. 10.1016/S0032-9592(99)00028-X.
Ishiwa H, Shibahara H: New shuttle vectors for Escherichia coli and Bacillus subtilis. II. Plasmid pHY300PLK, a multipurpose cloning vector with a polylinker, derived from pHY460. Jpn J Genet. 1985, 60: 235-43. 10.1266/jjg.60.235. 10.1266/jjg.60.235.
Niu D, Wang Z-X: Development of a pair of bifunctional expression vectors for Escherichia coli and Bacillus licheniformis. J Ind Microbiol Biotechnol. 2007, 34: 357-62. 10.1007/s10295-007-0204-x.
Sambrook J, Fitsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbour, Cold Spring Harbor Press
Niu D, Xu M, Ma J, Wang Z-X: Cloning of the gene encoding a thermostable alpha-amylase from Bacillus licheniformis CICIM B0204 and functional identification of its promoter. Acta Microbiol Sin. 2006, 46: 576-80.
Xu M, Ma J, Wang Z-X: Effect of high osmolarity on electro-transformation efficiency of bacteria. J Wuxi Uni Light Ind. 2004, 23 (4): 98-100.
Zhuge J, Wang Z-X: Industrial Microbiology: A Laboratory Manual. 1994, Beijing, China Light Industry Press
Hollo J, Szeitli J: The reaction of starch with iodine. Starch and its derivatives. Edited by: Rodley JA. 1968, 203-46. Chapman & Hall, 4
Acknowledgements
This work was financially supported by China High-Tech Research and Development Program 2006AA020204 and partly granted by Sino-South Africa Cooperation Program 2009DFA31300 and NRF67185. The authors thankfully thank Dr. B.A. Prior from University of Stellenbosch for the manuscript preparation and language revision.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DN carried out the molecular genetic studies. ZZ participated in the design of the study and performed the statistical analysis. G-YS participated fermentation experiments. Z-XW conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Niu, D., Zuo, Z., Shi, GY. et al. High yield recombinant thermostable α-amylase production using an improved Bacillus licheniformis system. Microb Cell Fact 8, 58 (2009). https://doi.org/10.1186/1475-2859-8-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-8-58