
Abstract
Background
Plasmid-based overexpression of genes has been the principal strategy for metabolic engineering. However, for biotechnological applications, plasmid-based expression systems are not suitable because of genetic instability, and the requirement for constant selective pressure to ensure plasmid maintenance.
Results
To overcome these drawbacks, we constructed an Escherichia coli lycopene production strain that does not carry a plasmid or an antibiotic marker. This was achieved using triclosan-induced chromosomal evolution, a high gene copy expression system. The engineered strain demonstrated high genetic stability in the absence of the selective agent during fermentation. The replacement of native appY promoter with a T5 promoter, and the deletion of the iclR gene in E. coli CBW 12241 further improved lycopene production. The resulting strain, E. coli CBW 12241(Δ iclR, PT5-appY), produced lycopene at 33.43 mg per gram of dry cell weight.
Conclusions
A lycopene hyper-producer E. coli strain that does not carry a plasmid or antibiotic marker was constructed using triclosan-induced chromosomal evolution. The methods detailed in this study can be used to engineer E. coli to produce other metabolites.
Similar content being viewed by others


Background
Carotenoids are a diverse class of C40 isoprenoids that have multiple physiological and nutritional functions in many organisms. Carotenoids have received considerable attention from the food industry, medicine and cosmetics because of their interesting pigment properties, and more importantly, their potential beneficial effects on human health. Lycopene is an effective antioxidant [1] and has beneficial biological and pharmaceutical activities, including anti-cancer [2], anti-inflammatory [3], and antioxidative activities [4]. Lycopene is widely used as a supplement in functional foods, animal feed, nutraceuticals, pharmaceuticals and as an additive in cosmetics.
Potential commercial applications mean that efficient biotechnological production of lycopene has become increasingly necessary, and a number of reports have described its production using metabolic engineering [5–21]. However, all of these above studies focused on the deletion of undesirable genes and plasmid overexpression of key genes. Such plasmid-based expression systems have several drawbacks, including structural instability, segregational instability or allele segregation [22–24]. These plasmid instabilities cause genetic instability, which reduces the production of the compound of interest. Tyo et al. reported that plasmid-carrying strains lost poly-3-hydroxybutyrate (PHB) productivity after 40 generations with antibiotics [25]. Moreover, antibiotic resistance genes are the most commonly used markers for selecting and maintaining plasmids in hosts during cultivation. However, antibiotics are both costly and banned from food and pharmaceutical production processes. There is also a potential risk of the spread of antibiotic-resistant marker to other microbes in nature, leading to the rapid emergence of multidrug-resistant organisms (e.g., superbacteria) [26, 27].
These drawbacks of using plasmids can be overcome by integration of genes into the chromosome. Recently, Tyo et al. developed a plasmid-free method for the over-production of metabolites that can achieve high copy numbers of the desired genes, termed chemically induced chromosomal evolution (CIChE) [25]. Genes of interest were inserted into chromosome of Escherichia coli by the λInCh genomic integration method, and then evolved to the desired gene copy number by chemical induction. However, the λInCh genomic integration protocol is complicated and time-consuming, because it contains three steps that involve two recombination steps. Chiang et al. modified the conditional-replication, integration, and modular plasmid system produced by Haldimann and Wanner [28], and developed a replicon-free and markerless method (RMM) for the chromosomal insertion of genes [26]. Genes of interest can be directly integrated into the bacterial attachment site of the E. coli chromosome as single copies, through transformation. However, the CIChE strains reported by Tyo et al. still have an antibiotic resistance marker (chloramphenicol resistance) [25]. To avoid the use of antibiotic resistance genes and antibiotics, Goh and Good developed a novel system using the widely used the biocide triclosan as the selective agent and the essential growth gene fabI of E. coli as the selective marker [27]. Thus, to overcome the drawbacks of CIChE as originally developed by Tyo et al., we developed a series of integration expression vectors, pXKF3T5b, for triclosan induction chromosomal evolution in our previous paper [29]. Using these vectors, genes of interest can be inserted into E. coli site-specifically by transformation using RMM. The gene copy number can then be evolved to the desired value by triclosan induction. In this study, we constructed a lycopene hyper-producer E. coli that does not carry plasmid or antibiotic marker, using the CIChE integration expression vector. To the best of our knowledge, this is the first report of metabolic engineering of an E. coli that does not carry a plasmid or antibiotic marker using a multiple gene expression system for lycopene production.
Results and discussion
CIChE of E. coli for lycopene production
To overcome the drawbacks of CIChE as originally devised by Tyo et al., we developed a series of integration expression vectors, pXKF3T5b, for triclosan induction chromosomal evolution in our previous paper [29]. Using these vectors, genes of interest can be inserted into E. coli site-specifically by transformation using RMM. The gene copy number can then be evolved to the desired value by triclosan induction. Thus, strains constructed using our CIChE integration expression vectors have no antibiotic resistance and are environmentally safe.
The CIChE integration expression vector containing the lycopene biosynthetic gene cluster (pP21KF3T5b-EIBipi) was transferred into E. coli BW25113 (ΔgdhAΔaceE, PT5dxs) and then increasing triclosan concentration was used to induce CIChE. Figure 1 shows the results of lycopene production in CIChE strains resistant to different triclosan concentrations. Lycopene production in CIChE strains increases with increasing triclosan concentration during chromosomal evolution. The maximum lycopene production of 9.55 ± 0.23 mg/g dry cell weight (DCW) was obtained by the CIChE strains resistant to 8 μM triclosan cultured in 2YT medium. Figure 2 shows that the crtI gene copy number of the CIChE strains increases with triclosan concentration during chromosomal evolution. At a triclosan concentration of 8 μM, the copy number reached about 30 in the CIChE strains, which is the equivalent copy number of a medium to high copy plasmid. When the triclosan concentration was above 8 μM, the gene copy number still increased; however, the lycopene production of the CIChE strains did not increase. The results indicated that there is an optimal copy number of the crt genes for efficient production of lycopene. Thus, the recA gene of the CIChE strain that was resistant to 8 μM triclosan was deleted to obtain E. coli CBW12241, in which homologous recombination, which could reduce the copy number, is inhibited. The crt gene number of the recA-deleted strain did not change (data not shown). The genetic stability was also assayed (Table 1). The level of lycopene production in E. coli CBW12241 remained constant after 30 rounds of subculturing without triclosan. However, the plasmid-bearing strain E. coli BW25113 (ΔgdhAΔaceE, PT5dxs, pBAD24-WZM1) lost lycopene productivity sharply, whether cultured with antibiotics or not. With antibiotics, the level of lycopene production in the plasmid-bearing strain, E. coli BW25113 (ΔgdhAΔaceE, PT5dxs, pBAD24-WZM1), was only about 30% of that of the parent strain after 30 rounds of subculturing. Without antibiotics, the lycopene productivity of the plasmid-bearing strain E. coli BW25113 (ΔgdhAΔaceE, PT5dxs, pBAD24-WZM1) dropped to zero after 30 rounds of subculturing. These results are consistent with previous reports wherein the gene copy number and PHB production of a CIChE strain remained constant after 40 rounds of subculturing [25]. They also found that PHB productivity of the plasmid-carrying strain cultured with antibiotics was completely lost after 40 generations. The loss of productivity in the plasmid system with antibiotics may be due to allele segregation. It is likely to occur regularly, despite antibiotic selection, particularly in subculturing experiments or in chemostats [25].

Lycopene production of CIChE strains at different triclosan concentrations cultured in 2YT medium supplemented with 5 g/L KAc.

Copy number of the crtI gene in CIChE strains at different triclosan concentrations.
Optimization of the lycopene synthetic pathway
Overexpression of the idi, dxs, pck, pps, rpoS, appY, yjiD, ycgW, wrbA and atpE genes are reported to improve lycopene production in E. coli[8–19, 21]. Thus, the effects of over-expression of these genes in E. coli CBW12241 on lycopene production were investigated. The results are presented in Table 2. Surprisingly, only the over-expression of appY slightly enhanced the specific lycopene content. This may reflect the different genetic backgrounds of the strains. In E. coli CBW12241, isopentenyl diphosphate isomerase is overexpressed by chromosomal evolution of ipiHPI. Thus, overexpression of the idi gene in E. coli CBW12241 was not beneficial to lycopene production. Zhao et al. [30] reported that the dxs gene from Bacillus subtilis functioned more efficiently to enhance isoprene production in E. coli than the native dxs gene. However, overexpression of the dxs gene from Bacillus subtilis significantly inhibited lycopene production in E. coli CBW12241, possibly because the T5 promoter has replaced the native promoter of the dxs gene in E. coli CBW12241.
To reduce the metabolic burden caused by the plasmid, the native promoter of the appY gene was replaced with the T5 promoter to obtain E. coli CBW12241 (PT5appY). As shown in Table 3, the replacement improved cell growth and lycopene concentration. Kang et al. used a shotgun approach and found that overexpression of the appY gene improved lycopene production [12]. The appY gene encodes a transcriptional activator of two regulator energy metabolism operons, hya and cbdAB-appA, which are induced by anaerobiosis. They reasoned that the appY gene might help rescue the strain from energy insufficiency caused by lower ubiquinone levels.
A comparative transcriptome and proteome analysis demonstrated that the deletion of the transcriptional repressor iclR of the glyoxylate pathway increased lycopene production [31]. Thus, we deleted the iclR gene and examined lycopene production. The deletion of the iclR gene in E. coli CBW12241 caused a slight inhibition of growth and did not affect the specific lycopene content. The deletion of the iclR gene in the promoter replacement strain, E. coli CBW12241 (PT5appY), slightly improved specific lycopene content, from 28.97 ± 0.67 mg/g DCW to 33.43 ± 0.81 mg/gDCW, and improved the lycopene concentration from 54.15 ± 3.44 mg/L to 77.85 ± 1.42 mg/L (Table 3). In the iclR gene knockout strain, the glyoxylate pathway is constitutively active [32]. Furthermore, there are other examples of metabolic engineering involving deleting iclR to increase productions of compounds. Sanchez et al. successfully knocked out iclR to boost the glyoxylate pathway flux for succinate overproduction [33]. Lee et al. observed aceBA upregulation in the threonine-overproducing E. coli, and when iclR deleted, a 30% higher threonine production was recorded [34].
Many reports have been published about lycopene production using recombinant E. coli[5–21]. However, these strains contain plasmids, which can cause genetic instability and can have a negative environmental impact. Chiang et al. [26] constructed a lycopene hyper-producer E. coli strain that did not carry the replicon or a selective marker using RMM. However, RMM is a single gene copy expression system. Tyo et al. [25] constructed a lycopene producer E. coli using CIChE, a plasmid-free, high gene copy expression system. However, this strain still contained a chloramphenicol resistance marker. In the present study, we engineered a lycopene producer E. coli that does not carry a plasmid or an antibiotic marker. The CIChE strain only contains the fab I gene, which is essential for the growth of E. coli. The strain does not present the safety problems associated with antibiotic resistance genes in plasmids. Moreover, the strain does not require the presence of the resistance compound (triclosan) during the fermentation process. To the best of our knowledge, this is the first report of engineering an E. coli that does not carry a plasmid or antibiotic marker using a high gene copy expression system.
The maximum specific lycopene content (33.43 mg/g DCW) of E. coli CBW12241 (ΔiclR, PT5appY) was higher than that of the CIChE strain reported by Tyo et al. (about 11 mg/gDCW) [25]. The value was also higher than other plasmid-carrying strains, which achieved 22 mg/g DCW [21], 18 mg/g DCW [6] or 32 mg/g DCW produced by the engineered E. coli containing heterologous lycopene and mevalonate pathways [35]. However, the yield is slightly lower than that reported by Chiang et al. (38.5 mg/g DCW) [26]. In their study, the RMM strain E. coli BL21-CrtD1K has an additional chromosomal copy of dxs fused to the T7 promoter. However, plasmid-based overexpression of the dxs gene in the CIChE strain did not further enhance lycopene production. Moreover, E. coli CBW12251, containing an additional chromosomal copy of the dxs gene, produced lycopene at 31.73 ± 0.48 mg/gDCW, which was lower than E. coli CBW12241 (ΔiclR, PT5appY) (Table 3).
Although the yield of our engineered E. coli harboring only a heterologous lycopene pathway achieved the advanced levels quoted in the literature, many papers have reported that the introduction of a heterologous mevalonate pathway improved production of isoprenoids such as lycopene [16, 17, 20, 35], CoQ10[36], α-farnesene [37], terpenoid [38], taxol [39] and amorphadiene [40]. Thus, integration of a heterologous mevalonate pathway into the chromosome of the engineered E. coli CBW12241 (ΔiclR, PT5appY) may further improve lycopene production.
Conclusions
We constructed a lycopene producer E. coli strain that does not carry a plasmid or antibiotic marker using CIChE, replacement of a promoter as well as knockout of a gene. No resistance compound was required during the fermentation process using this strain. This is the first report of engineering an E. coli that lacks a plasmid and an antibiotic marker using a high gene copy expression system. The engineered strain remained stable, as determined by its lycopene production, after 30 sequential transfers.
Methods
Strains, primers and plasmids
The strains and plasmids used in this study are listed in Table 4. E. coli DH5α was used for plasmid construction. E. coli BW2511 (ΔaceFΔgdhA, PT5dxs) [19] was used as the parent strain for chromosomal integration. The primers used in this study are listed in Table 5.
Chemically induced chromosomal evolution
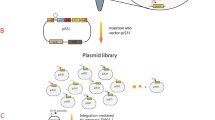
The crtE, crtI, crtB and ipi gene cluster was cut from pBAD24-WZM1 [19] using restriction enzymes ClaI and HindIII and treated with Primestar HS DNA polymerase (TaKaRa, China) and digested with EcoRI. The resulting gene cluster was cloned into the EcoRI/BamHI sites of pP21KF3T5b [29] to obtain pP21KF3T5b-crtEIBipi. The resulting integration vector was inserted into the bacterial attachment (attB) site of E. coli using a helper plasmid, pAH121, expressing the phage integrase, by direct transformation, as described by Chiang et al. [26]. The general procedure for using the integration vector is illustrated in Figure 3. In brief, strains containing the helper plasmid pAH121 were first cultured in super optimal broth (SOB) medium with 100 μg/mL of ampicillin (Amp) at 30°C to an optical density at 600 nm of approximately 0.6. The cells were made electrocompetent and transformed with the integration vector pP21KF3T5b-crtEIBipi. Following electroporation, the cells were suspended in SOB plus glucose medium without Ampicillin, incubated at 37°C for 1 h and at 42°C for 30 min, and then spread onto Luria-Bertani (LB) agar plates containing 25 μg/mL of kanamycin (Kan) and incubated overnight at 37°C. Colonies were verified by colony PCR using primer sets BLP1 and CGA, or BLP4 and CGB (Table 5). Positive colonies were cultured in 5 mL LB medium with 25 μg/mL kanamycin at 42°C overnight and then spread onto an LB medium with Kan agar plate at 37°C overnight. Stable integration was scored by antibiotic resistance (conferring Kan resistance) and loss of the helper plasmid (conferring Amp sensitivity), as well as by colony PCR using primer sets BLP1 and CGA, or BLP4 and CGB (Table 5). To eliminate the region containing the selective marker (Kan resistance) and the replication origin, the resulting integrants bearing the inserted DNA were transformed with pCP20 expressing FLP recombinase [23]. Colonies only resistant to triclosan were verified by colony PCR using primer sets BLP1 and CGA, or BLP4 and CGB.

Change in genomic structure induced by integration of pP21KF3T5b-crtEIBipi at the attP P21 site. Primers BLP1, BLP4, CGA, CGB (Table 5) were used to verify the genomic structure by colony PCR. The numbers beside the bars represent the expected sizes of PCR fragments amplified using corresponding primer sets (shown by arrows).
CIChE of the above construct was carried out by subculturing the resulting strains in 5 mL SOB medium with increasing concentrations of triclosan, in 15 mL culture tubes, as described by Tyo et al. [25]. The strains were grown to stationary phase in 1 μM triclosan. Fifty milliliters of the culture was subcultured into a new culture tube. In the new tube, the triclosan concentration was doubled from 2 μM to 64 μM and allowed to grow to stationary phase. The process was repeated until the desired concentration was reached. The recA gene of the CIChE strain was then deleted (see below).
Construction of the plasmid
The appY, idi, pck, pps, rpoS, ycgW and yjiD genes were amplified from E. coli genomic DNA using the primers shown in Table 5, and ligated into pBAD24 [41], respectively. The wrbA, ydeO and atpE genes were amplified from genomic DNA of E. coli using the primers shown in Table 5 and ligated into pQE30, respectively. The dxs gene was amplified from genomic DNA of B. subtilis ATCC 6633 using primers dxsF and dxsR (Table 5) and ligated into pQE30 (Qiagen).
Knockout of genes
Gene knockouts and replacement of the native promoter of the appY gene with the T5 promoter were carried out by PCR product recombination [42] using the pSIM6 plasmid [43] expressing the lambda red recombination system and pKD4 [42] as the template for PCR. Gene knockouts were verified using colony PCR using the appropriate primers (Table 5).
Quantitative PCR (qPCR) measurement of gene copy number
Gene copy numbers were measured by qPCR on genomic DNA isolated from the appropriate CIChE strains. qPCR was performed with an iCycler iQ5 Real Time PCR system (Bio-Rad Laboratories, USA) using the SYBR Premix Ex Taq II (TaKaRa, China), following the manufacturer’s protocol. PCR conditions were as follows: 30 s at 95°C; 40 cycles of at 95°C for 5 s, 60°C for 30 s, and 95°C for 60 s; followed by melting curve analysis. The copy numbers of the crtI gene were detected and compared to the copy number of minD, a nearby native gene in the chromosome. The primers QCF and QCR (Table 5) were used to measure the copy number of crtI. The primers NDF and NDR (Table 5) were used to measure the copy number of minD.
Lycopene production
For lycopene production, 5 mL of LB medium supplement with 5 g/L KAc was used for overnight precultivation of E. coli in a falcon tube at 37°C. The main cultures were in 2YT medium or SBMSN medium supplemented with 5 g/L KAc. SBMSN medium (pH7.0) contains (g/L): peptone 12, yeast extract 24, KH2PO4 1.7, K2HPO4 11.42, MgCl2·6H2O 1, ammonium oxalate 1.42, Tween-80 2. The main cultures were inoculated with a starting OD600 of 0.1 and incubated at 37°C for 24 h in a rotary shaking incubator at 150 rpm. Cell growth was measured by optical density at 600 nm and converted into DCW (g/L) using a standard curve.
Extraction and measurement of lycopene
The lycopene content of recombinant E. coli strains was quantified as previously reported [5]. Two hundred fifty microliters of E. coli cells were harvested by centrifugation at 12000 rpm for 5 min. The cell pellet was washed with water and then extracted in 1 mL of acetone at 55°C for 15 min with intermittent vortexing. The mixture was then centrifuged at 12000 rpm for 10 min, and the acetone supernatant was transferred to a new tube. The absorbance of the resulting extract was measured at 474 nm and converted to lycopene concentration (μg/mL) using a standard curve obtained using commercial lycopene (Sigma).
Statistical analysis
All experiments were conducted in triplicate, and data were averaged and presented as the mean ± standard deviation. One-way analysis of variance followed by Tukey’s test was used to determine significant differences using the OriginPro (version 7.5) package. Statistical significance was defined as p < 0.05.
References
Sies H, Stahl W: Lycopene: antioxidant and biological effects and its bioavailability in the human. Proc Soc Exp Biol Med. 1998, 218: 121-124.
Giovannucci E: A review of epidemiologic studies of tomatoes, lycopene, and prostate cancer. Exp Biol Med (Maywood). 2002, 227: 852-859.
Bignotto L, Rocha J, Sepodes B, Eduardo-Figueira M, Pinto R, Chaud M, de Carvalho J, Moreno H, Mota-Filipe H: Anti-inflammatory effect of lycopene on carrageenan-induced paw oedema and hepatic ischaemia-reperfusion in the rat. Br J Nutr. 2009, 102: 126-133. 10.1017/S0007114508137886.
Erdman JW, Ford NA, Lindshield BL: Are the health attributes of lycopene related to its antioxidant function?. Arch Biochem Biophys. 2009, 483: 229-235. 10.1016/j.abb.2008.10.022.
Alper H, Jin YS, Moxley JF, Stephanopoulos G: Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab Eng. 2005, 7: 155-164. 10.1016/j.ymben.2004.12.003.
Alper H, Miyaoku K, Moxley JF, Stephanopoulos G: Construction of lycopene-overproduction E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005, 23: 612-616. 10.1038/nbt1083.
Alper H, Stephanopoulos G: Uncovering the gene knockout landscape for improved lycopene production in E. coli. Appl Microbiol Biotechnol. 2008, 78: 801-810. 10.1007/s00253-008-1373-x.
Choi HS, Lee SY, Kim TY, Woo HM: In Silico identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol. 2010, 76: 3097-3105. 10.1128/AEM.00115-10.
Farmer WR, Liao JC: Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol. 2000, 18: 533-537. 10.1038/75398.
Farmer WR, Liao JC: Precursor balancing for metabolic engineering of lycopene production in Escherichia coli. Biotechnol Prog. 2001, 17: 57-61. 10.1021/bp000137t.
Jin YS, Stephanopoulos G: Multi-dimensional gene target search for improving lycopene biosynthesis in Escherichia coli. Metab Eng. 2007, 9: 337-347. 10.1016/j.ymben.2007.03.003.
Kang MJ, Lee YM, Yoon SH, Kim JH, Ock SW, Jung KH, Shin YC, Keasling JD, Kim SW: Identification of genes affecting lycopene accumulation in Escherichia coli using a shot-gun method. Biotechnol Bioeng. 2005, 91: 636-642. 10.1002/bit.20539.
Kang MJ, Yoon SH, Lee YM, Lee SH, Kim JE, Jung KH, Shin YC, Kim SW: Enhancement of lycopene production Escherichia coli by optimization of the lycopene synthetic pathway. J Microbiol Biotechnol. 2005, 15: 880-886.
Kim SW, Keasling JD: Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng. 2001, 72: 408-415. 10.1002/1097-0290(20000220)72:4<408::AID-BIT1003>3.0.CO;2-H.
Kim SW, Kim JB, Ryu JM, Jung JK, Kim JH: High-level production of lycopene in metabolically engineered E. coli. Process Biochem. 2009, 44: 899-905. 10.1016/j.procbio.2009.04.018.
Rad SA, Zahiri HS, Noghabi KA, Rajaei S, Heidari R, Mojallali L: Type 2 IDI performs better than type 1 for improving lycopene production in metabolically engineered E. coli strains. World J Microbiol Biotechnol. 2012, 28: 313-321. 10.1007/s11274-011-0821-4.
Rodríguez-Villalón A, Pèrez-Gil J, Rodríguez-Concepción M: Carotenoid accumulation in bacteria with enhanced supply of isoprenoid precursors by upregulation of exogenous or endogenous pathways. J Biotechnol. 2008, 135: 78-84. 10.1016/j.jbiotec.2008.02.023.
Wang CW, Oh MK, Liao JC: Directed evolution of metabolically engineered Escherichia coli for carotenoid production. Biotechnol Prog. 2000, 16: 922-926. 10.1021/bp000124f.
Weng ZM, Wang Y, Liu JZ: Overproduction of lycopene by metabolic engineering Escherichia coli. Bioprocess. 2012, 2: 51-57. 10.4236/bp. 2012.22009.
Yoon SH, Lee YM, Kim JE, Lee SH, Lee JH, Kim JY, Jung KH, Shin YC, Keasling JD, Kim SW: Enhanced lycopene production in Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate. Biotechnol Bioeng. 2006, 94: 1025-1032. 10.1002/bit.20912.
Yuan LZ, Rouvière PE, LaRossa RA, Suh W: Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng. 2006, 8: 79-90. 10.1016/j.ymben.2005.08.005.
Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS: Plasmid-encoded protein: the principal factor in the metabolic burden associated with recombinant bacteria. Biotechnol Bioeng. 1990, 35: 668-681. 10.1002/bit.260350704.
Noack D, Roth M, Geuther R, Muller G, Undisz K, Hoffmeier C, Gaspar S: Maintenance and genetic stability of vector plasmids pBR322 and pBR325 in Escherichia coli K12 strains grown in a chemostat. Mol Gen Genet. 1981, 184: 121-124. 10.1007/BF00271207.
O’Connor M, Peifer M, Bender W: Construction of large DNA segments in Escherichia coli. Science. 1989, 244: 1307-1312. 10.1126/science.2660262.
Tyo KEJ, Ajikumar PK, Stephanopoulos G: Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat Biotechnol. 2009, 27: 760-765. 10.1038/nbt.1555.
Chiang C-J, Chen PT, Chao YP: Replicon-free and markerless methods for genomic insertion of DNAs in phage attachment sites and controlled expression of chromosomal genes in Escherichia coli. Biotechnol Bioeng. 2008, 101: 985-995. 10.1002/bit.21976.
Goh S, Good L: Plasmid selection in Escherichia coli using an endogenous essential gene marker. BMC Biotechnol. 2008, 8: 61-10.1186/1472-6750-8-61.
Haldimann A, Wanner BL: Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001, 183: 6384-6393. 10.1128/JB.183.21.6384-6393.2001.
Liu JZ, Huang MT, Cui YY, Chen YY: A series of expression plasmids for chromosomal integration and evolution. Chinese patent 201210060042.5. 2012
Zhao Y, Yang J, Qin B, Li Y, Sun Y, Su S, Xian M: Biosynthesis of isoprene in Escherichia coli via methylerythritol phosphate (MEP) pathway. Appl Microbiol Biotechnol. 2011, 90: 1915-1922. 10.1007/s00253-011-3199-1.
Mickus BE: PhD thesis. Transcriptomic and proteomic analysis of lycopene-overproducing Escherichia coli strains. 2009, US: Massachusetts Institute of Technology
Maloy SR, Nunn WD: Genetic regulation of the glyoxylate shunt in Escherichia coli K-12. J Bacteriol. 1982, 149: 173-180.
Sánchez AM, Bennett GN, San K: Novel pathway engineering design of the aerobic center metabolic pathway in Escherichia coli to increase succinate yield and productivity. Metab Eng. 2005, 7: 229-239. 10.1016/j.ymben.2005.03.001.
Lee KH, Park JH, Kim TY, Kim HU, Lee SY: Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Sys Biol. 2007, 3: 149-
Kim Y-S, Lee J-H, Kim N-H, Yeom S-J, Kim S-W, Oh D-K: Increase of lycopene production by supplementing auxiliary carbon sources in metabolically engineered Escherichia coli. Appl Microbiol Biotechnol. 2011, 90: 489-497. 10.1007/s00253-011-3091-z.
Zahiri HS, Yoon SH, Keasling JD, Lee SH, Kim SW, Yoon SC, Shin YC: Coenzyme Q10 production in recombinant Escherichia coli strains engineered with a heterologous decaprenyl diphosphate synthase gene and foreign mevalonate pathway. Metab Eng. 2006, 8: 406-416. 10.1016/j.ymben.2006.05.002.
Wang C, Yoon SH, Jang HJ, Chung YR, Kim JY, Choi ES, Kim SW: Metabolic engineering of Escherichia coli for α-farnesene production. Metab Eng. 2011, 13: 648-655. 10.1016/j.ymben.2011.08.001.
Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD: Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol. 2003, 21: 796-802. 10.1038/nbt833.
Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G: Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science. 2010, 330: 70-74. 10.1126/science.1191652.
Anthony JR, Anthony LC, Farnaz N, Kwon G, Newman JD, Keasling JD: Optimization of the mevalonate-based isoprenoids biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab Eng. 2009, 11: 13-19. 10.1016/j.ymben.2008.07.007.
Guzman LM, Belin D, Carson MJ, Beckwith J: Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995, 177: 4121-4130.
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000, 97: 6640-6645. 10.1073/pnas.120163297.
Sharan SK, Thomason LC, Kuznetsov SG, Court DL: Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009, 4: 206-223. 10.1038/nprot.2008.227.
Acknowledgments
We are grateful to both the National Natural Science Foundation of China (Grant No. 30970089, 20876181, 21276289) and the Natural Science Foundation of Guangdong Province (No. 9351027501000003, S2011010001396) for their financial support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YY C carried out most of the experiments. HJ S constructed E. coli CBW12251 and investigated the effect of the dxs gene. YY C constructed the integration expression vectors. SG C carried out fermentation experiments. ZM W constructed expression vectors for some of the genes. M Z guided the project. JZ L developed the concept and designed the method, led the project and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, YY., Shen, HJ., Cui, YY. et al. Chromosomal evolution of Escherichia coli for the efficient production of lycopene. BMC Biotechnol 13, 6 (2013). https://doi.org/10.1186/1472-6750-13-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6750-13-6