
Abstract
Background
Inflammation within the oral cavity occurs due to dysregulation between microbial biofilms and the host response. Understanding how different oral hygiene products influence inflammatory properties is important for the development of new products. Therefore, creation of a robust host-pathogen biofilm platform capable of evaluating novel oral healthcare compounds is an attractive option. We therefore devised a multi-species biofilm co-culture model to evaluate the naturally derived polyphenol resveratrol (RSV) and gold standard chlorhexidine (CHX) with respect to anti-biofilm and anti-inflammatory properties.
Methods
An in vitro multi-species biofilm containing S. mitis, F. nucleatum, P. gingivalis and A. actinomycetemcomitans was created to represent a disease-associated biofilm and the oral epithelial cell in OKF6-TERT2. Cytotoxicity studies were performed using RSV and CHX. Multi-species biofilms were either treated with either molecule, or alternatively epithelial cells were treated with these prior to biofilm co-culture. Biofilm composition was evaluated and inflammatory responses quantified at a transcriptional and protein level.
Results
CHX was toxic to epithelial cells and multi-species biofilms at concentrations ranging from 0.01-0.2%. RSV did not effect multi-species biofilm composition, but was toxic to epithelial cells at concentrations greater than 0.01%. In co-culture, CHX-treated biofilms resulted in down regulation of the inflammatory chemokine IL-8 at both mRNA and protein level. RSV-treated epithelial cells in co-culture were down-regulated in the release of IL-8 protein, but not mRNA.
Conclusions
CHX possesses potent bactericidal properties, which may impact downstream inflammatory mediators. RSV does not appear to have bactericidal properties against multi-species biofilms, however it did appear to supress epithelial cells from releasing inflammatory mediators. This study demonstrates the potential to understand the mechanisms by which different oral hygiene products may influence gingival inflammation, thereby validating the use of a biofilm co-culture model.
Similar content being viewed by others


Background
Periodontal disease occurs from a dysregulation between the bacterial biofilm at the gum margin and the immune response, resulting in irreversible destruction of both soft and hard tissues supporting the teeth, ultimately leading to tooth loss. In health, innate and adaptive immune molecules mediate the equilibrium between the host and the predominantly Gram positive heterogeneous bacterial suspensions in saliva and adherent biofilm communities on soft and hard tissue surfaces throughout the oral cavity. In periodontal disease the shift in the microbiota from Gram positive to Gram negative species leads to an dysregulated host response both from local tissues and immune cells which induces inflammation and creates a niche at the root for anaerobic species to survive, further exacerbating the disease [1].
Numerous prokaryote species have been identified within the oral cavity existing within complex biofilm ecosystems either as supra- or sub-gingival plaque. Many are uncultivated and unnamed, but all playing important structural and functional roles [2]. Microarray and next generation sequencing studies of the oral microbiota has allowed classification of some species of bacteria into complexes based on associations with health and disease [3, 4]. These have allowed studies to investigate species, such as the disease-associated bacteria Porphyromonas gingivalis, and understand their role in challenging the host response [5]. However, oral biofilms are complex and within these polymicrobial biofilm structures a myriad of intimate interactions occur, making it difficult to delineate the precise triggers for the pathogenic outcomes associated with immune dysregulation. Therefore, creating a simplified model with several of these key pathogens is an attractive proposition to evaluate host-pathogen interactions and test actives for treatment potential.
Treating periodontal disease is difficult; dentists perform debridement on teeth, administration of antimicrobial mouthwashes to target biofilms and in advanced periodontitis, surgery. However, successes of treatment are variable and on an individual basis and suggesting biofilm composition may influence the outcome of treatment [6]. Antimicrobial mouthwashes currently employed to manage oral diseases include a variety of compounds with chlorhexidine (CHX) being considered the ‘gold standard’ due to its bactericidal and bacteriostatic properties [7]. Additionally, polyphenols, found naturally occurring in plants such as grapes have become a focus for oral therapies due to their anti-inflammatory and anti-bacterial properties [8]. Resveratrol (RSV) is a naturally derived polyphenol, which has been shown to have potent anti-inflammatory properties in a variety of cancer cells and recently periodontitis in rats [9, 10].
Both CHX and RSV have desirable properties for periodontitis prevention, being antimicrobial or anti-inflammatory and antimicrobial, respectively. The purpose of this study was therefore to create and validate a multi-species biofilm model to be used in co-culture with host epithelial cells in order to test the actives CHX and RSV in order to validate whether the model system could be used as a sensitive method of delineating their basic modes of action. Here we report that oral epithelial cells produce a reproducible pro-inflammatory response to our multi-species biofilm at both gene and protein level. Additionally the treatment of either epithelial cells or multi-species biofilms with actives resulted in an alteration of inflammation within the co-culture model.
Methods
Growth and standardisation of bacteria
Porphyromonas gingivalis ATCC 33277, Fusobacterium nucleatum ATCC 10953, Aggregatibacter actinomycetemcomitans ATCC 43718 and Streptococcus mitis ATCC 12261 were used in the course of these studies. P. gingivalis ATCC 33277 and F. nucleatum ATCC 10596 were grown at 37°C in Schaedler anaerobe broth (Oxoid, Cambridge, UK) for 2 days and 1 day, respectively, in an anaerobic chamber (85% N2, 10% CO2 and 5% H2, [Don Whitley Scientific Limited, Shipley, UK]). A. actinomycetemcomitans ATCC 43718 and Streptococcus mitis ATCC 12261 were grown at 37°C in tryptic soy broth (Sigma, Poole, UK) supplemented with 0.8% w/v glucose (BDH, Poole, UK) and 0.6% w/v yeast extract (Oxoid, Cambridge, UK) for 1 day in 5% CO2. The bacteria were washed with PBS then standardized to an OD550 of 0.2, except for S. mitis, which was standardized to an OD550 of 0.5, in a colorimeter to obtain approximately 1 × 108 cfu/mL of each bacterial species on their specified day of use.
Active compounds
Chlorhexidine (CHX [Sigma]) and resveratrol (RSV [Sigma]) were used throughout this study. CHX solution was prepared at 0.01, 0.05 and 0.2% v/v in keratinocyte serum free media (KSFM) and used for subsequent antimicrobial testing. RSV powder was solubilised in ddH2O prior to preparation in KSFM at 0.01, 0.05 and 0.5% v/v and used for subsequent cell stimulation studies.
Development of multi-species biofilms
Bacteria were standardized (1 × 107 cfu/mL) in artificial saliva (AS), which contained the following constituents, as described previously [11]. This included porcine stomach mucins (0.25% w/v), sodium chloride (0.35 w/v), potassium chloride (0.02 w/v), calcium chloride dihydrate (0.02 w/v), yeast extract (0.2 w/v), lab lemco powder (0.1 w/v), proteose peptone (0.5 w/v) in ddH2O (Sigma, Poole, UK). Urea was then added to independently to a final concentration of 0.05% (v/v). To initiate multispecies biofilm development the pioneer species S. mitis biofilm were first formed for 24 h in 5% CO2 on 13 mm diameter Thermanox™ coverslips within 24 well plates (Corning, NY, USA). The supernatant was then removed and F. nucleatum added, which was incubated anaerobically at 37°C for a further 24 h. The supernatant was removed and P. gingivalis and A. actinomycetemcomitans added to the dual species biofilm, which was incubated anaerobically at 37°C for a further 4 days, replacing the AS daily to produce a mixed four species biofilm (Figure 1A).

Development of multi-species biofilm co-culture model. A. Multi-species biofilm culture protocol. Thermanox™ coverslips were placed in 24 well plates for biofilm culture. Bacteria were grown on appropriate agar plates and cultured in broth for 1–2 days. Cultures were then washed three times in PBS and standardized at 1×109 CFU/ ml, added to artificial saliva to make a 1×108 CFU/ml final volume and added to the biofilm on the appropriate days and cultured in aerobic and anaerobic conditions. After 7 days culture artificial saliva is removed from mature biofilms which are then washed with PBS and stored at -80°C. B. Hanging basket co-culture model. Multi-species biofilms were grown on Thermanox™ coverslips and previously described. Epithelial cells (OKF6-TERT2) were seeded in 24 well plates at 1×105 cells/ml in cell culture media. Biofilms were attached inversely to Millipore cell culture inserts using Vaseline® and placed over wells containing cells. Cells and biofilms were co-cultured for 4 and 24 h.
Quantitative analysis of biofilm composition
Real-time quantitative PCR (qPCR) was then performed to enumerate the definitive and relative composition of the biofilms. Briefly, bacterial biofilms were removed by sonication in a sonic bath at 35 kHz for 10 min, as previously described [12]. For qPCR the biofilm sonicate was used for DNA extraction using the MasterPure Gram Positive DNA Purificiation Kit (Epicentre®, Cambridge, UK), following manufacturers instructions, with the modification that the sonicate was incubated for 4 h to ensure cell lysis. The extracted DNA underwent quality checks using the NanoDrop spectrophotometer (Fischer Scientific, Loughborough). Briefly, 1 μL of extracted DNA was added to a mastermix containing 12.5 μL SYBR® GreenER™, 9.5 μL UV-treated RNase-free water and 1 μL of 10 μM forward/reverse primers for each bacterial species. The primers used were previously published, as listed in Table 1. Three independent replicates from each parameter were analysed in triplicate using MxProP Quantitative PCR machine and MxProP 3000 software (Stratagene, Amsterdam, Netherlands). Samples were quantified to calculate the colony forming equivalent (CFE) based upon a previously established standard curve of bacterial colony forming units ranging from 1 × 103 to 108 cfu/mL. The R2 values for these standard curves ranged from 0.956 to 0.994. Melting curve analysis was performed for all primer sets to ensure a single peak, which was indicative of primer specificity. Biofilm architecture was subsequently analysed by scanning electron microscopy (SEM). For this biofilm specimens were washed in PBS, fixed in 2% para-formaldehyde, 2% gluteraldehyde, 0.15% w/v Alcian Blue in 0.15 M sodium cacodylate (pH 7.4) [13]. The fixed and dried samples were sputter-coated with gold and viewed under a JEOL JSM-6400 scanning electron microscope [14].
Development of an epithelial biofilm co-culture model
OKF6-TERT2 cells (kind gift of the Rheinwald laboratory, Brigham and Woman’s Hospital, Boston) are an immortalized human oral keratinocyte cell line [20] was used throughout these investigations. OKF6 cells were cultured in keratinocyte serum-free medium (KSFM) as previously described [21]. At 90% confluence the cells were trypsinised, washed in Hanks balanced salt solution then re-seeded to approximately 1 × 105 cells/mL and seeded onto a Thermanox™ coverslip within a 24 well cell culture plate (Corning, NY, USA). The epithelial cells were washed and then subject to challenge with inverted biofilms, attached using Vaseline® to hanging cell culture inserts (Millipore, MA, USA) as illustrated in Figure 1B. The biofilms on the Thermanox™ discs were separated from the epithelial cells on the bottom of the well by a small space of <0.5 mm, representative of a gingival crevice. Multispecies biofilms were incubated in the co-culture model for 4 and 24 h and the viability of the epithelial cells ± active treatments (CHX and RSV) evaluated using a metabolic assay of 10% v/v alamarBlue®, according to the manufacturer’s instructions (Life Technologies, Paisley, UK). After 4 h incubation the absorbance was read at 570 nm and the reference wavelength at 600 nm, and the percentage reduction in biofilm viability calculated using the manufacturer’s formula.
Assessing inflammatory changes during biofilm co-culture
Supernatants and cell lysates were collected from OKF6/TERT2 cells stimulated with different multispecies biofilms, which were used for protein and transcriptional analysis, respectively, to assess the regulation of pro-inflammatory mediators. Initial gene expression analysis was carried out using a custom designed RT2 Profiler PCR Array (Qiagen, Crawley, UK). RT2 Profiler arrays are a SYBR® GreenER™ based real-time PCR that allow for the detection of several genes of interest, simultaneously. Briefly, RNA was extracted from cell lysates (Qiagen, Crawley, UK) and 55 ng/μl of cDNA synthesised using the RT2 First Strand cDNA synthesis kit (Qiagen, Crawley, UK), as per manufacturers instructions. Briefly, 24 μl of a mastermix containing SYBR® GreenER™, cDNA synthesised using the RT2 First Strand kit (Qiagen) and RNase-free water was added to each well of the RT2 Profiler plate, which already contained the forward and reverse primers for the genes of interest (IL-1α, IL-1β, IL-6, TNF, CSF2, CSF3, IL-8, CXCL1, CXCL3, CXCL5, CCL1 and GAPDH), based on an experimental gingivitis model [22]. Two replicates of each condition were used in the RT2 Profiler, which was carried out on two separate occasions.
Further verification was performed using IL-8 gene expression analysed using SYBR® Green based qPCR (Invitrogen), using GAPDH as a housekeeping gene. Primer sequences and reference sources are listed in Table 1. All primers were tested against each bacterial species to ensure specificity, which was the case (data not shown). RNA was synthesised into cDNA then added to a mastermix containing 12.5 μl SYBR® GreenER™, 10.5 μl UV-treated RNase-free water and 0.5 μl of forward/reverse primers. Three independent replicates from each parameter were analysed in duplicate using MxProP Quantitative PCR machine and MxProP 3000 software (Stratagene, Amsterdam, Netherlands) and gene expression normalised to the housekeeping gene GAPDH according to the 2-ΔΔCT method [23]. IL-8 release into cell culture supernatants was assessed by ELISA (Invitrogen, Paisley, UK), as per manufacturer’s instructions. Results were calculated using a 4-parameter curve fit, quantifying colometric changes at 630 nm (BMG-Labtech, Ortenberg, Germany).
Statistical analysis
Graph production, data distribution and statistical analysis were performed using GraphPad Prism (version 4; La Jolla, CA, USA). After assessing whether data conformed to a normal distribution by before and after data transforms, One-way Analysis of Variance (ANOVA) and t tests were used to investigate significant differences between independent groups of data that approximated to a Gaussian distribution. A Bonferroni correction was applied to the p value to account for multiple comparisons of the data. Non-parametric data was analysed using the Mann–Whitney U-test to assess differences between two independent sample groups. Student t-tests were used to measure statistical differences between the ΔCt values of the two independent groups assessed in gene expression studies, although data may be represented as percentage or fold change in the figures. Statistical significance was achieved if P < 0.05.
Results
Quantitative analysis of a multi-species periodontal biofilm model
Sonicated multi-species biofilms were quantified by qPCR (Figure 2A). It was shown that quantitatively that S. mitis was the most dominant species within the mature biofilm (1.36 × 107 CFE/mL; 83.24%), followed by F. nucleatum (2.28 × 106; 15.16%), P. gingivalis (3.53 ×10 4; 1.01%) and A. actinomycetemcomitans (1.13 × 105; 0.58%). The biofilm composition was then examined using SEM analysis. The biofilm was shown to be a dense complex of different morphotypes dominated by F. nucleatum (Figure 2B and C).

Analysis of multi-species biofilms in anaerobic and aerobic conditions. Multi-species biofilm were grown on Thermanox™ coverslips in 24 well plates for 6 days as previously described. After maturation DNA was extracted using Masterpure™ Gram-positive DNA purification kit for quantification of each species using SYBR® GreenER™ based qPCR (A). Biofilm morphology was analyzed by SEM at 2000x (B) and 5000x (C). Samples were processed and viewed on a JEOL JSM-6400 scanning electron microscope and images assembled using Photoshop software. Biofilms can be seen to be complex (B). At higher magnifications different morphologies can be identified with S. mitis and F. nucleatum making up the majority of the biofilm (C). Mature biofilms were also cultured in cell culture media in 5% CO2 for 4 and 24 h before DNA extraction and quantification of each species using using SYBR® GreenER™ based qPCR (D). All samples were assayed in triplicate on three separate occasions. Data are mean ± SD.
In order to test the multi-species biofilms in co-culture with epithelial cells, we examined the impact of moving the biofilm from anaerobic conditions within AS to 5% CO2 in KSFM. To do this we evaluated the biofilm composition as described above. No significant differences were observed in the composition and quantity of the 4 species within the CO2 biofilm at 4 and 24 h (Figure 2D). There was also no significant difference between 24 h biofilms formed under anaerobic conditions and those then placed in CO2 for a further 24 h. These biofilms were then used in a co-culture system to investigate the biological properties of two different bioactive agents.
Investigating the effects of bioactive agents in a multi-species biofilm epithelial cell co-culture model
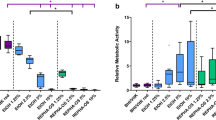
First, to optimise the concentrations of RSV and CHX to be tested in this model we undertook cytoxicity tests on both epithelial cells and on biofilms.Oral epithelial cells were treated for 30 min with three concentration of the antimicrobial active CHX (0.01, 0.05, 0.2% v/v) and anti-inflammatory active RSV (0.01, 0.05, 0.5% w/v) before washing with PBS and incubated for 4 and 24 h before cell viability was measured assessed using an AlamarBlue® assay. The data showed a significant decrease (p < 0.001) in cell viability compared to the untreated media controls when epithelial cells are incubated with CHX at any concentration for both 4 and 24 h (Figure 3A). When epithelial cells were co-cultured with RSV significant decreases in cell viability compared with the media control were observed using 0.05 and 0.5% w/v RSV concentrations at both 4 (p < 0.001) and 24 h (p < 0.01), with an approximately 50% decrease in cell viability at each time (Figure 3B).Next, from these data the concentrations taken forward for the remainder of the study were 0.2% v/v CHX and 0.01% w/v RSV. Multi-species biofilms were treated with the chosen concentrations of actives for 30 min and biofilm viability measured using an AlamarBlue® assay (Figure 3C). Treatment of the multi-species biofilm with RSV did not significantly affect biofilm viability compared to the untreated biofilm control. However, treatment with CHX showed a significant reduction (p < 0.001) of 75% in bacteria viability compared with the untreated biofilm.To determine if treatment with these actives altered the species composition of the biofilm, they were treated for 30 min then DNA extracted and quantification of each species performed by qPCR (Figure 3D). The data show no significant change in the composition following a 30 min treatment with CHX or RSV compared with the untreated biofilm. To further investigate this effect SEM analysis was performed to examine the impact of each active on the architecture of the biofilms post-treatment (Figure 3E-J). CHX appeared to destabilise the biofilm, as the complexity of the biofilm was reduced, whereas RSV appeared to have no visual effect on architecture.

Direct effect of actives on oral epithelial cells and multi-species biofilms. The oral epithelial cell line OKF6-TERT2 was seeded at 1×105 cells/ ml in 24 well plates for toxicity studies. Cells were treated with concentrations of CHX (0.01, 0.05, 0.2% v/v) (A) and RSV (0.01, 0.05, 0.5% w/v) (B) for 30 minutes before washing with PBS. Cell viability was assessed using the Alamarblue® assay with absorbance read at 570 nm and 600 nm. Following this concentrations chosen for the remainder of the study were CHX 0.2% (v/v) and RSV 0.01% (w/v). To investigate the role of actives in biofilm treatment biofilms were grown on coverslips in 24 well plates for 6 days as previously described. Mature biofilms were treated with 0.2% v/v CHX and 0.01% w/v RSV for 30 minutes before washing with PBS. Biofilm viability was measured using Alamarblue® read at 570 nm and 600 nm (C). Bacterial DNA was extracted and each species quantified using SYBR® GreenER™ based qPCR (D). Biofilms were also analyzed by SEM at both 2000x (E, G, I) and 5000× (F, H, J). Samples were processed and viewed on a JEOL JSM-6400 scanning electron microscope and images assembled using Photoshop software. Untreated biofilms (E, F) were compared with biofilms treated with 0.2% (v/v) CHX (G, H) and and 0.01% (w/v) RSV (I, J). Samples were assayed in triplicate on three separate occasions and data are mean ± SD (**p < 0.01, ***p < 0.001).
Next, we evaluated the effects of the untreated multi-species biofilm stimulated OKF6 cells to ensure the biofilm induced inflammatory mediators. Using the RT2 Profiler we compared biofilm stimulated cells to media control cells after 4 h to determine the inflammatory characteristics of the model (Table 2). Significant increases were observed for all the genes upon the RT2 profiler selected based on their expression during induced experimental gingivitis in human subjects, ranging from 4.39 fold change (CCL1) to 249.8 fold change (IL-8), with an mean fold change of 60 compared to the unstimulated media control. These data were verified by investigating IL-8 using specific primers within a reverse transcriptase (RT) qPCR assay (Figure 4B). A significant increased was observed of a 15.57 fold change (p < 0.001) after 4 h and 312.88 fold change (p < 0.001) after 24 h when compared to the media control (Table 2). Finally, we evaluated the cell supernatants for the release of IL-8 within this system, where it was shown that after 4 h (535 ng) and 24 h (450 ng) that IL-8 release was significantly increased (p < 0.001) compared to the untreated media controls (Figure 4C). Based on this collective data, we have demonstrated that the 4-species biofilm has reproducible inflammatory properties within an epithelial co-culture model.

CHX and RSV immunomodulate epithelial cell responses to multi-species biofilm co-culture in vitro. The oral epithelial cell line OKF6-TERT2 was seeded at 1×105 cells/ml in 24 well plates for co-culture with multi-species biofilms. Biofilm were pre treated with 0.2% (v/v) CHX or cells were treated with 0.01% (w/v) RSV for 30 minutes before washing with PBS and then co-cultured with untreated biofilms or cells respectively for 4 and 24 h. Untreated control co-culture were also included. RNA was extracted from the cell lysates at 4 h, cDNA was synthesized and pro-inflammatory gene expression quantified on an RT2 profiler plate (A). Duplicate samples from three independent experiments were used and data are mean ± SD relative to media control. Samples were also assayed for IL-8 relative to housekeeping gene GAPDH at 4 and 24 h using using SYBR® GreenER™ based qPCR (B). Supernatants were also analyzed for exogenous IL-8, measured by ELISA (C). Samples were assayed in triplicate from three independent experiments, data are mean ± SD (**p < 0.01, ***p < 0.001).
We then determined how CHX treatment of the biofilm affected inflammation. The co-culture model was utilised with biofilms treated with 0.2% CHX for 30 min. Following co-culture epithelial cell lysates and supernatants were removed to be tested for increased gene and protein inflammatory markers. An untreated biofilm and epithelial cells not co-cultured with the multi-species biofilm were used as controls. After 4 h co-stimulation with the CHX treated biofilms we showed no significant difference in gene expression using the RT2 profiler, though IL-8 and CXCL1 were notably decreased (Figure 4A). Further analysis of IL-8 using qPCR again showed no significant difference in expression at 4 h, whereas at 24 h a significant decrease of 5.65 fold was observed in the CHX treated biofilm stimulated cells (p > 0.001) (Figure 4B). IL-8 protein expression was also measured at 4 and 24 h by ELISA. A significant reduction (p < 0.001) of IL-8 protein was observed at 4 h (10 fold) and at 24 h (13 fold) when compared to the untreated control (Figure 4C).
Finally, we evaluated the effect of RSV treated epithelial cells in the co-culture model. Epithelial cells were treated for 30 min with RSV, washed and co-incubated with untreated biofilms for 4 and 24 h. The RT2 profiler showed no significant differences in gene expression after 4 h compared with untreated biofilms (Figure 4A). Further analysis of IL-8 at 4 and 24 h also showed no significant differences in expression levels (Figure 4B). However, a significant decrease in IL-8 protein levels at 4 h (25 fold; p < 0.001) and 24 h (13 fold; p < 0.01) were observed when compared to the untreated biofilm co-culture model (Figure 4C).
Discussion
Using experimental in vitro biofilm models to investigate interactions between biofilms and host epithelial cells is essential to understanding the mechanisms of disease pathology and how actives can influence the host response. Using this bespoke multi-species biofilm model we have demonstrated that the epithelial inflammatory response to multi-species biofilms can be altered in the presence of antimicrobial and anti-inflammatory compounds.
The data show co-culture of epithelial cells and untreated multi-species biofilms produce a significant increase in both gene and protein expression at 4 and 24 h. IL-8 protein levels were significantly increased at both 4 and 24 h in co-culture of epithelial cells and untreated multi-species biofilms compared with the cells only control. Additionally, increases in gene expression of a variety of pro-inflammatory chemokines and cytokines including IL-6, IL-8, TNF, CSF-2, CXCL1 and CXCL3 at 4 h compared with the cells only control were observed. These dynamic changes in pro-inflammatory mediators demonstrate that there is interplay between the complex biofilm and the epithelial cells. Notably, at 24 h the RNA quality from the co-culture model was poor and therefore not used to test gene expression using the RT2 profiler assay, however IL-8 was able to be measured at 24 h using SYBR® GreenER™ based qPCR, and was significantly increased compared to media controls. Pro-inflammatory responses by epithelial cells after challenge with oral biofilms have been previously reported using a variety of oral associated bacteria [24, 25]. Recently, it has been shown that biofilms including the ‘red complex’ organisms such as P. gingivalis or F. nucleatum increased IL-8 production during early host interaction, yet with longer exposure the chemotactic factors are downregulated, presumably through the release of proteolytic gingipains [25]. These studies also highlight the essential role of studying epithelial responses to bacteria in a biofilm model compared with planktonic bacteria and suggest biofilms behave differently to their single species counterparts, which may be in part due to the complex biofilm interaction that occur in a microbial community [26].
We have also demonstrated how antimicrobial and anti-inflammatory actives can influence the host response in this co-culture model. CHX while a potent antimicrobial, also has a high toxicity level when used to treat epithelial cells and in this study it was therefore decided to treat the biofilms with CHX before co-culture. Our data show that biofilms treated for 30 min with CHX have significantly decreased bacterial viability. However, differences were observed between the quantitative analysis of the biofilm composition by qPCR and SEM after 30 min treatment with CHX. These are most likely due to the substantivity of CHX where straight after treatment bacteria are dead but not dissociated from the biofilm, which is shown in the qPCR composition. After treatment and 4 h co-culture images by SEM show the biofilm is less complex which may be due to the loss of the dead bacteria from the biofilm as the effects of CHX decrease over time. This data agrees with previous work of the mechanisms of action of CHX which show an immediate bacteriocidal action combined with a prolonged bacteriostatic action due to absorption of the active to the surface [27]. Epithelial cells co-cultured with CHX treated multi-species biofilms did not show a significant change in overall gene expression at 4 h but a significant decrease in IL-8 levels was observed at 24 h. However, many of the other pro-inflammatory molecules did tend to decrease at 4 h, such as IL-6 and IL-1β, though increased others such as CCL1. Levels of IL-8 protein were significantly decreased at both 4 and 24 h. Clinical data of studies using CHX mouthwashes show significant reductions in inflammation following CHX treatment than control groups [28]. This suggests that reduction in inflammatory markers may be time dependent and linked to the disaggregation of dead bacteria from the surface of the biofilm, though the release of bacterial products may inadvertently activate other pathways.
Testing the anti-inflammatory active RSV we observed no significant decrease in biofilm viability or epithelial cell viability at 0.01%. However a significant decrease in epithelial cell viability was observed at concentrations of 0.05% and 0.5%. RSV has been reported to have antimicrobial effects both against planktonic and bacterial biofilms, with most studies focusing on bacteria such as P. aeruginosa and E. coli with MIC ranging from 5-50 μg/ml [29, 30]. There are no previous studies investigating the toxicity of RSV on epithelial cells. However. studies have shown RSV induces apoptosis in cancer cell lines at 0.1-0.3% [31]. Epithelial cells treated with RSV and co-cultured with multi-species biofilms showed no significant differences in overall gene expression compared with the untreated controls, though CXCL5, the neutrophil activating peptide was decreased transcriptionally, which may be involved in reducing neutrophil activation [32]. Moreover, a significant decrease in IL-8 protein expression was observed at 4 and 24 h compared with the untreated control. This suggests that RSV may disrupt the signalling pathway and result in reduced chemokine and cytokine production. A variety of studies investigating the role of RSV and inflammation have found RSV modulates a variety of cell responses by interfering with signalling pathways [33, 34]. With regards to our own work previous studies have reported RSV modulates pro-inflammatory responses by modulating NF-κB and AP-1 [35, 36].
Conclusions
This study has investigated the inflammatory response by epithelial cells to oral biofilms in co-culture and how actives can mediate this response. We have shown that multi-species biofilm treatment with the antimicrobial CHX is able to reduce inflammation produced by epithelial cells at both a gene and protein level. Treatment of epithelial cells with the anti-inflammatory compound RSV results in a reduction of epithelial cell inflammation at protein level. Although the main limitation of this model may be the number of species within the biofilm consortium, this does allow more control and reproducibility in the model. However, increasing the complexity of this model is subject to further work by our group. Collectively, this work supports the generation of a validated multi-species periodontal biofilm model suitable for use in the screening and testing of potential oral hygiene actives.
Authors’ information
Infection and Immunity Research Group, Glasgow Dental School, School of Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow.
References
Darveau RP: Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010, 8 (7): 481-490.
Wade WG: The oral microbiome in health and disease. Pharmacol Res. 2013, 69 (1): 137-143.
Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL: Microbial complexes in subgingival plaque. J Clin Periodontol. 1998, 25 (2): 134-144.
Zarco MF, Vess TJ, Ginsburg GS: The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Dis. 2012, 18 (2): 109-120.
Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA: Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011, 10 (5): 497-506.
Fujise O, Hamachi T, Inoue K, Miura M, Maeda K: Microbiological markers for prediction and assessment of treatment outcome following non-surgical periodontal therapy. J Periodontol. 2002, 73 (11): 1253-1259.
Herrera D: Chlorhexidine mouthwash reduces plaque and gingivitis. Evid Based Dent. 2013, 14 (1): 17-18.
Palaska I, Papathanasiou E, Theoharides TC: Use of polyphenols in periodontal inflammation. Eur J Pharmacol. 2013, 720 (1–3): 77-83.
Casati MZ, Algayer C, Cardoso da Cruz G, Ribeiro FV, Casarin RC, Pimentel SP, Cirano FR: Resveratrol decreases periodontal breakdown and modulates local levels of cytokines during periodontitis in rats. J Periodontol. 2013, 84 (10): e58-e64.
Shan Z, Yang G, Xiang W, Pei-Jun W, Bin Z: Effects of resveratrol on oral squamous cell carcinoma (OSCC) cells in vitro. J Cancer Res Clin Oncol. 2014, 140 (3): 371-374.
Pratten J, Smith AW, Wilson M: Response of single species biofilms and microcosm dental plaques to pulsing with chlorhexidine. J Antimicrob Chemother. 1998, 42 (4): 453-459.
Tunney MM, Patrick S, Gorman SP, Nixon JR, Anderson N, Davis RI, Hanna D, Ramage G: Improved detection of infection in hip replacements. A currently underestimated problem. J Bone Joint Surg (Br). 1998, 80 (4): 568-572.
Sherry L, Millhouse E, Lappin DF, Murray C, Culshaw S, Nile CJ, Ramage G: Investigating the biological properties of carbohydrate derived fulvic acid (CHD-FA) as a potential novel therapy for the management of oral biofilm infections. BMC Oral Health. 2013, 13 (1): 47-
Erlandsen SL, Kristich CJ, Dunny GM, Wells CL: High-resolution visualization of the microbial glycocalyx with low-voltage scanning electron microscopy: dependence on cationic dyes. J Histochem Cytochem. 2004, 52 (11): 1427-1435.
Ramage G, Milligan S, Lappin DF, Sherry L, Sweeney P, Williams C, Bagg J, Culshaw S: Antifungal, cytotoxic, and immunomodulatory properties of tea tree oil and its derivative components: potential role in management of oral candidosis in cancer patients. Front Microbiol. 2012, 3: 220-
Periasamy S, Chalmers NI, Du-Thumm L, Kolenbrander PE: Fusobacterium nucleatum ATCC 10953 requires Actinomyces naeslundii ATCC 43146 for growth on saliva in a three-species community that includes Streptococcus oralis 34. Appl Environ Microbiol. 2009, 75 (10): 3250-3257.
Sanchez MC, Marin MJ, Figuero E, Llama-Palacios A, Leon R, Blanc V, Herrera D, Sanz M: Quantitative real-time PCR combined with propidium monoazide for the selective quantification of viable periodontal pathogens in an in vitro subgingival biofilm model. J Periodontal Res. 2014, 49 (1): 20-28.
Boutaga K, van Winkelhoff AJ, Vandenbroucke-Grauls CM, Savelkoul PH: Comparison of real-time PCR and culture for detection of Porphyromonas gingivalis in subgingival plaque samples. J Clin Microbiol. 2003, 41 (11): 4950-4954.
van der Reijden WA, Brunner J, Bosch-Tijhof CJ, van Trappen S, Rijnsburger MC, de Graaff MP, van Winkelhoff AJ, Cleenwerck I, de Vos P: Phylogenetic variation of Aggregatibacter actinomycetemcomitans serotype e reveals an aberrant distinct evolutionary stable lineage. Infect Genet Evol. 2010, 10 (7): 1124-1131.
Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG: Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000, 20 (4): 1436-1447.
Wollert T, Rollenhagen C, Langford GM, Sundstrom P: Human oral keratinocytes: a model system to analyze host-pathogen interactions. Methods Mol Biol. 2012, 845: 289-302.
Offenbacher S, Barros SP, Paquette DW, Winston JL, Biesbrock AR, Thomason RG, Gibb RD, Fulmer AW, Tiesman JP, Juhlin KD, Wang SL, Reichling TD, Chen KS, Ho B: Gingival transcriptome patterns during induction and resolution of experimental gingivitis in humans. J Periodontol. 2009, 80 (12): 1963-1982.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25 (4): 402-408.
Dickinson BC, Moffatt CE, Hagerty D, Whitmore SE, Brown TA, Graves DT, Lamont RJ: Interaction of oral bacteria with gingival epithelial cell multilayers. Mol Oral Microbiol. 2011, 26 (3): 210-220.
Belibasakis GN, Thurnheer T, Bostanci N: Interleukin-8 responses of multi-layer gingival epithelia to subgingival biofilms: role of the “red complex” species. Plos One. 2013, 8 (12): e81581-
Peyyala R, Kirakodu SS, Novak KF, Ebersole JL: Oral epithelial cell responses to multispecies microbial biofilms. J Dent Res. 2013, 92 (3): 235-240.
Jenkins S, Addy M, Wade W: The mechanism of action of chlorhexidine. A study of plaque growth on enamel inserts in vivo. J Clin Periodontol. 1988, 15 (7): 415-424.
Turkoglu O, Becerik S, Emingil G, Kutukculer N, Baylas H, Atilla G: The effect of adjunctive chlorhexidine mouthrinse on clinical parameters and gingival crevicular fluid cytokine levels in untreated plaque-associated gingivitis. Inflamm Res. 2009, 58 (5): 277-283.
Lee JH, Kim YG, Ryu SY, Cho MH, Lee J: Resveratrol Oligomers Inhibit biofilm formation of Escherichia coli O157:H7 and Pseudomonas aeruginosa. J Nat Prod. 2014, 77 (1): 168-172.
Chan MM: Antimicrobial effect of resveratrol on dermatophytes and bacterial pathogens of the skin. Biochem Pharmacol. 2002, 63 (2): 99-104.
Joe AK, Liu H, Suzui M, Vural ME, Xiao D, Weinstein IB: Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res. 2002, 8 (3): 893-903.
Lappin DF, Murad M, Sherrabeh S, Ramage G: Increased plasma levels epithelial cell-derived neutrophil-activating peptide 78/CXCL5 in periodontitis patients undergoing supportive therapy. J Clin Periodontol. 2011, 38 (10): 887-893.
Vergara D, Simeone P, Toraldo D, Del Boccio P, Vergaro V, Leporatti S, Pieragostino D, Tinelli A, De Domenico S, Alberti S, Urbani A, Salzet M, Santino A, Maffia M: Resveratrol downregulates Akt/GSK and ERK signalling pathways in OVCAR-3 ovarian cancer cells. Mol Biosyst. 2012, 8 (4): 1078-1087.
Herbert KJ, Snow ET: Modulation of arsenic-induced epidermal growth factor receptor pathway signalling by resveratrol. Chem Biol Interact. 2012, 198 (1–3): 38-48.
Kundu JK, Shin YK, Surh YJ: Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-kappaB and AP-1 as prime targets. Biochem Pharmacol. 2006, 72 (11): 1506-1515.
Jeong JY, Silver M, Parnes A, Nikiforow S, Berliner N, Vanasse GJ: Resveratrol ameliorates TNFalpha-mediated suppression of erythropoiesis in human CD34(+) cells via modulation of NF-kappaB signalling. Br J Haematol. 2011, 155 (1): 93-101.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6831/14/80/prepub
Acknowledgements
We are grateful to GlaxoSmithKline for supporting the studentship of EM and AJ. Margaret Mullin (University of Glasgow) for her assistance in scanning electron microscopy techniques.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
EM, LS, DFL, SC and GR have no competing interests. AJ, NP, AMM and JP are all employees of GlaxoSmithKline, the sponsor of the study.
Authors’ contributions
EM, AJ and LS participated in the study design, carried out the experimental studies on biofilms, performed statistical analysis and were responsible for the manuscript. DFL participated in study design, assisted with statistical support and helped draft the manuscript. NP, AMM, JP and SC contributed to study design and supervised manuscript writing. GR conceived the study, participated in study design, data analysis and was responsible for writing and submission of the final manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Millhouse, E., Jose, A., Sherry, L. et al. Development of an in vitroperiodontal biofilm model for assessing antimicrobial and host modulatory effects of bioactive molecules. BMC Oral Health 14, 80 (2014). https://doi.org/10.1186/1472-6831-14-80
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6831-14-80