
Abstract
Background
Matriptase is a type II transmembrane serine protease that is found on the surfaces of epithelial cells and certain cancer cells. Matriptase has been implicated in the degradation of certain extracellular matrix components as well as the activation of various cellular proteins and proteases, including hepatocyte growth factor and urokinase. Sunflower trypsin inhibitor-1 (SFTI-1), a cyclic peptide inhibitor originally isolated from sunflower seeds, exhibits potent inhibitory activity toward matriptase.
Results
We have engineered and produced recombinant proteins of the matriptase protease domain, and have determined the crystal structures of the protease:SFTI-1 complex at 2.0 Å as well as the protease:benzamidine complex at 1.2 Å. These structures elaborate the structural basis of substrate selectivity of matriptase, and show that the matriptase S1 substrate specificity pocket is larger enough to allow movement of benzamidine inside the S1 pocket. Our study also reveals that SFTI-1 binds to matriptase in a way similar to its binding to trypsin despite the significantly different isoelectric points of the two proteins (5.6 vs. 8.2).
Conclusions
This work helps to define the structural basis of substrate specificity of matriptase and the interactions between the inhibitor and protease. The complex structure also provides a structural template for designing new SFTI-1 derivatives with better potency and selectivity against matriptase and other proteases.
Similar content being viewed by others


Background
Matriptase is a type II transmembrane serine protease of the S1 trypsin-like family. Matriptase activity is down-regulated by its physiological inhibitor, hepatocyte growth factor activator inhibitor-1 (HAI-1) [1–3]. Matriptase is expressed in most epithelial cells and plays essential roles in the establishment and maintenance of epithelial integrity. New evidence suggests that matriptase is also expressed on mast cells, peripheral blood monocytes and B cells, implicating matriptase in the physiological and pathologic functions of these cells [4–6]. Knock down studies in mice have shown that the protease is important in postnatal survival, epidermal barrier formation, hair follicle growth and thymichomeostasis [7]. At the same time, genetic studies using zebra fish and mice have indicated that the activity of matriptase is critical for tissue-integrity and function, and must be strictly controlled by HAI-1 [8–11].
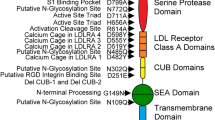
The catalytic domain of matriptase is tethered to the cell surface via its N-terminal signal anchor, linked by a sea urchin sperm protein/enterokinase/agrin (SEA) domain, two tandem complement/urchin embryonic growth factor/bone morphogenetic protein (CUB) domains, and four tandem low-density lipoprotein receptor class A (LDLRA) domains. Interestingly, matriptase activation does not depend on other active proteases. Instead, several lines of evidence have indicated that matriptase undergoes autoactivation through a mechanism relying on its own catalytic triad and requires its non-catalytic domains as well as the presence of its cognate inhibitor HAI-1 [12, 13]. Although the autoactivation mechanism is not fully understood, one study has showed that matriptase could be activated by acidification, and suggested that matriptase might act as an early response to cellular acidosis [14]. Once activated, matriptase has only short time to cleave and activate its substrates since the protease will be quickly inhibited by HAI-1.
Matriptase activates a number of substrates, including G-protein-coupled protease-activated receptor 2, urokinase plasminogen activator and pro-hepatocyte growth factor [15, 16]. Recently, it has been demonstrated that matriptase could also activate prekallikren either in vitro or in vivo[17]. Matriptase is recognized as a cancer-associated protease since the activation of urokinase plasminogen activator and/or pro-hepatocyte growth factor has been implicated in cancer invasion and metastasis (reviewed in [18]). In addition, matriptase has been found to be upregulated in various forms of cancers including breast, cervical, ovarian, liver, and prostate cancers. It has been demonstrated that the level of expression of matriptase correlates with the tumor stage and malignancy of breast, cervical, ovarian and prostate cancers [19–21]. In some of these cancers, the ratios of the protease relative to its inhibitor HAI-1 are unbalanced; suggesting that strict regulation of matriptase by HAI-1 is required to prevent carcinogenesis. A recent study showed that matriptase orthotopically overexpressed at modest levels in the skin of transgenic mice caused spontaneous squamous cell carcinoma, potentiated chemical carcinogenesis, and supported both ras-dependent and -independent carcinogenesis, whereas the overexpression of HAI-1 could nullify these oncogenic effects [22]. In addition to its role in cancers, recent studies have suggested that matriptase also has potential implications in a variety of diseases including osteroarthritis, atherosclerosis, and skin disorders like autosomal recessive ichthyosis and hypotrichosis (ARIH) [4, 23–26]. Taken together, matriptase has emerged as an attractive target for the development of anti-metastasis therapy as well as treatment for many other diseases.
Sunflower trypsin inhibitor-1 (SFTI-1), a 14-amino acid cyclic peptide, is originally isolated from sunflower seeds and characterized as the most potent peptidic inhibitor of trypsin (Ki = 0.1 nM and 1 nM from two independent studies) [27, 28]. A later study finds that synthetic SFTI-1 also exhibits very potent matriptase inhibitory activity (Ki = 0.92 nM) [27]. To evaluate the structural basis of the high inhibitory effect of SFTI-1 to matriptase, we have determined the X-ray structure of matriptase in complex with SFTI-1. We have also determined the high-resolution structure of matriptase:benzamidine complex for structural comparison. The crystal structures provide new insights into the molecular basis of matriptase inhibition and this information might facilitate future design of more potent and selective peptide inhibitors using SFTI-1 as template.
Results and Discussion
Engineering of recombinant matriptase catalytic domain in P. pastoris for structural study
For our structural studies, we constructed a recombinant protease domain of matriptase (residue 615 to 854 of the EXPASY entry Q9Y5Y6) with a point mutation N164Q (chymotrypsin numbering will be used throughout the paper starting from here), which is referred as β-matriptase-N164Q. The point mutation removes a glycosylation site (N164) and allows the protein to be purified to homogeneity. Another unique feature of the current design of the expression scheme is that the secreted recombinant matriptase protease domain is an active serine protease without the need of being activated. This is due to the processing of the secreted protein by an endogenous kex2 enzyme of P. pastoris that generates the genuine N-terminus of matriptase protease domain and allows the correct folding of the protease into its active form. We have used such approach to generate a number of active proteases including urokinase-type plasminogen activator [29], tissue-type plasminogen activator and coagulation factor XIa (to be published), suggesting that our method can be widely adapted for the expression of different active proteases.
Structure of β-matriptase-N164Q:benzamidine shows benzamidine mobility
In the structure of β-matriptase-N164Q complexed with benzamidine, the benzamidine binds to the specificity pocket S1 right next to the active site and is sandwiched by the segments of Ser190-Gln192 and Trp215-Gly216 as expected. This structure is similar to the previously reported structure of the matriptase protease domain in complex with benzamidine (PDB ID = 1EAX, RMSD = 0.1 Å for the Cα atoms of 227 residues) [30]. However, there are interesting differences between these two structures. The phenyl group of the benzamidine in our structure shows a 35° rotation when compared to the 1EAX structure. While the amidino group of the inhibitor juxtaposes the side chain of Asp189 and forms favorable ionic interaction with its carboxylate group as in the 1EAX structure, the bis-amine in our structure is rotated for ~20° and makes an extra hydrogen bond with the backbone carbonyl of Ser190, in addition to those formed with the Gly219 carboxylate and a conserved solvent molecule in 1EAX (Figure 1). Considering the high resolutions of both matriptase-benzamidine structures (1.2 Å in our structure) and the relatively small errors of structures (0.4 Å in our structure), such differences in benzamidines are significant, and indicates that S1 pocket is larger than the molecular size of benzamidine and allows movement of benzamidine inside the S1 specificity pocket. This is consistent with the observed higher temperature factor of benzamidine (40 Å2) comparing to the average temperature factor of protein (15 Å2).

Comparison of benzamidine orientations in two matriptase:benzamidine complexes. Benzamidine mediates extra hydrogen bonding and van der Waals interactions to the peptide segment of Ser190-Gln192 in our structure (blue) when compared to 1EAX (yellow).
Interactions between matriptase and SFTI-1 at the active site
Since SFTI-1 is a highly potent inhibitor to either matriptase or trypsin with comparable potencies, we want to investigate if the inhibitory mechanisms are the same for the two proteases. The crystal structure of β-matriptase-N164Q in complex with SFTI-1 was solved at 2.0 Å (Figure 2A). In this structure, SFTI-1 forms a bicyclic structure where two antiparallel β-strands are constrained by a disulfide bond between Cys3 and Cys11 (Figure 2B). The cyclic peptidic inhibitor docks onto matriptase's P4-P2' specificity sites with good shape and charge complementarity. This resembles the SFTI-1 binding to trypsin and other BBI to serine proteases, as well as the reactive site loop of the bovine pancreatic trypsin inhibitor (BPTI) to matriptase [28, 30–33]. Like the equivalent side chains in the other complexes, Lys5 from the "reactive loop" of SFTI-1 extends into the S1 pocket of matriptase to form a hydrogen bond directly with Ser190 and may also interact with Asp189 at the bottom of the pocket (Figure 2C and Table 1; Table 1 summarizes hydrogen bonds formed between matriptase and SFTI-1). The SFTI-1 scissile bond (Lys5-Ser6) is in close proximity to the catalytic residue Ser195 where the Ser195 hydroxyl group hydrogen bonds with the main-chain amide of Ser6 and makes close contacts with the backbone carbonyl and amide of Lys5 (2.9Å and 3.0 Å respectively). The side chain oxygen atom of Ser195 is also in close contact with the carbonyl carbon of P1 residue (Lys5) (2.6 Å) at an angle of 88.6° (Lys5O-C-Ser195OG), suggesting the protease is in an active conformation capable of initiating bond scission. Furthermore, His57 is suitably oriented to interact with Ser195 to support catalysis (at 2.6 Å and with Ser195CB-OG-His57NE2 angle of 101.4°). These observations support that SFTI-1 functions as a substrate variant of matriptase in a similar manner to other BBIs. Thus, the cyclic peptidic inhibitor is likely to inhibit the enzyme through high affinity binding to the active site and through excluding water molecules from the active site to destabilize the reaction-path transition state.

Crystal structure of matriptase:SFTI-1 complex. A) Overall structure of SFTI-1-bound matriptase in surface rendition (grey) with SFTI-1 in cyan. B) SFTI forms a bicyclic structure. 2Fo-Fc electron density map of the peptide inhibitor contoured at 1σ is shown. C) Interactions of matriptase (grey) and SFTI-1 in stereo view. Intermolecular hydrogen bonds formed between the protease and inhibitor are denoted by dashed lines.
Comparison of the conformations of matriptase in the benzamidine- and SFTI-1-bound forms reveals that they are nearly identical except the side chain of Phe99 at the S2 subsite, which will be discussed later, and Gln192 at the S3 subsite. Gln192 side chain is solvent-exposed in the benzamidine-bound structure and forms the lining of the extended active site groove. However, it undergoes a major conformational change to "bend inward" to accommodate SFTI-1 and hydrogen bonds with the backbone carbonyl of Thr4 of the cyclic inhibitor in a manner similar to that in the BPTI-bound matriptase structure.
Substrate selectivity of matriptase
Previous studies using positional scanning-synthetic combinatorial library and substrate phage display have revealed that matriptase also strongly prefers substrates with small residues including glycine and serine, or phenylalanine at the P2 position, in addition to the protease specificity for basic residue at the P1 pocket [16]. Moreover, the most effective substrate has been found to contain basic residues at the P3 or P4 sites, but not both simultaneously [16]. Indeed, the sequence of SFTI-1 matches these findings except the presence of a threonine at the P2 position. Our structure explains how SFTI-1 achieves such substrate selectivity. In the structure, the S2 subsite of matriptase is occupied by SFTI-1 Thr4 with Thr4 side chain flanked by His57 and Phe99. The methyl group of Thr4 makes CH-π interaction with both flanking side chains and docks snugly in the pocket. Comparing to the benzamidine-bound structure, we observed that the benzyl ring of Phe99 undergoes a rotation to widen the pocket to better accommodate the P2 residue (Figure 3A). Similar observation has also been made in the BPTI:matriptase complex [30]. This structural explanation for the suitability of threonine at P2 position suggests that the S2 pocket is not rigid and its size is controlled by the conformation of Phe99. A large residue at substrate P2 position will inevitably cause steric hindrance with His57 and alter its position, and will disrupt the hydrogen bond between Asp102 and His57 and hinder catalysis. Yet, despite its large size, a phenylalanine at P2 would make favorable π-π interactions with Phe99 and His57, and/or cation-π interaction when the histidine is protonated during catalysis, explaining why phenylalanine is also preferred in addition to small residues at this site [16].

Comparison of matriptase:SFTI-1 and trypisn:SFTI-1 complexes. A) The matriptase Phe99 side chains (grey) and SFTI Phe12 -1 (cyan) undergo large rotation to widen the S2 subsite when compared to the structures of matriptase in benzamidine-bound form (light blue) and trypsin: SFTI-1 complex (deep blue). B) SFTI-1 Arg2 is accommodated at the S4 subsite (in surface representation and colored by electrostatic potential, ±7 kT/e).
Comparison with the trypsin:SFTI-1 complex (PDB ID = 1SFI) reveals that the β-turn consisted of Phe12, Pro13, Asp14 and Gly1 of SFTI-1 adopts a slightly different conformation upon binding to matriptase [27, 28]. This is to avoid steric hindrance with the matriptase 99 loop. Particularly, both side chains of SFTI-1 Phe12 and matriptase Phe99 (Leu99 in trypsin) undergo large rotational changes to avoid clashing with each other (Figure 3A). In its new conformation, Phe12 is now stabilized by van der Waals interaction with the benzyl rings of Phe97 and Phe99. Gly1 consequently shifts slightly closer to the active site groove by ~0.6 Å, allowing its main-chain carbonyl to make an extra contact with the amide of Gly219. Interaction between Asp14 and Gln175 of trypsin is replaced with intramolecular interaction between Asp14 and Arg2 of SFTI-1, which stabilizes the conformation of Arg2 side chain to interact with the S4 subsite (Figure 3B). The backbone carbonyls of Asp95, Phe97 and Thr98 form the S4 subsite, which is confined and protected from the solvent by the side chains of Phe97 and Phe99 [30]. Previous positional scanning and substrate phage display studies performed by Takeuchi et al[16] had revealed that the S4 subsite of matriptase strongly prefers basic residues. Our structure provides a clear explanation on this selectivity. Firstly, unlike in the trypsin-SFTI-1 complex, the guanidinium group of Arg2 is anchored by Asp14 to form two hydrogen bonds with the carbonyl of Phe97. Secondly, the rearrangement of the benzyl groups of Phe97 and Phe99 results in a solvent-shielding environment that lowers the dielectric constant and thus stabilizes the hydrogen bonding interactions mediated by Arg2 (Figure 3B). More importantly, the guanidinium group of Arg2 forms cation-π interactions with both phenylalanines as well as Trp215 in T-shaped geometries and provides further stabilization [34, 35]. To confirm these interactions, we used the program CaPTURE to estimate the stabilizing energies from the cation-π interactions (Table 2) [34]. The interaction between Arg2 and the S4 subsite alone is estimated to contribute ~12 kcal/mol, suggesting that the S4 subsite may play a key role in matriptase specificity. Indeed, inhibitory assays with different SFTI-1 derivatives have shown that the S4 subsite strongly prefers arginine [36]. Minor modifications of the Arg2 guanidinium in SFTI-1 result in significant loss of matriptase inhibition, and replacement of Arg2 with a Phe(Glu) decreases the inhibitory activity by 900-fold [36].
Comparison of SFTI-1 binding to matriptase and trypsin
Matriptase protease domain contains many acidic residues, leading to a lower theoretical pI in comparison with bovine trypsin (5.6 vs. 8.2). In addition, the electrostatic surface potential of the active site groove of the matriptase protease domain is much more negatively charged than that of trypsin (Figure 4A and 4B). Thus, the basic SFTI-1 (pI = 8.1) would presumably favor matriptase binding than trypsin binding. However, SFTI-1 has been shown to inhibit trypsin and matriptase with comparable affinities [27], and we observed here that the binding interface of SFTI-1 to matriptase is structurally very similar to trypsin. Such apparent discrepancy may be due to the lower mobility of SFTI-1, and thus stronger binding, in the current matriptase:SFTI-1 crystal structure when compared to the trypsin:SFTI-1 structure (relative mobility of 0.7 vs. 1.5). Here the relative mobility is defined by the average temperature factor of SFTI-1 in the structure of the complexes divided, by the average temperature factor of the rest of protein in order to cancel out the effect of resolution on the temperature factor. Another possible explanation is to take into account the penalty of the loss of configurational entropy of both protein and ligand upon SFTI-1 binding [37]. For instance, Phe12 of SFTI-1 is more constrained in the matriptase complex than in the trypsin complex due to the presence of Phe97 and Phe99 that surround it, which might result in a lower torsional degree of freedom and yield an entropy penalty, compensating the gain of enthalpy from the charge complementarity.

Solvent-accessible surfaces of (A) matriptase and (B) trypsin are colored by electrostatic potential (-7 to +7 kT/e in blue to red) computed by APBS [48]. The matriptase active site surface is more acidic than that of trypsin.
Implication for future inhibitor design
The current structure provides a template for further improvement of SFTI-1. For instance, non-polar residues like Ile7 and Ile10 are good candidates for modifications of this bicyclic peptide to improve its binding enthalpy. Ile10 is located in a cavity formed by the surface exposed insertion loops (loops 66 and 99) of matriptase and is proximal to the active site. However, it does not make any direct contacts with residues from the protease. Previous work by Li et al. shows that Ile10 plays an important role in the selectivity of the inhibitor as its replacement by a more polar and bulky glutamine improves the compound selectivity for matriptase versus thrombin by >1073 fold [36]. Based on the electrostatic surface potential of matriptase calculated from our structure, the modification of SFTI-1 Ile10 to a positively charged amino acid might fit the cavity more tightly and provide a favorable enthalpy. However, increasing flexibility of the inhibitor might result in loss of configurational entropy upon binding, as illustrated by the higher Ki of the Gln10 derivative of SFTI-1 [36]. Therefore, flexible amino acids like lysine and arginine should be avoided for the substitution and small positively charged unnatural amino acids such as diaminopropionic acid or diaminobutyric acid may be used instead. These small side chains will likely favor the interaction with Asp96 without disrupting the conformation of the catalytic triad and improves the enthalpy of binding while minimizing the entropy penalty [38]. Similarly, Ile7 lies on top of a groove adjacent to the catalytic cleft of matriptase and its sole direct interaction with matriptase is through weak van der Waals interaction with Ile41. We believe our suggested strategy for the modification of Ile10 can also be applied to Ile7. Together, the combination of modifications at Ile7 and Ile10 should improve the inhibitory activity of SFTI-1 towards matriptase.
Conclusions
SFTI-1 is originally isolated and characterized as a potent trypsin inhibitor. It has also been synthesized by Roller's group and shown to exhibit potent inhibitory effect against matriptase. The same group also investigated the structural basis of the high inhibitory activity of SFTI-1 using molecular modeling study and obtained information that aids the design and synthesis of new SFTI-1 analogs. While modification to stabilize the disulfide bond within the cyclic peptide maintains the compound's inhibitory potency and selectivity of matriptase versus thrombin, replacement of Ile10 with the more polar glutamine improves selectivity towards matriptase at the expense of weakening its inhibitory activity. Nevertheless, none of the modified inhibitors show improvement in binding affinity to matriptase. This major drawback can now be overcome by better aid from the structural information of an experimentally obtained structure of matriptase:SFTI-1 complex. This work helps to define the structural basis of substrate specificity of matriptase and provides more details in the interactions between the inhibitor and protease. Our structure also reveals the structural difference between the SFTI-1 bound matriptase and trypsin complexes to allow development of more potent and selective inhibitors for matriptase.
Methods
Recombinant protein expression and Purification
The matriptase N164Q (chymotrypsin numbering) catalytic domain mutant was first generated by site-directed mutagenesis using cDNA encoding the entire protease domain of human matriptase as template. The cDNA was then amplified by PCR using primers containing XhoI and SalI restriction sites. The purified PCR products were digested with XhoI and SalI and subcloned into the XhoI-SalI sites of the Pichia pastoris (P. pastoris) expression vector pPICZαA (Invitrogen). Plasmid DNAs were linearized with the restriction enzyme SacI prior to transformation into P. pastoris strain X-33. Recombinant matriptases were expressed in P. pastoris according to the manufacturer's recommendations. P. pastoris expression medium was concentrated 10-20 fold using a Millipore concentrator (8000 Da MWCO membrane) and pH was adjusted to 7.4. The concentrated medium was applied onto a benzamidine column (GE Healthcare) equilibrated with 50 mM Tris, 0.5 M NaCl, pH7.4, and eluted with 100 mM glycine, pH 3.0. The elution fractions were neutralized with 1 M Tris pH 9 immediately. Fractions containing matriptase activity were pooled and concentrated, and passed through MonoQ column (Amersham Biosciences, Inc.) pre-equilibrated with 40 mM Tris. The protein was eluted in a buffer containing 40 mM Tris, pH7.4 with a 0-0.4 M NaCl gradient. Fractions containing protein were pooled and concentrated to 5 mg/ml. Aliquots of the purified protein were frozen at 193 K for crystallization experiments.
Crystallization of β-matriptase-N164Q complex with its inhibitors
For protein crystallization, β-matriptase-N164Q:benzamidine complexes were mixed at 1:10 ratio, and crystallized by the hanging-drop vapor diffusion method with a precipitant solution of 0.1 M Tris, pH 8.0, 1.5 M ammonium sulfate, 3% ethanol. SFTI-1 was synthesized by solid state synthesis as previously reported [39]. The complex of β-matriptase-N164Q with SFTI-1 was crystallized with a precipitant condition of 22% polyethylene glycol 8000, 0.1 M Tris pH 8 and 20 mM CaCl2.
Data collection, structure solution and refinement
X-ray diffraction data were collected at APS to 1.2 Å and 2.0 Å for β-matriptase-N164Q:benzamidine and β-matriptase-N164Q:SFTI-1 complexes, respectively (Table 3). The crystal structures of the complexes were solved using the program MOLREP [40] from the CCP4 program suite [41] and the published β-matriptase structure (Protein Data Bank (PDB) ID: 1EAX) [30] as the search model. After refinement using the REFMAC program [42], the Fo-Fc difference electron density showed extra electron density attaching to Cys122. Based on the contour of this extra electron density, a molecule of reduced glutathione (GSH) was modeled covalently linking to Cys122 (Additional file 1). In the structure of β-matriptase-SFTI-1 complex, SFTI-1 was modeled into the extra electron density around matriptase active pocket. The resulting models were refined using Phenix [43], and manual model fitting was carried out using the program COOT [44]. In the final cycles of refinement, TLS & restrained refinement with twenty TLS groups which generated by the TLS motion determination (TLSMD) server [45] was applied. Detailed refinement and structure statistics are listed in Table 3. The final structures were analyzed by PROCHECK [46] and PYMOL [47].
Abbreviations
- BBI:
-
Bowman-Birk inhibitor
- BPTI:
-
bovine pancreatic trypsin inhibitor
- HAI-1:
-
hepatocyte growth factor activator inhibitor-1
- PDB:
-
Protein Data Bank
- RMSD:
-
root mean square deviation
- SFTI-1:
-
Sunflower trypsin inhibitor-1.
References
Tanimoto H, Underwood LJ, Wang Y, Shigemasa K, Parmley TH, O'Brien TJ: Ovarian tumor cells express a transmembrane serine protease: a potential candidate for early diagnosis and therapeutic intervention. Tumour Biol 2001, 22(2):104–114. 10.1159/000050604
Lin CY, Anders J, Johnson M, Sang QA, Dickson RB: Molecular cloning of cDNA for matriptase, a matrix-degrading serine protease with trypsin-like activity. J Biol Chem 1999, 274(26):18231–18236. 10.1074/jbc.274.26.18231
Takeuchi T, Shuman MA, Craik CS: Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci USA 1999, 96(20):11054–11061. 10.1073/pnas.96.20.11054
Milner JM, Patel A, Davidson RK, Swingler TE, Desilets A, Young DA, Kelso EB, Donell ST, Cawston TE, Clark IM, et al.: Matriptase is a novel initiator of cartilage matrix degradation in osteoarthritis. Arthritis Rheum 2010, 62(7):1955–1966.
Kilpatrick LM, Harris RL, Owen KA, Bass R, Ghorayeb C, Bar-Or A, Ellis V: Initiation of plasminogen activation on the surface of monocytes expressing the type II transmembrane serine protease matriptase. Blood 2006, 108(8):2616–2623. 10.1182/blood-2006-02-001073
Cheng MF, Jin JS, Wu HW, Chiang PC, Sheu LF, Lee HS: Matriptase expression in the normal and neoplastic mast cells. Eur J Dermatol 2007, 17(5):375–380.
List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, Engelholm LH, Behrendt N, Bugge TH: Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 2002, 21(23):3765–3779. 10.1038/sj.onc.1205502
Szabo R, Molinolo A, List K, Bugge TH: Matriptase inhibition by hepatocyte growth factor activator inhibitor-1 is essential for placental development. Oncogene 2007, 26(11):1546–1556. 10.1038/sj.onc.1209966
Szabo R, Kosa P, List K, Bugge TH: Loss of matriptase suppression underlies spint1 mutation-associated ichthyosis and postnatal lethality. Am J Pathol 2009, 174(6):2015–2022. 10.2353/ajpath.2009.090053
Fan B, Brennan J, Grant D, Peale F, Rangell L, Kirchhofer D: Hepatocyte growth factor activator inhibitor-1 (HAI-1) is essential for the integrity of basement membranes in the developing placental labyrinth. Dev Biol 2007, 303(1):222–230. 10.1016/j.ydbio.2006.11.005
Carney TJ, von der Hardt S, Sonntag C, Amsterdam A, Topczewski J, Hopkins N, Hammerschmidt M: Inactivation of serine protease Matriptase1a by its inhibitor Hai1 is required for epithelial integrity of the zebrafish epidermis. Development 2007, 134(19):3461–3471. 10.1242/dev.004556
Oberst MD, Williams CA, Dickson RB, Johnson MD, Lin CY: The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J Biol Chem 2003, 278(29):26773–26779. 10.1074/jbc.M304282200
Lee MS, Tseng IC, Wang Y, Kiyomiya K, Johnson MD, Dickson RB, Lin CY: Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am J Physiol Cell Physiol 2007, 293(1):C95–105. 10.1152/ajpcell.00611.2006
Tseng IC, Xu H, Chou FP, Li G, Vazzano AP, Kao JP, Johnson MD, Lin CY: Matriptase activation, an early cellular response to acidosis. J Biol Chem 2010, 285(5):3261–3270. 10.1074/jbc.M109.055640
Lee SL, Dickson RB, Lin CY: Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem 2000, 275(47):36720–36725. 10.1074/jbc.M007802200
Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS: Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem 2000, 275(34):26333–26342. 10.1074/jbc.M002941200
Sales K, Masedunskas A, Bey A, Rasmussen A, Weigert R, List K, Szabo R, Overbeek P, Bugge T: Matriptase initiates activation of epidermal pro-kallikrein and disease onset in a mouse model of Netherton syndrome. Nat Genet 2010, 42(8):676–683. 10.1038/ng.629
List K: Matriptase: a culprit in cancer? Future Oncol 2009, 5(1):97–104. 10.2217/14796694.5.1.97
Saleem M, Adhami VM, Zhong W, Longley BJ, Lin CY, Dickson RB, Reagan-Shaw S, Jarrard DF, Mukhtar H: A novel biomarker for staging human prostate adenocarcinoma: overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiol Biomarkers Prev 2006, 15(2):217–227. 10.1158/1055-9965.EPI-05-0737
Lee JW, Yong Song S, Choi JJ, Lee SJ, Kim BG, Park CS, Lee JH, Lin CY, Dickson RB, Bae DS: Increased expression of matriptase is associated with histopathologic grades of cervical neoplasia. Hum Pathol 2005, 36(6):626–633. 10.1016/j.humpath.2005.03.003
Tanimoto H, Shigemasa K, Tian X, Gu L, Beard JB, Sawasaki T, O'Brien TJ: Transmembrane serine protease TADG-15 (ST14/Matriptase/MT-SP1): expression and prognostic value in ovarian cancer. Br J Cancer 2005, 92(2):278–283.
List K, Szabo R, Molinolo A, Sriuranpong V, Redeye V, Murdock T, Burke B, Nielsen BS, Gutkind JS, Bugge TH: Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev 2005, 19(16):1934–1950. 10.1101/gad.1300705
Seitz I, Hess S, Schulz H, Eckl R, Busch G, Montens HP, Brandl R, Seidl S, Schomig A, Ott I: Membrane-type serine protease-1/matriptase induces interleukin-6 and -8 in endothelial cells by activation of protease-activated receptor-2: potential implications in atherosclerosis. Arterioscler Thromb Vasc Biol 2007, 27(4):769–775. 10.1161/01.ATV.0000258862.61067.14
Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, Rainshtein L, Ben Amitai D, Lurie R, Pasmanik-Chor M, Indelman M, Zvulunov A, Saban S, et al.: Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am J Hum Genet 2007, 80(3):467–477. 10.1086/512487
Avrahami L, Maas S, Pasmanik-Chor M, Rainshtein L, Magal N, Smitt J, van Marle J, Shohat M, Basel-Vanagaite L: Autosomal recessive ichthyosis with hypotrichosis syndrome: further delineation of the phenotype. Clin Genet 2008, 74(1):47–53. 10.1111/j.1399-0004.2008.01006.x
Alef T, Torres S, Hausser I, Metze D, Tursen U, Lestringant GG, Hennies HC: Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. J Invest Dermatol 2009, 129(4):862–869. 10.1038/jid.2008.311
Long YQ, Lee SL, Lin CY, Enyedy IJ, Wang S, Li P, Dickson RB, Roller PP: Synthesis and evaluation of the sunflower derived trypsin inhibitor as a potent inhibitor of the type II transmembrane serine protease, matriptase. Bioorg Med Chem Lett 2001, 11(18):2515–2519. 10.1016/S0960-894X(01)00493-0
Luckett S, Garcia RS, Barker JJ, Konarev AV, Shewry PR, Clarke AR, Brady RL: High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol 1999, 290(2):525–533. 10.1006/jmbi.1999.2891
Zhao G, Yuan C, Wind T, Huang Z, Andreasen PA, Huang M: Structural basis of specificity of a peptidyl urokinase inhibitor, upain-1. J Struct Biol 2007, 160(1):1–10. 10.1016/j.jsb.2007.06.003
Friedrich R, Fuentes-Prior P, Ong E, Coombs G, Hunter M, Oehler R, Pierson D, Gonzalez R, Huber R, Bode W, et al.: Catalytic domain structures of MT-SP1/matriptase, a matrix-degrading transmembrane serine proteinase. J Biol Chem 2002, 277(3):2160–2168. 10.1074/jbc.M109830200
Li Y, Huang Q, Zhang S, Liu S, Chi C, Tang Y: Studies on an artificial trypsin inhibitor peptide derived from the mung bean trypsin inhibitor: chemical synthesis, refolding, and crystallographic analysis of its complex with trypsin. J Biochem 1994, 116(1):18–25.
Tsunogae Y, Tanaka I, Yamane T, Kikkawa J, Ashida T, Ishikawa C, Watanabe K, Nakamura S, Takahashi K: Structure of the trypsin-binding domain of Bowman-Birk type protease inhibitor and its interaction with trypsin. J Biochem 1986, 100(6):1637–1646.
Werner MH, Wemmer DE: Three-dimensional structure of soybean trypsin/chymotrypsin Bowman-Birk inhibitor in solution. Biochemistry 1992, 31(4):999–1010. 10.1021/bi00119a008
Gallivan JP, Dougherty DA: Cation-pi interactions in structural biology. Proc Natl Acad Sci USA 1999, 96(17):9459–9464. 10.1073/pnas.96.17.9459
Crowley PB, Golovin A: Cation-pi interactions in protein-protein interfaces. Proteins 2005, 59(2):231–239. 10.1002/prot.20417
Li P, Jiang S, Lee SL, Lin CY, Johnson MD, Dickson RB, Michejda CJ, Roller PP: Design and synthesis of novel and potent inhibitors of the type II transmembrane serine protease, matriptase, based upon the sunflower trypsin inhibitor-1. J Med Chem 2007, 50(24):5976–5983. 10.1021/jm0704898
Chang CE, Chen W, Gilson MK: Ligand configurational entropy and protein binding. Proc Natl Acad Sci USA 2007, 104(5):1534–1539. 10.1073/pnas.0610494104
Shakkottai VG, Regaya I, Wulff H, Fajloun Z, Tomita H, Fathallah M, Cahalan MD, Gargus JJ, Sabatier JM, Chandy KG: Design and characterization of a highly selective peptide inhibitor of the small conductance calcium-activated K+ channel, SkCa2. J Biol Chem 2001, 276(46):43145–43151. 10.1074/jbc.M106981200
Korsinczky MLJ, Schirra HJ, Rosengren KJ, West J, Condie BA, Otvos L, Anderson MA, Craik DJ: Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J Mol Biol 2001, 311(3):579–591. 10.1006/jmbi.2001.4887
Vagin A, Teplyakov A: MOLREP: an automated program for molecular replacement. J Appl Cryst 1997, 30: 1022–1025. 10.1107/S0021889897006766
CCP4: The CCP4 suite; Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994, 50: 760–763. 10.1107/S0907444994003112
Murshudov GN, Vagin AA, Dodson EJ: Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 1997, 53(Pt 3):240–255.
Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al.: PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66(2):213–221. 10.1107/S0907444909052925
Emsley P, Cowtan K: Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60: 2126–2132. 10.1107/S0907444904019158
Painter J, Merritt EA: TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr 2006, 39: 109–111. 10.1107/S0021889805038987
Laskowski RA, MacArthur MW, Mass DS, Thornton JM: PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993, 26: 283–291. 10.1107/S0021889892009944
DeLano WL: The PyMol Molecular Graphics system. DeLano scientific, San Carlos, CA, USA; 2002.
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA: Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA 2001, 98: 10037–10041. 10.1073/pnas.181342398
Jin L, Pandey P, Babine RE, Weaver DT, Abdel-Meguid SS, Strickler JE: Mutation of surface residues to promote crystallization of activated factor XI as a complex with benzamidine: an essential step for the iterative structure-based design of factor XI inhibitors. Acta Crystallogr D Biol Crystallogr 2005, 61: 1418–1425. 10.1107/S0907444905024340
Acknowledgements
The work was supported by Natural Science Foundation of China (30770429, 30811130467) and Fujian Province (2009J05091).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
MH designed the study, MH and JCN wrote and revised the manuscript, CY participated in the design of the study, carried out all experiments in molecular biology, protein chemistry, structure refinement and drafted the manuscript, ND and DJ synthesized SFTI-1 peptide, LC and EJM collected the X-ray data, and all authors read and approved the final manuscript.
Electronic supplementary material
12900_2011_374_MOESM1_ESM.TIFF
Additional file 1: Fig. S1. The extra electron density (in stereo representation) around the Cys122 of the β-matriptase-N164Q:benzamidine structure suggests the Cys122 is conjugated by a disulfide bond (yellow) to a glutathione (GSH, in green sticks). GSH might have attached to the protein during protein synthesis because GSH is one of the abundant intracellular sulfhydryl antioxidants in yeast as a similar observation was made in other structures including the rhFXI-benzamidine complex 49 where the protein was also expressed in P. pastoris . 2Fo-Fc electron density map is contoured at 1s at 1.2 Å. (TIFF 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yuan, C., Chen, L., Meehan, E.J. et al. Structure of catalytic domain of Matriptase in complex with Sunflower trypsin inhibitor-1. BMC Struct Biol 11, 30 (2011). https://doi.org/10.1186/1472-6807-11-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6807-11-30