
Abstract
Background
Loss of DNA mismatch repair (MMR) in humans, mainly due to mutations in the hMLH1 gene, is linked to hereditary nonpolyposis colorectal cancer (HNPCC). Because not all MLH1 alterations result in loss of MMR function, accurate characterization of variants and their classification in terms of their effect on MMR function is essential for reliable genetic testing and effective treatment. To date, in vivo assays for functional characterization of MLH1 mutations performed in various model systems have used episomal expression of the modified MMR genes. We describe here a novel approach to determine accurately the functional significance of hMLH1 mutations in vivo, based on co-expression of human MLH1 and PMS2 in yeast cells.
Methods
Yeast MLH1 and PMS1 genes, whose protein products form the MutLα complex, were replaced by human orthologs directly on yeast chromosomes by homologous recombination, and the resulting MMR activity was tested.
Results
The yeast strain co-expressing hMLH1 and hPMS2 exhibited the same mutation rate as the wild-type. Eight cancer-related MLH1 variants were introduced, using the same approach, into the prepared yeast model, and their effect on MMR function was determined. Five variants (A92P, S93G, I219V, K618R and K618T) were classified as non-pathogenic, whereas variants T117M, Y646C and R659Q were characterized as pathogenic.
Conclusion
Results of our in vivo yeast-based approach correlate well with clinical data in five out of seven hMLH1 variants and the described model was thus shown to be useful for functional characterization of MLH1 variants in cancer patients found throughout the entire coding region of the gene.
Similar content being viewed by others


Background
Mismatch repair (MMR) genes are genome-stability genes. Mutations in these genes cause defects in the MMR pathway that can lead to carcinogenesis, as a result of accumulation of non-repaired post-replicational mis-incorporations in cancer contributing genes. Inherited germline mutations of MMR genes cause a predisposition to cancer, which can develop at an early age, as the next somatic mutation needs to affect only one, wild-type allele of a gene [1, 2]. It has been established that many families with hereditary non-polyposis colorectal cancer (HNPCC) harbour potentially pathogenic mutations in MMR genes, particularly in hMLH1 and hMSH2 [3]. The disease manifests with high mortality rate if not detected and treated early, therefore presymptomatic genetic testing is required [4]. However, the functional effects of MMR mutations need to be characterized first. This is essential, not only for effective diagnosis of cancer predisposition, but also for choosing the appropriate chemotherapy, since it is known that MMR mutations can cause resistance to chemotherapeutic agents by insufficient induction of apoptosis [5].
There are over 450 germline allelic variants of hMLH1 and hMSH2 genes known to date and more are being found, but the functional relevance of each is very difficult to establish on clinical samples alone. Therefore many in vitro and in vivo assays have been developed for MMR gene mutation analysis, using various molecular genetics tools [6–16]. An in vitro functional analysis - involving Western immunoblotting - of four hMLH1 mutations found in HNPCC patients was described by Belvederesi et al. [6]. Several other strategies have been based on analysing the interaction between MMR variants, using the yeast two-hybrid system [7, 8]. One recent study described the characterization of MLH1 variants in terms of multiple functional properties (e.g. protein expression/stability, nuclear localization, protein-protein interaction and MMR efficiency) [9]. In vivo assays have been performed in bacterial and yeast model systems, all of them using expression vectors with human MMR gene variants [10–13]. An alternative, commonly reported approach in yeast is the use of vectors with yeast MMR orthologs harbouring a mutation corresponding to those found in HNPCC patients, but the method is limited by the degree of homology between human and yeast genes [14, 15]. MLH1 variants were also introduced into human cell lines to analyze their effects on apoptosis, proliferation, and regulation of mRNA expression, as well as expression of interacting proteins [16].
Although these studies have provided valuable clues to a better understanding of MLH1 variants, it is difficult to establish and compare their clinical relevance since a variety of strategies have been used. We believe that for reliable interpretation of functional effects of MLH1 gene mutations and their classification, all MLH1 mutations extending within the whole gene coding region should be analyzed using the same approach. In this study we are proposing a novel in vivo approach to determine the functional significance of hMLH1 mutations throughout the whole coding region of the gene. The hPMS2 and hMLH1 genes, whose proteins form the hMutLα complex, were each introduced into the yeast chromosome, replacing the yeast orthologs. Direct chromosomal integration enabled just one copy of a target gene to be introduced per cell. Both human orthologs were under control of the yeast promoter, and the original yeast genetic background was preserved. The functionality of mismatch repair was examined using a quantitative in vivo DNA MMR assay. To assess the functionality of the novel test system, nine different MLH1 amino acid replacements were introduced in vivo and evaluated for their pathogenicity. Results show that our in vivo yeast-based approach can help assess the pathogenic potential of cancer-related hMLH1 variants.
Methods
Yeast strain and vectors
The yeast Saccharomyces cerevisiae haploid strain used was W303-1A/RE966 (MATa ade2 can1-100 his3-11,15 leu2-3, 112 trp1-1 ura3-1) [17]. Cells were maintained in standard rich medium (YPD) [yeast extract/peptone/dextrose] and in selective synthetic medium (SD) [YNB: 0,67% w/v yeast nitrogen base, 2% w/v dextrose] supplemented with amino acids, depending on the auxotrophic requirements, and grown at 30°C. Media were solidified by addition of 2% w/v bacto-agar. The E. coli DH5α strain was used as bacterial host for plasmid propagation. Plasmid pCORE with KlURA3-kanMX4 counter-selectable reporter marker cassette (CORE) and plasmid pSH91 were provided by Francesca Storici [18] and Thomas D. Petes [19], respectively. The yeast expression vector pSH91 used for in vivo MMR assay contains an in-frame (GT)16.5 tract in the URA3 coding region. hMLH1 [GeneBank: NM000249] and hPMS2 [GeneBank: NM000535] cDNAs were obtained from pCMV-SPORT6 vector (ATCC, MGC-5172) and pSG-PMS2-wt, respectively. The latter plasmid was provided by Bert Vogelstein and Kenneth W. Kinzler [20]. Expression of yeast URA3 gene in pSH91 is under LEU2 gene promoter, so all test strains were transformed with Yep181 vector expressing LEU2 gene for restoration of leucine prototrophy [21].
Construction of humanized yeast strains
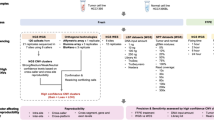
A »delitto perfetto« method was used for constructing humanized yeast strains, as described by Storici et al. [18]. Delitto Perfetto is a two step approach for in vivo site-directed mutagenesis in yeast, using highly proficient homologous recombination system of S cerevisiae. The first step involves integration of a counterselectable (CORE) cassette in the region of interest. Subsequently, the CORE cassette is replaced with DNA fragment containing the mutation or the gene of interest (Figure 1). The CORE cassette allows the selection of yeast cells that receive the cassette during the first step, as well as selection of cells that lose the cassette in the second step of the process.

Constructing humanized yeast strains with delitto perfetto system. A. Replacing yeast MMR (yMMR) genes (i.e. yMLH1 and yPMS1) with their human orthologs (hMMR). First, the CORE cassette was introduced into yeast chromosome (line) after the stop codon of a yeast MMR gene, enabling the yeast gene to be still active during the second step of the process. Next, together with CORE cassette, the yeast gene was replaced with its human ortholog. B. Introducing missense alterations into hMLH1 gene in strain co-expressing hMLH1 and hPMS2 genes. First, the CORE cassette was introduced into hMLH1, replacing nucleotide of interest (black vertical line). Next, the CORE cassette was replaced by DNA fragment harbouring a single-nucleotide alteration (yellow vertical line). Genetic technique, delitto perfetto, is based on recombination event (grey area) between two identical strand of DNA.
In this study, the counter-selectable reporter (CORE) cassette with KlURA3 and kanMX4 was amplified from pCORE and integrated into yeast chromosome XIII after the stop codon of the yMLH1 coding region (Figure 1A). 60- to 70-meric oligonucleotides (Sigma-Aldrich), consisting of 40- to 50-nucleotide flanking regions homologous with the appropriate yeast genomic target locus and a 20-nucleotide tract required for amplification from the vector were used for the amplification. DNA fragments were amplified by the polymerase chain reaction (PCR) using Platinum® Pfx DNA polymerase (Invitrogen) and introduced into yeast cells with the LiAc transformation protocol [22]. Yeast clones that integrated CORE cassette into their genome were selected on YPD plates containing 200 μg/mL of geneticin (G418; Sigma-Aldrich). The CORE cassette was then completely removed by introducing complete hMLH1 cDNA into the yeast cells. Human MLH1 cDNA was previously amplified from pCMV-SPORT6 vector using 60-meric oligonucleotides. After transformation, the resulting Ura-cells, which lost CORE cassette, were isolated from synthetic complete medium containing 1 mg/L 5-fluororotic acid monohydrate (5-FOA; Toronto Research Chemicals Inc.). Isolated clones (strain MV200) were identified by PCR, using Taq polymerase (Eppendorf® Master Mix), and the nucleotide sequences of the constructs were confirmed by DNA sequencing (Sequiserve).
To replace yPMS1 by hPMS2 directly on the chromosome (strain MV300), the CORE cassette was amplified with 60-meric oligonucleotides from pCORE and introduced after the stop codon of the yPMS1 gene in the yeast chromosome. The integrated CORE cassette, together with the yPMS1 gene, was then replaced by hPMS2 cDNA, previously amplified from vector pSG-PMS2-wt with oligonucleotides targeting yeast chromosome XIV. PCR products were introduced into the yeast cells and clones were isolated as described above. In order to obtain a yeast strain containing both hMLH1 and hPMS2 (strain MV400), the replacement of yMLH1 with hMLH1 was done in the strain MV300 already harbouring hPMS2.
The yeast mlh1::kanMX gene disruption was constructed by replacing the entire yMLH1 gene coding region with kanMX gene amplified from pFA6aKanMX4. Clones (strain MV100) were selected on YPD plates containing G418 and identified by PCR.
When constructing MLH1 single-nucleotide substitutions (Figure 1B), the CORE cassettes were amplified from pCORE with oligonucleotides targeting the human ortholog at the relevant codons. Integrative 3'-overlapping recombinant oligonucleotides harbouring the desired single-nucleotide substitutions were extended in vitro as described by Storici et al [18]. PCR products were again transformed into appropriate geneticin resistant yeast cells and clones were isolated as described above. All oligonucleotide sequences are available upon request.
In vivo MMR assay and statistical analysis
The standardized in vivo MMR assay has been described in detail elsewhere [23]. The assay is based on stability of 33 bp GT tract, which is inserted in-frame the yeast URA3 gene in plasmid pSH91, as a measure of MMR efficiency. Instability of dinucleotide repeat is associated with DNA polymerase slipping during replication and alterations in tract length result in ura3 mutants, if not repaired by MMR. Mutant ura3 cells can be measured by their resistance to 5-FOA. Expression of the yeast URA3 gene in pSH91 is under LEU2 gene promoter, so all strains have to be leucine prototrophs.
In brief, plasmids pSH91 and Yep181 were first transformed [24] into the prepared yeast strains. At least 12 colonies of each analyzed yeast strain were picked from selective media plates (lacking tryptophan, leucine and uracil) and grown to saturation in SD supplemented with adenine and histidine to ensure that the starting cultures were homogeneous for URA3 cells with in-frame GT tracts. Cultures were diluted 1:1000 into SD supplemented with adenine, histidine and uracil, and grown to OD660 of 1. This allowed growth of all cells, including newly arising ura3 mutants. Each culture was then serially diluted and plated on SD plates supplemented with adenine, histidine and uracil, and the number of colonies was counted after a 3-day incubation to determine cell concentration. 100 μL aliquots of undiluted culture were plated on SD plates supplemented with adenine, histidine and uracil and containing 5-FOA. Colonies were counted after 3 days to calculate the concentration of ura3 mutants. Strains with increased instability of the GT tract, due to a disrupted MMR system, resulted in the ura3 mutant strain. Mutation rates were calculated using the method of the median [25]. For statistical analysis, Mann-Whitney tests were performed between all strains using GraphPad-InStat software. Differences in mutation rates were considered significant when P ≤ 0.05.
Results
Characterization of human MMR genes in yeast
For functional analysis of HNPCC-related hMLH1 variants we prepared several humanized yeast strains with hPMS2 and different hMLH1 variants inserted into the genome and replacing their yeast orthologs. The function of the human MLH1 protein variants was determined with a standardized in vivo MMR assay measuring stability of the (GT)16.5 tract in vivo. A MV100 strain was constructed by integrating kanMX gene into the coding region of the yeast MLH1. By this intervention the ability of MMR to repair insertion/deletion loops on microsatellite GT tract was completely disrupted, with 79-fold greater mutation rate than in the wild-type (Figure 2).

The MMR efficiency in yeast strains harbouring endogenous and/or human MMR genes. Each datum is the mean, with a 95% confidence interval indicated, of three independent experiments. Mean mutation rates are: Wild-type 2.19 × 10-6 ; MV100 1.74 × 104; MV200 6.05 × 10-5; MV300 8.86 × 10-5; MV400 2.25 × 10-6. Genotypes of strains are boxed. All prepared strains originated from W303 background [17].
For in vivo replacement of the yeast MLH1 gene by its human ortholog, we first integrated the CORE cassette into the yeast MLH1 gene. Experiments replacing the CORE cassette at codon 73 of the yeast MLH1 coding region with complete human MLH1 cDNA failed (data not shown). It was shown before in yeast, that MLH1 can play a role in regulating mitotic recombination [26] and we believe that its disruption, in our case by the integrated CORE cassette, could have a negative impact on recombination events needed for successful replacement of the cassette. For this reason we engineered an alternative gene fusion by integrating the CORE cassette after the yeast MLH1 stop codon, thus maintaining the integrity of the yeast gene. Replacement of this fusion construct with the complete human MLH1 cDNA was now feasible. However, human MLH1 alone did not complement the yeast ortholog and exhibited a 28-fold increase in mutation rate over the wild-type (Figure 2). Using the same strategy we then replaced the yeast PMS1 with the complete human PMS2 cDNA in the strain harbouring the hMLH1 gene. Co-expression of human MLH1 and PMS2 (strain MV400) indicated that the exogenous hMLH1-hPMS2 complex was able to fully restore the MMR function in yeast cells (Figure 2). Mutation rate was the same as observed in the wild-type strain (P = 0.89). However, the MV300 strain expressing hPMS2 alone exhibited a 40-fold higher mutation rate than that in the wild-type parental strain.
The described system is operational for functional characterization of hMLH1 alterations
Our approach was shown to be operational for functional analysis of hMLH1 mutations by assessing eight different hMLH1 missense mutations found in cancer patients. Two variants (T117M and I219V) have already been evaluated by several different functional assays (Table 1). Because of the consistency of data obtained from the assays and their correlation with clinical reports, these variants were included as experimental controls in our study. We further analyzed the impact on phenotype of six missense mutations (A92P, S93G, K618R, K618T, Y646C and R659Q). An additional, non-cancer-associated frameshift mutation (1955 del 1 bp), which determines an early stop codon at position 660, was used to evaluate the impact of a clear loss-of-function mutation. All nucleotide rearrangements resulting in relevant codon changes were introduced in vivo into strain MV400 co-expressing hMLH1 and hPMS2 genes.
The results of functional characterization of the hMLH1 variants are presented in Table 2. The amino acid alteration A92P did not alter MMR function, since the mutation rate was the same as observed in the wild-type parental MV400 strain. The 1.8-fold greater mutation rate in the strain expressing K618T variant, in comparison with the parental MV400 strain, was also not significant (P = 0.69). Six of the hMLH1 codon changes analyzed encode proteins which were observed in this study to support intermediate efficiencies of DNA MMR. The mutation rates measured in strains expressing hMLH1 variants S93G, T117M, I219V, K618R, Y646C and R659Q were significantly lower than in the mlh1 deficient yeast strain MV100 (P < 0.0001). Mutation rates of T117M, Y646C and R659Q were also clearly (P < 0,0001) greater (12.3-, 15.8- and 7.9-fold, respectively) than those obtained in the strain MV400 co-expressing wild-type human orthologs. A frameshift mutation 1955 del 1 bp, an experimental control never reported in HNPCC-patients, exhibited complete loss of MMR activity as expected. The alteration conferred a 82.7-fold higher mutation rate, than exhibited by the wild-type parental MV400 strain.
Discussion
Hundreds of hMLH1 alterations have been found in HNPCC patients and for many, especially those leading to amino acid substitutions, the pathogenicity is still difficult to interpret. These non-truncating mutations should be functionally characterized. For this purpose, a number of yeast-based assays evaluating the functional significance of MMR variants have been developed, based on the fact that MMR has been conserved throughout eukaryotic evolution. Because of conservation of cellular functions from yeast to mammals and the ease of genetic manipulation in yeast S cerevisiae, this unicellular organism has been used before for assessing patogenicity of other cancer-related genes (eg. BRCA1 and p53) [27, 28]. Besides many in vitro functional assays that require separate characterization of multiple functional properties [9, 12], in vivo tests have been developed which appear to be more reliable [7, 10, 11, 13]. However, it is important to note that, with known in vivo yeast assays, only mutations occurring in evolutionarily conserved regions in humans and yeast could be functionally analyzed, due to the lack of homology between human and yeast genes [14, 15]. Moreover, clinical data associated with the disease usually lack to correlate with functional analysis data (for references see Table 1). Considering the fact that the efficiency of mismatch repair is highly affected by the degree of expression of the MLH1 gene [15] (variable in an episomal way), we have proposed a yeast-based in vivo system co-expressing chromosome-integrated human MLH1 and human PMS2 genes. With this intervention, the expressions of both human genes were controlled by the orthologous yeast promoters. Besides that, the expression of gene in chromosome seems to be region-depended. Higher-expressed and lower-expressed regions have been reported along yeasts' chromosome III. Moreover, the stability of foreign gene in chromosome is better than that in plasmids [29].
The MLH1 protein contains an amino-terminal ATPase domain and a carboxy-terminal domain. According to Ellison's conclusion [10] and to our unpublished data, the human ATP domain is functional in yeast. The human ortholog alone does not restore MMR activity in mlh1-deficient yeast cells when expressed episomally [13]. The same appeared to be true with the chromosomal integration of the entire hMLH1 performed in our study (Figure 2). However, additional replacement of yeast PMS1 with hPMS2 (which encodes a heterodimeric partner of hMLH1) directly on the yeast chromosome, in a strain expressing human MLH1 protein, resulted in complete restoration of MMR function (Figure 2).
Co-expression of human MLH1 and PMS2 proved to be effective for evaluating the pathogenicity of alterations found throughout the coding region of human MLH1 gene. Nine different hMLH1 alterations were shown to bring about from little or no effect on protein function to complete loss of MMR activity (Table 2). Before interpreting the pathogenicity of these MLH1 alterations, the appropriate tolerance level for functional proficiency had to be established. For this reason, hMLH1 missense alteration I219V was included in the assay as the functional control. This hMLH1 modification is a known harmless polymorphism consistently reported to have no functional effect on MLH1 (for reference see Table 1). In our test system the variant exhibited a significantly greater mutation rate than the wild-type. We therefore propose that 3-fold increase in mutation rate over the wild-type is the reasonable threshold between harmless variants on one side, and the variants with reduced MMR activity that may contribute to cancer susceptibility on the other side of a threshold. The proposed treshold might change, as the pathogenicities of more variants with proven phenotypes are assessed with the described approach.
Accordingly, the amino acid changes A92P, S93G, K618R and K618T were classified as non-pathogenic variations (Table 2). S93G is associated with a late age of onset, but has been reported in biochemical studies to have no functional effect on MLH1. The latter was confirmed in our study. The alteration K618T has been functionally characterized with various biological tools, with controversial results as to its pathogenicity. Our results correlate with clinical studies, in which K618T was reported not to co-segregate with the disease and in which affected carriers showed microsatellite stability and normal immunohistochemical staining of MLH1. (Table 1). Modifications A92P and K618R were functionally characterized for the first time. The novel amino acid change A92P was found in a patient with gastric carcinoma, and reported as probably pathogenic from its predicted structural changes [30]. The K618R modification was identified in colorectal cancer patients, however the clinical data are rather incomplete. The results of our assay suggest that these two modifications may not be causally associated with the observed clinical phenotype in mutation carriers (Table 1). Nevertheless, A92P, S93G, K618R and K618T must not be considered as absolutely non-pathogenic. Some alterations, despite being functionally proficient, exhibited reduction of protein efficiency that was significant compared to the wild-type, and mutation carriers may still, with low penetrance, express a particular phenotype. Therefore functional data have to be considered carefully alondside clinical studies that need to be undertaken with much more caution.
According to our assay, a further four alterations, including the functional control - a frameshift mutation 1995 del 1 bp - should be considered as pathogenic variants. As assumed before, the missense mutations did not completely destroy protein function and are thus expected to result in incomplete clinical penetrance [31]. In this study, we confirmed previous reports showing that the T117M variant may be associated with pathogenicity in HNPCC (Table 1). Functional characterization of another two variants (Y646C and R659Q) was, however, not consistent with other functional studies, in which they were identified as non-pathogenic (Table 2). However, carriers of these variants developed cancer at early-middle age and showed HNPCC clinical features (Table 1). The discrepancy between our assay and previous functional studies can be explained by the conclusion that subtle functional alterations may not be revealed with in vitro biological tools [9]. In our study, complete elimination of MMR function was only established with the frameshift mutation 1995 del 1 bp.
Conclusion
We have shown, in contrast to in vitro studies, that co-employing the chromosome-integrated hMLH1 and hPMS2 genes in yeast not only enabled pathogenic MLH1 variants to be discriminated from polymorphisms that have no effect on MMR function, but also enabled variants resulting in reduced MMR activity to be identified and functionally classified. Moreover, the results correlated with clinical data in five out of seven hMLH1 variants. Our yeast-based in vivo system may thus be used for functional characterization of variants with incomplete clinical data, since precise quantification of MLH1 variants is critical for effective early diagnosis and prognosis, as well as for genetic counselling of affected individuals.
References
Vogelstein B, Kinzler KW: Cancer genes and the pathways they control. Nat Med. 2004, 10: 789-799.
Loeb LA, Loeb KR, Anderson JP: Multiple mutations and cancer. Proc Natl Acad Sci USA. 2003, 100: 776-781.
Nystrom-Lahti M, Perrera C, Raschle M, Panyushkina-Seiler E, Marra G, Curci A, Quaresima B, Costanzo F, D'Urso M, Venuta S, Jiricny J: Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer. 2002, 33: 160-167.
Jiricny J, Nystrom-Lahti M: Mismatch repair defects in cancer. Curr Opin Genet Dev. 2000, 10: 157-161.
Clodfelter JE, M BG, Drotschmann K: MSH2 missense mutations alter cisplatin cytotoxicity and promote cisplatin-induced genome instability. Nucleic Acids Res. 2005, 33: 3323-3330.
Belvederesi L, Bianchi F, Loretelli C, Gagliardini D, Galizia E, Bracci R, Rosati S, Bearzi I, Viel A, Cellerino R, Porfiri E: Assessing the pathogenicity of MLH1 missense mutations in patients with suspected hereditary nonpolyposis colorectal cancer: correlation with clinical, genetic and functional features. Eur J Hum Genet. 2006, 14: 853-859.
Kondo E, Suzuki H, Horii A, Fukushige S: A yeast two-hybrid assay provides a simple way to evaluate the vast majority of hMLH1 germ-line mutations. Cancer Res. 2003, 63: 3302-3308.
Mac Partlin M, Homer E, Robinson H, McCormick CJ, Crouch DH, Durant ST, Matheson EC, Hall AG, Gillespie DA, Brown R: Interactions of the DNA mismatch repair proteins MLH1 and MSH2 with c-MYC and MAX. Oncogene. 2003, 22: 819-825.
Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, et al: Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005, 129: 537-549.
Ellison AR, Lofing J, Bitter GA: Functional analysis of human MLH1 and MSH2 missense variants and hybrid human-yeast MLH1 proteins in Saccharomyces cerevisiae. Hum Mol Genet. 2001, 10: 1889-1900.
Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C: Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007, 67: 4595-4604.
Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, Plotz G, Jiricny J, Marra G: Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology. 2002, 122: 211-219.
Shimodaira H, Filosi N, Shibata H, Suzuki T, Radice P, Kanamaru R, Friend SH, Kolodner RD, Ishioka C: Functional analysis of human MLH1 mutations in Saccharomyces cerevisiae. Nat Genet. 1998, 19: 384-389.
Drotschmann K, Clark AB, Kunkel TA: Mutator phenotypes of common polymorphisms and missense mutations in MSH2. Curr Biol. 1999, 9: 907-910.
Shcherbakova PV, Hall MC, Lewis MS, Bennett SE, Martin KJ, Bushel PR, Afshari CA, Kunkel TA: Inactivation of DNA mismatch repair by increased expression of yeast MLH1. Mol Cell Biol. 2001, 21: 940-951.
Brieger A, Trojan J, Raedle J, Plotz G, Zeuzem S: Transient mismatch repair gene transfection for functional analysis of genetic hMLH1 and hMSH2 variants. Gut. 2002, 51: 677-684.
Rothstein RJ: One-step gene disruption in yeast. Methods Enzymol. 1983, 101: 202-211.
Storici F, Lewis LK, Resnick MA: In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001, 19: 773-776.
Strand M, Prolla TA, Liskay RM, Petes TD: Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993, 365: 274-276.
Parsons R, Li GM, Longley M, Modrich P, Liu B, Berk T, Hamilton SR, Kinzler KW, Vogelstein B: Mismatch repair deficiency in phenotypically normal human cells. Science. 1995, 268: 738-740.
Gietz RD, Sugino A: New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988, 74: 527-534.
Wach A, Brachat A, Pohlmann R, Philippsen P: New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994, 10: 1793-1808.
Polaczek P, Putzke AP, Leong K, Bitter GA: Functional genetic tests of DNA mismatch repair protein activity in Saccharomyces cerevisiae. Gene. 1998, 213: 159-167.
Gietz RD, Woods RA: Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350: 87-96.
Lea DE, Coulson CA: The contribution of the number of mutants in bacterial populations. Journal of Genetics. 1948, 49: 264-285.
Nicholson A, Hendrix M, Jinks-Robertson S, Crouse GF: Regulation of mitotic homeologous recombination in yeast. Functions of mismatch repair and nucleotide excision repair genes. Genetics. 2000, 154: 133-146.
Caligo MA, Bonatti F, Guidugli L, Aretini P, Galli A: A yeast recombination assay to characterize human BRCA1 missense variants of unknown pathological significance. Hum Mutat. 2009, 30: 123-133.
Ishioka C, Frebourg T, Yan YX, Vidal M, Friend SH, Schmidt S, Iggo R: Screening patients for heterozygous p53 mutations using a functional assay in yeast. Nat Genet. 1993, 5: 124-129.
Yamane S, Yamaoka M, Yamamoto M, Maruki T, Matsuzaki H, Hatano T, Fukui S: Region specificity of chromosome III on gene expression in the yeast Saccharomyces cerevisiae. J Gen Appl Microbiol. 1998, 44: 275-281.
Hudler P, Vouk K, Liovic M, Repse S, Juvan R, Komel R: Mutations in the hMLH1 gene in Slovenian patients with gastric carcinoma. Clin Genet. 2004, 65: 405-411.
Tomlinson IP, Beck NE, Homfray T, Harocopos CJ, Bodmer WF: Germline HNPCC gene variants have little influence on the risk for sporadic colorectal cancer. J Med Genet. 1997, 34: 39-42.
Hendriks Y, Franken P, Dierssen JW, De Leeuw W, Wijnen J, Dreef E, Tops C, Breuning M, Brocker-Vriends A, Vasen H, et al: Conventional and tissue microarray immunohistochemical expression analysis of mismatch repair in hereditary colorectal tumors. Am J Pathol. 2003, 162: 469-477.
Quaresima B, Grandinetti C, Baudi F, Tassone P, Barbieri V, Conforti S, Avvedimento EV, Costanzo F, Venuta S: Hereditary nonpolyposis coloretal cancer: identification of novel germline mutations in two kindreds not fulfulling the Amsterdam criteria. Mutations in brief no. 203. Online. Hum Mutat. 1998, 12: 433-
Raedle J, Brieger A, Trojan J, Hardt T, Herrmann G, Zeuzem S: Evaluation of rapid microsatellite analysis of paraffin-embedded specimens in screening for hereditary nonpolyposis colorectal cancer. Mod Pathol. 1999, 12: 485-491.
Buerstedde JM, Alday P, Torhorst J, Weber W, Muller H, Scott R: Detection of new mutations in six out of 10 Swiss HNPCC families by genomic sequencing of the hMSH2 and hMLH1 genes. J Med Genet. 1995, 32: 909-912.
Ward R, Meldrum C, Williams R, Mokany E, Scott R, Turner J, Hawkins N, Burgess B, Groombridge C, Spigelman A: Impact of microsatellite testing and mismatch repair protein expression on the clinical interpretation of genetic testing in hereditary non-polyposis colorectal cancer. J Cancer Res Clin Oncol. 2002, 128: 403-411.
Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, et al: Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996, 2: 169-174.
Dieumegard B, Grandjouan S, Sabourin JC, Le Bihan ML, Lefrere I, Bellefqih , Pignon JP, Rougier P, Lasser P, Benard J, et al: Extensive molecular screening for hereditary non-polyposis colorectal cancer. Br J Cancer. 2000, 82: 871-880.
Plevova P, Krepelova A, Papezova M, Sedlakova E, Curik R, Foretova L, Navratilova M, Novotny J, Zapletalova J, Palas J, et al: Immunohistochemical detection of the hMLH1 and hMSH2 proteins in hereditary non-polyposis colon cancer and sporadic colon cancer. Neoplasma. 2004, 51: 275-284.
Plaschke J, Kruger S, Pistorius S, Theissig F, Saeger HD, Schackert HK: Involvement of hMSH6 in the development of hereditary and sporadic colorectal cancer revealed by immunostaining is based on germline mutations, but rarely on somatic inactivation. Int J Cancer. 2002, 97: 643-648.
Yuan Z, Legendre B, Sreeramoju P, Lowes C, Reynolds D, Bennett A, Kent TS, Miller A, Zhu J, Weber TK: A novel mutation detection approach of hMLH1 and hMSH2 genes for screening of colorectal cancer. Cancer Detect Prev. 2006, 30: 333-340.
Guerrette S, Acharya S, Fishel R: The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999, 274: 6336-6341.
Genuardi M, Carrara S, Anti M, Ponz de Leon M, Viel A: Assessment of pathogenicity criteria for constitutional missense mutations of the hereditary nonpolyposis colorectal cancer genes MLH1 and MSH2. Eur J Hum Genet. 1999, 7: 778-782.
Cravo M, Afonso AJ, Lage P, Albuquerque C, Maia L, Lacerda C, Fidalgo P, Chaves P, Cruz C, Nobre-Leitao C: Pathogenicity of missense and splice site mutations in hMSH2 and hMLH1 mismatch repair genes: implications for genetic testing. Gut. 2002, 50: 405-412.
Rossi BM, Lopes A, Oliveira Ferreira F, Nakagawa WT, Napoli Ferreira CC, Casali Da Rocha JC, Simpson CC, Simpson AJ: hMLH1 and hMSH2 gene mutation in Brazilian families with suspected hereditary nonpolyposis colorectal cancer. Ann Surg Oncol. 2002, 9: 555-561.
Scartozzi M, Bianchi F, Rosati S, Galizia E, Antolini A, Loretelli C, Piga A, Bearzi I, Cellerino R, Porfiri E: Mutations of hMLH1 and hMSH2 in patients with suspected hereditary nonpolyposis colorectal cancer: correlation with microsatellite instability and abnormalities of mismatch repair protein expression. J Clin Oncol. 2002, 20: 1203-1208.
Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, Thibodeau SN, de la Chapelle A: Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res. 2004, 64: 4721-4727.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/9/382/prepub
Acknowledgements
We thank Drs. Francesca Storici and Thomas D. Petes for kindly providing pCORE and pSH91 plasmids, and Drs. Bert Vogelstein and Kenneth W. Kinzler for providing pSG-PMS2-wt plasmid. We thank Jelka Lenarcic for excellent technical assistance. The authors are grateful to Professor Bill Milne and Professor Roger Pain for critical reading of the manuscript. This work was supported by grant P1-0104 from ARRS and a JI grant to author MV.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MV participated in the design of the study, performed the statistical analysis and drafted the manuscript. MV and NZ carried out the experiments. AC and PH participated in the design of the study. RK coordinated the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vogelsang, M., Comino, A., Zupanec, N. et al. Assessing pathogenicity of MLH1 variants by co-expression of human MLH1 and PMS2genes in yeast. BMC Cancer 9, 382 (2009). https://doi.org/10.1186/1471-2407-9-382
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-9-382