
Abstract
Background
TGFβ has pleiotropic effects that range from regulation of proliferation and apoptosis to morphological changes and epithelial-mesenchymal transition (EMT). Some evidence suggests that these effects may be interconnected. We have recently reported that P53, P21Cip1 and pRB, three critical regulators of the G1/S transition are variably involved in TGFβ-induced cell cycle arrest in hepatocytes. As these proteins are also involved in the regulation of apoptosis in many circumstances, we investigated their contribution to other relevant TGFβ-induced effects, namely apoptosis and EMT, and examined how the various processes were interrelated.
Methods
Primary mouse hepatocytes deficient in p53, p21 and/or Rb, singly or in combination were treated with TGFβ for 24 to 96 hours. Apoptosis was quantified according to morphology and by immunostaining for cleaved-capsase 3. Epithelial and mesenchymal marker expression was studied using immunocytochemistry and real time PCR.
Results
We found that TGFβ similarly induced morphological changes regardless of genotype and independently of proliferation index or sensitivity to inhibition of proliferation by TGFβ. Morphological changes were accompanied by decrease in E-cadherin and increased Snail expression but the mesenchymal markers (N-cadherin, SMAα and Vimentin) studied remained unchanged. TGFβ induced high levels of apoptosis in p53-/-, Rb-/-, p21 cip1-/- and control hepatocytes although with slight differences in kinetics. This was unrelated to proliferation or changes in morphology and loss of cell-cell adhesion. However, hepatocytes deficient in both p53 and p21 cip1were less sensitive to TGFβ-induced apoptosis.
Conclusion
Although p53, p21 Cip1 and pRb are well known regulators of both proliferation and apoptosis in response to a multitude of stresses, we conclude that they are critical for TGFβ-driven inhibition of hepatocytes proliferation, but only slightly modulate TGFβ-induced apoptosis. This effect may depend on other parameters such as proliferation and the presence of other regulatory proteins as suggested by the consequences of p53, p21 Cip1 double deficiency. Similarly, p53, p21 Cip1 and pRB deficiency had no effect on the morphological changes and loss of cell adhesion which is thought to be critical for metastasis. This indicates that possible association of these genes with metastasis potential would be unlikely to involve TGFβ-induced EMT.
Similar content being viewed by others


Background
TGFβ is known to have pleiotropic effects which differ according to cell state and differentiation (reviewed in reference [1]). This includes regulation of proliferation and apoptosis, and stimulation of epithelial-mesenchymal transition (EMT) which together are critical for the development of invasive and metastasis potential.
In the liver TGFβ is released in many settings to act as a critical mediator of responses to injury [2]. It controls the proliferation of hepatocytes [3], induces hepatocytes apoptosis [4] and activates EMT [5–8] which, in certain contexts can actually protect hepatocytes from TGFβ-induced apoptosis [9].
In hepatocellular carcinoma (HCC), there is often production of TGFβ from both malignant hepatocytes and the non-parenchymal cells [10, 11]. However, evidence suggests that HCC have acquired a resistance to TGFβ inhibition of proliferation through mechanisms that include decreased TGFβ-receptor II expression [12] or induction of the inhibitory SMAD7 in advanced HCC [12, 13]. In HCC of different aetiologies, particularly in livers with chronic viral hepatitis B or hepatitis C infection, P53, P21Cip1 and pRb function are altered [14–21]. We have recently reported that deficiency in these proteins significantly affects hepatocyte responses to TGFβ-induced inhibition of proliferation, and may therefore contribute to resistance to TGFβ [22]. As these proteins are also key regulators of hepatocyte apoptosis [23] and given the association of p53 with metastasis potential [24], we determined whether deficiency in these genes also altered TGFβ effects on morphology, EMT and apoptosis, and investigated the possible relationships between those changes.
Methods
Hepatocyte culture
Mouse primary hepatocytes (male, 6–12 weeks old), were isolated by liver perfusion of Rb-floxed mice (homozygous for exon 19 of Rb flanked by LoxP sequences)[25]p53-/- [26] and p21 Cip1-null mice [18] using a standard two-step retrograde procedure [27]. Hepatocytes were purified using Percoll gradient [28] and cultured in supplemented serum-free medium selecting against survival of non-parenchymal cells. [29, 30]. Rb-floxed hepatocytes were infected at the time of plating with either a replication-deficient adenovirus expressing Cre-recombinase (Ad-Cre) or with the control adenovirus (Ad-Dl70) using a multiplicity of infection of 10 [23]. This gave rise to isogenic hepatocytes either homozygous for the floxed-Rb allele (phenotypically wild-type) or homozygous for deletion of exon 19 of Rb (Rb-null). Where indicated, hepatocytes were treated daily with 160pM of TGF-β 1 (TGFβ) for the indicated time starting from 24 hours of culture.
All animals used in this study received humane care. The study protocols are in compliance with the UK Home Office regulation and the local institutional policies.
Quantification of apoptosis
Cells were prepared using Feulgen stain and light green counterstain [31, 32]. Briefly, cells were fixed in Boum's fixative, treated with 5M HCl for 45 minutes then stained with Schiff reagent. Apoptosis was quantified according the morphological characteristics of the cells as previously described [33]. Normal hepatocytes are flat, with big pale pink nuclei and green cytoplasm while necrotic hepatocytes have shrunken and distorted nuclei darker pink and no condensation of the chromatin (see additional file 1). Apoptotic cells have condensed, uniformly refractile chromatin with retracted condensed cytoplasm (dark green) often with blebbing. Typically apoptotic bodies are best seen at a different plane of focus (above) normal cells. For each time point of each experiment 500 hepatocytes were counted from 2 independent cultures allowing us to plot a percentage of apoptotic cells +/- SD. Each experiment was repeated at least 3 times with similar results.
Statistical analyses were performed using Minitab 13.0. The proportion of apoptotic cells was arcsine transformed, and differences between means were evaluated with Analysis of Variance (ANOVA). Differences were taken to be significant when p < 0.05. Satisfactory homogeneity of variances was determined with Bartlett's test. Where a significant difference between means was identified with ANOVA, pairwise comparisons were performed using Bonferroni tests for multiple comparisons.
In addition to morphological assessment, activation of apoptosis was confirmed biochemically by quantification of cleaved-caspase 3. Briefly, hepatocytes in culture for 72 and 96 hours, treated or not with TGFβ, were fixed in acetone/methanol. Cleaved-caspase 3 was labelled using a rabbit polyclonal antibody (9661S, New England Biolabs, UK) (1/100 overnight) and red-fluorescent secondary antibody (Alexa555 goat antirabbit, molecular probes, UK).
Immunohistochemistry
Control (wt) and Rb -/- hepatocytes in culture for 24 hours on Permanox 2-well chamberslides (LabTech, UK) were treated daily or not with TGFβ(160pM of TGF-β1). 72 hours after treatment (96 hours after plating) the hepatocytes were fixed in Acetone/Methanol (50/50 V/V). After Avidin/Biotin block (Vector, UK), the slides were incubated with a rabbit polyclonal anti E-cadherin antibody (clone NCH38, DAKO UK), rabbit monoclonal anti CK18 (clone E431-1, AbCam, UK), mouse monoclonal anti-SMAα (clone 1A4, Sigma UK) or rabbit polyclonal anti N-cadherin (AbCam, UK), followed by the appropriate biotinylated secondary antibody (Dako, UK) and revealed using ready-to-use peroxidase stain (Vector, UK) and counterstained with haematoxylin.
Real time RT-PCR
RNA was extracted using Qiagen RNeasy mini kit according to manufacturers' instructions. The concentration and purity of RNA was determined using a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies DE, USA). Total RNA (up to 1 μg) was reverse transcribed using Superscript II reverse transcriptase (Invitrogen, UK) in a reaction mix consisting of: 4 μl of 5× buffer, 2 μl DTT (0.1 M), 0.5 μl dNTPs (10 mM), 2 μg of random hexamers, 0.5 μl (100 U) Superscript II and 0.5 μl (20 U) RNAse inhibitor (all Invitrogen, UK). The resulting cDNA was diluted 1:100 ready for PCR.
Expression of both E-cadherin and Snail was determined using SYBR green master mix (Invitrogen, UK) and the following primers: E-Cadherin: Forward primer ACT GTG AAG GGA CGG TCA AC; Reverse primer GGA GCA GCA GGA TCA GAA TC. Snail: Forward primer CCA CTG CAA CCG TGC TTT T; Reverse primer TCT TCA CAT CCG AGT GGG TTT. Pre-designed 18S primer/probe (Applied Biosystems, UK) was run as an internal control using a TaqMan master mix (Invitrogen, UK). Each system was run under standard conditions on an ABI 7500 detection system
Results
TGFβ induces a high level of apoptosis in hepatocytes deficient in p53, p21 and/or Rb, but the sensitivity is genotype-dependent
TGFβ can induce apoptosis in certain cell-types, including hepatocytes [4, 34, 35]. Given the role of P53 and pRb, respectively as a positive and a negative regulator of apoptosis induced by a variety of stimuli [36, 37], we investigated whether the absence of either gene alters the susceptibility of hepatocytes to TGFβ-induced apoptosis.
We found that hepatocytes of all tested genotypes undergo apoptosis after treatment with TGFβ. Interestingly, apoptotic cells were observed earlier in p53-/-, Rb-/- and double null cultures (p53Rb-/-) compared with wild-type (4.41; 4.26 and 2.71% versus 0.52% in untreated wild type control 48 hours after treatment) (figure 1A) indicating that the loss of Rb or p53 sensitises hepatocytes to TGFβ-induced death.

Effect of p53 , p21 Cip1 and Rb deficiency on hepatocyte sensitivity to TGFβ-induced apoptosis. Where appropriate cells were treated with TGFβ from 24 hours in culture. For apoptosis curves in A, B & C, each time point is the average percentage of apoptotic cells +/- SDV obtained from two cultures where 500 cells were counted. The experiment was repeated 3 times with similar results. Control cells are shown in all graphs; in blue control cells treated with TGFβ. Untreated controls are shown with open symbols. The tables show the p values of statistical analysis for pairwise comparison of apoptosis in the genotype of interest (see text and methods). A: p53 &Rb deficiency increase sensitivity to apoptosis. The curves show the % of apoptotic cells in control (▲,△); p53-/- (□,■); Rb-/- (○,●); and p53-/- Rb-/-(◇, ◆) cells treated (plain) or not (open) with TGFβ. B: p21 Cip1-deficiency has no effect on Rb -/- cells. The curve shows the percentage of apoptosis in cells of indicated genotypes either wild type (black/blue) or deleted for p21 Cip1 (red) treated or not with TGFβ. C: Cells deficient in both p53 and p21 Cip1 are less sensitive to TGFβ-induced apoptosis. Compare black symbols (single nulls for p53 or p21 Cip1) with red symbols (deficient in both p53 and p21 Cip1).
We then studied the effect of p21 Cip1 deficiency (figure 1B). The absence of functional P21Cip1 had no effect on TGFβ-induced apoptosis in hepatocytes with functional P53 (control and Rb-/-) (figure 1B). By contrast, regardless of Rb status, deficiency in both P53 and P21Cip1 decreased the sensitivity to TGFβ (decreased apoptosis both in double p53-/-p21 Cip1-/- and triple-nulls p53-/-Rb-/-p21 Cip1-/-) (figure 1C). These differences in apoptosis levels were confirmed by labelling the cells with an antibody specific for cleaved caspase 3 (ASP175) (figure 2). The results were concordant with the morphology data: after 48 hours of treatment with TGFβ (72 hours in culture) control, p53 p21 cip1 -/- and TRPL hepatocytes had similar low levels of cleaved-caspase 3 (figure 2A). Hepatocytes of all other genotypes exhibited higher levels of cleaved-caspase 3, with the highest level observed in Rb-/- cells. This is in accordance with the kinetics of appearance of cells with apoptotic morphology (figure 1). After 72 hours of treatment (96 hours in culture) the levels of cleaved-capsase 3 increased in hepatocytes of all genotypes but control, p53p21-/- and TRPL showed the lowest levels of cleaved-caspase 3 positivity.

Caspase 3 cleavage in hepatocytes of different genotypes. Cleaved-caspase 3 was labelled using red immunofluorescence and the proportion of positive cells quantified by counting 3 × 20 fields of view per genotype. B: The figure shows representative photos of (a, b) untreated hepatocytes 72 hours after plating (here Rb-/-); (c, d) hepatocytes in culture for 72 hours treated with TGFβ for 48 hours (p 53-/- & control); (e) untreated hepatocytes 96 hours after plating (p21 cip1-/-); (f, g, h) hepatocytes in culture for 96 hours treated with TGFβ for 72 hours (p53 p21 cip1 -/-, control &p21 cip1-/-).
We have previously reported that p53, p21 Cip1 and Rb deficiencies lead to increased proliferation albeit of different magnitude depending on the genotype [23]. More recently, we have shown that these genes are also involved in the regulation of TGFβ-induced cell cycle arrest, but again to various degrees [22]. It has been suggested that increased cell cycle activity can lower the apoptotic threshold which would explain the early onset of apoptosis in p53-/- and Rb-/-. The current observation that apoptosis starts when the first untreated cells enter mitosis (90 to 96 hours after plating in wild-type, and around 72 hours in p53-/- and Rb-/- cells) might indicate that TGFβ induces a coordinated regulation of growth and apoptosis. However the level of proliferation and apoptosis were found to be unrelated in these cells (additional file 2A). In fact, regardless of p53 and Rb genotypes, hepatocytes deficient in p21 Cip1 always proliferate more but showed less apoptosis after TGFβ-treatment than their "wild type for p21 Cip1 "counterpart (additional file 2B follow blue arrows). By contrast deletion of Rb always increased proliferation and apoptosis after TGFβ, regardless of the other genes' status (additional file 2B, follow purple arrows).
TGFβ induces morphological changes regardless of genotype
TGFβ stimulates production of extracellular matrix (ECM) proteins and their integrin receptors [38], thus affecting cytoskeletal structure, cell shape, and cell spreading, which in turn regulate gene expression and cell growth [39, 40]. We observed that TGFβ-treated hepatocytes exhibited dramatic changes in cell shape and spreading. Untreated hepatocytes cultured on plastic coated with fibronectin spread to form a monolayer of cobblestone epithelioid cells. By contrast, TGFβ-treated cells became elongated with reduced cytoplasm and assumed a fibroblast-like morphology (figure 3). These changes occurred regardless of genotype and are similar to those described as epithelial to mesenchymal transition (EMT). We therefore investigated the expression of various proteins that characterise EMT [41].

TGFβ induces morphological changes regardless of genotype. The figure shows representative photos of control, Rb-/-, p53-/- and p21 Cip1-/- cells 72 hours after plating, TGFβ-treated or not for 48 hours. The cells were cultured in the presence of BrdU to assess proliferation indexes for another study [23]. The dark nuclei represent immunopositivity for BrdU. Negative nuclei are paler.
TGFβ does not induce complete EMT
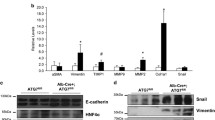
EMT is characterised by changes in expression of various proteins, in which downregulation of E-cadherin and concomitant expression of N-cadherin represents defining events ([42] and therein). We analysed the expression of these proteins, together with the expression of vimentin and alpha-smooth muscle actin (SMAα), also expected to increase during EMT (figure 4A) and cytokeratin 18 (CK18), an epitheial hepatocyte marker. As expected, primary hepatocytes expressed E-cadherin and CK18 but not vimentin, N-cadherin or SMAα. After TGFβ-treatment, E-cadherin staining was negative in all genotypes. By contrast there were no changes in expression of N-cadherin, SMAα and vimentin (all negative) or cytokeratin 18 (positive). To confirm these data, we studied E-cadherin, Snail (a key regulator of E-cadherin expression during EMT) and N-cadherin gene expression. Using real time PCR we found that E-cadherin expression decreased with TGFβ-treatment while Snail expression increased in cells of all genotypes (figure 4B). N-cadherin expression was barely detectable and no increase in expression was observed after TGFβ-treatment (data not shown).

TGFβ decreases E-cadherin expression. A: The figure shows representative photos of control, Rb-/-, p53-/- and p21 Cip1-/- cells 96 hours after plating, TGFβ-treated or not for 72 hours labelled for E-cadherin, N-cadherin, SMAα, vimentin and CK18. Positive controls: E-cadherin: membrane staining of hepatocytes; N-cadherin: mouse primary hepatic stellate cells; SMAα: mouse primary hepatic stellate cell; cytokeratin 18: staining of hepatocytes but not sinusoidal cells; vimentin: intense staining of sinusoidal cells but not hepatocytes. B: E-cadherin and snail expression corrected for 18S amplification in hepatocytes treated or not with TGFβ for 72 hours. Repeat real time RT-PCR with independent samples showed similar results except for snail expression in p21-/- which was already high and did not further increase after TGFβ-treatment.
Discussion
TGFβ has pleiotropic effects ranging from regulation of proliferation and apoptosis to morphological changes and mesenchymal transition (EMT). Our evidence suggests that these effects may be interconnected: for example, TGFβ stimulates the production of extracellular matrix proteins and their receptors [38], affecting cytoskeletal structure, cell shape, and cell spreading, which in turn regulates gene expression and cell proliferation [39, 43].
We have recently reported that P53, P21Cip1 and pRB, three critical regulators of the G1/S transition are variably involved in TGFβ-induced cell cycle arrest in hepatocytes [22]. As these proteins are also involved in the regulation of apoptosis in many circumstances [36, 37], we investigated their contribution to other TGFβ-induced effects, namely apoptosis and EMT, and examined how these different processes were interrelated.
One of TGFβ properties is to induce morphological changes associated with epithelial to mesenchymal transition (EMT), which is characterised by downregulation of epithelial markers concomitant with upregulation of mesenchymal markers [44–46]. Mouse primary hepatocytes of all genotypes expressed high levels of E-cadherin and CK18, two epithelial markers. After treatment with TGFβ, the morphological changes observed were consistent with that seen during EMT: E-cadherin expression decreased sharply while snail expression increased. However, mesenchymal markers expression was not significantly increased (N-cadherin, vimentin, SMAα) and CK18 remained unchanged. Therefore the morphological changes were only associated with changes in E-cadherin expression, and this is most probably driven by snail [47] and reflect loss of epithelial differentiation rather than mesenchymal transition. Why mesenchymal markers were not expressed is unclear, but longer exposure (4 to 6 days) to several signals may be required for completion of EMT [48]. In any case, this occurred for all genotypes suggesting that P53, P21Cip1 and pRB are not involved in these changes. Nevertheless, morphological changes and proliferation have been shown to be correlated. For example, in rat hepatocytes, morphological changes induced by TGFβ-treatment govern proliferation through alteration of p27KIP1 and p21Cip1 expression [49]. In our cells, TGFβ-treatment did not upregulate p27KIP1 [22]. The morphological alterations also occurred similarly without p21 Cip1, suggesting that these cyclin-dependent kinases (CDKI) are not required for this TGFβ-effect in primary adult mouse hepatocytes. Furthermore, the morphological changes occurred irrespective of genotypes which we have previously reported have differing proliferation index [23] and responses to inhibition of proliferation by TGFβ [22]. This demonstrates that TGFβ-signalling to morphological changes is different to its growth inhibitory signalling in primary mouse hepatocytes. A similar observation has recently been reported for human keratinocytes [50].
TGFβ is well described as a trigger for apoptosis in hepatocytes [35] and high level of apoptosis was observed regardless of genotype studied. Apoptosis appeared somewhat later than in other published studies where high levels are usually reported after 48 hours of TGFβ; the fact that primary hepatocytes in our system are almost synchronous in terms of entry from quiescence to cell cycle on plating [22, 23, 32] may be a factor and supports the hypothesised influence of cell cycle stage on TGFβ-induced apoptosis. In our study, apoptosis triggered by TGFβ occurred earlier in p53 and Rb-deficient cells than in control cells, showing an increased sensitivity. It has been suggested that increased cell cycle activity may lower the threshold for apoptosis. More specifically, the abundance of pRb and its level of phosphorylation have been shown to correlate with the threshold for TGFβ-induced apoptosis in hepatocytes [51]. In that study, Fan et al reported an enrichment of the G2/M population after TGFβ-treatment and proposed that TGFβ-induced apoptosis occurred in G1 or S phase [51]. In our system, using near synchronous adult primary hepatocytes, TGFβ-treatment triggers a strong and sustained G1-arrest in control cells [22] (less than 1% cells entering S phase) supporting the hypothesis that TGFβ-induced apoptosis occurs in G1. However, the earlier onset of apoptosis in p53 and Rb-null cells correlates with the peak of S phase and the appearance of the first mitosis suggesting that in cells escaping TGFβ-induced cell cycle arrest, another exit to apoptosis may exist in S phase and G2/M. Although this could contribute to the higher level of apoptosis in Rb-/- and p53-/- hepatocytes [23] it is clearly not the case in p21-/- hepatocytes: we have previously reported that p21 cip1-/- hepatocytes proliferate more and enter S phase earlier than p53 or Rb-/- cells [23] and that a substantial proportion of p21 cip1-/- cells treated with TGFβ are able to enter S phase (10.3% +/- 3.3 in p21-/- versus 0.70% +/- 0.42 in control hepatocytes; ([22] and additional file 2). Despite this sustained proliferation in p21 cip1-/- cells, apoptosis was of similar level to that of control cells. Similarly, p21 cip1 Rb-/- cells proliferate more than Rb-/- cells after TGFβ-treatment (21.1% +/- 0.26 versus 9.05% +/- 3.17 respectively, additional file 2), yet apoptosis was not significantly different in those cells. This may suggest that the presence of p21 cip1, and more particularly expression of cytoplasmic P21Cip1after TGFβ-treatment [22] which has been associated with apoptosis [52], sensitises hepatocytes to TGFβ-induced apoptosis independently to the proliferation index.
In fact, comparison of all genotypes revealed no simple relationship between the level of proliferation and the amount of apoptosis (additional file 2). This therefore suggests that other parameters are important and that higher proliferation itself can not predict cells' sensitivity to TGFβ.
Interestingly, apoptosis induced by TGFβ in p53-null cells was at least partially, dependent on P21Cip1 (regardless of Rb genotype) as double deficiency in p53 and p21 cip1decreases the level of apoptosis. This effect is specific to TGFβ as it is not observed when apoptosis is induced by other means such as UV-induced DNA-damage (data not shown).
Various recent reports have shown an association between resistance to apoptosis and EMT in hepatocytes [8, 9, 53]. In one study, TGFβ treatment of a subpopulation of fetal rat hepatocytes resulted in EMT which was associated with resistance to apoptosis. By contrast, adult rat hepatocytes which did not undergo EMT, died by apoptosis [54]. In our system, the morphological changes and loss of cell adhesion may be insufficient to provide resistance, or the pathway may be different in adult primary mouse hepatocytes. This is suggested by the recent study from Ju et al. [55] that showed that Smad2 deficiency leads to EMT but does not affect apoptosis in adult primary hepatocytes.
Conclusion
During carcinogenesis, growth inhibitory responses to TGFβ are often lost, and insufficient apoptosis has been associated with the development of hepatocellular carcinoma [56]. P53, P21Cip1 and pRb are well known regulators of both proliferation and apoptosis in response to a multitude of stresses. In a previous study, we found that p53, p21 cip1and Rb deficiencies decreased the sensitivity of primary hepatocytes to TGFβ-driven cell cycle arrest. Rb deficiency had the strongest effect which was independent of the presence of p53 and p21 cip1[22]. Our present results reveal a more subtle involvement of theses genes in the regulation of TGFβ-induced apoptosis. Although differences in the onset of apoptosis were observed in the different genotypes, all but p53 p21 cip1 double null remained very sensitive to apoptosis induced by TGFβ. We therefore propose that p53 and Rb have only slight modulatory effect on TGFβ-induced apoptosis and that other parameters including proliferation index, presence of P21CIP1 and double deficiency in p53 and p21 cip1are likely to be more important.
EMT is thought to be critical for metastasis. Increased snail expression and decreased E-cadherin, associated with changes of morphology occurred irrespective of p53, p21 Cip1 and pRb genotypes without mesenchymal transition, suggesting that the association of p53 deficiency with metastasis in the liver [24] for example does not correlate with a change in the ability of hepatocytes to undergo loss of cell adhesion induced by TGFβ.
Abbreviations
- EMT:
-
Epithelial-Mesenchymal transition
- CK18:
-
cytokeratin 18
- HCC:
-
Hepatocellular carcinoma
- SD:
-
Standard deviation
- SMAα:
-
Alpha-smooth muscle actin
- TGFβ:
-
Transforming growth factor β
- BrdU:
-
5-bromo-2-deoxyuridine
- TRPL:
-
p53 p21 cip1 Rb triple null genotype
References
Massague J, Blain SW, Lo RS: TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000, 103: 295-309. 10.1016/S0092-8674(00)00121-5.
Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S: TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006, 44: 57-66. 10.1055/s-2005-858989.
Oe S, Lemmer ER, Conner EA, Factor VM, Leveen P, Larsson J, Karlsson S, Thorgeirsson SS: Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology. 2004, 40: 1098-1105. 10.1002/hep.20426.
Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R: Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci U S A. 1992, 89: 5408-5412. 10.1073/pnas.89.12.5408.
Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S: Laminin-5 with transforming growth factor-beta 1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005, 129: 1375-1383. 10.1053/j.gastro.2005.09.055.
Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A: Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007, 282: 22089-22101. 10.1074/jbc.M700998200.
Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD, Kirby JA: Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest. 2008, 88: 112-123. 10.1038/labinvest.3700704.
Caja L, Conrad O, Esther B, Murillo MM, Miro-Obradors MJ, Palacios E, Fabregat I: Differential intracellular signalling induced by TGF-beta in rat adult hepatocytes and hepatoma cells: Implications in liver carcinogenesis. Cellular Signalling. 2007, 19: 683-694. 10.1016/j.cellsig.2006.09.002.
Del Castillo G, Murillo MM, Alvarez-Barrientos A, Bertran E, Fernandez M, Sanchez A, Fabregat I: Autocrine production of TGF-beta confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: Role of EGF receptor ligands. Exp Cell Res. 2006, 312: 2860-2871. 10.1016/j.yexcr.2006.05.017.
Bedossa P, Peltier E, Terris B, Franco D, Poynard T: Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology. 1995, 21: 760-766.
Paik SY, Park YN, Kim H, Park C: Expression of transforming growth factor-beta1 and transforming growth factor-beta receptors in hepatocellular carcinoma and dysplastic nodules. Mod Pathol. 2003, 16: 86-96. 10.1097/01.MP.0000047308.03300.9C.
Halder SK, Beauchamp RD, Datta PK: Smad7 induces tumorigenicity by blocking TGF-beta-induced growth inhibition and apoptosis. Exp Cell Res. 2005, 307: 231-246. 10.1016/j.yexcr.2005.03.009.
Park YN, Chae KJ, Oh BK, Choi J, Choi KS, Park C: Expression of Smad7 in hepatocellular carcinoma and dysplastic nodules: resistance mechanism to transforming growth factor-beta. Hepatogastroenterology. 2004, 51: 396-400.
Cho J, Baek W, Yang S, Chang J, Sung YC, Suh M: HCV core protein modulates Rb pathway through pRb down-regulation and E2F-1 up-regulation. Biochim Biophys Acta. 2001, 1538: 59-66. 10.1016/S0167-4889(00)00137-3.
Choi BH, Choi M, Jeon HY, Rho HM: Hepatitis B viral X protein overcomes inhibition of E2F1 activity by pRb on the human Rb gene promoter. DNA Cell Biol. 2001, 20: 75-80. 10.1089/104454901750070274.
Munakata T, Nakamura M, Liang Y, Li K, Lemon SM: Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2005, 102: 18159-18164. 10.1073/pnas.0505605102.
Deng L, Nagano-Fujii M, Tanaka M, Nomura-Takigawa Y, Ikeda M, Kato N, Sada K, Hotta H: NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J Gen Virol. 2006, 87: 1703-1713. 10.1099/vir.0.81735-0.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P: Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995, 82: 675-684. 10.1016/0092-8674(95)90039-X.
Gong GZ, Jiang YF, He Y, Lai LY, Zhu YH, Su XS: HCV NS5A abrogates p53 protein function by interfering with p53-DNA binding. World J Gastroenterol. 2004, 10: 2223-2227.
Han HJ, Jung EY, Lee WJ, Jang KL: Cooperative repression of cyclin-dependent kinase inhibitor p21 gene expression by hepatitis B virus X protein and hepatitis C virus core protein. FEBS Lett. 2002, 518: 169-172. 10.1016/S0014-5793(02)02694-7.
Kwun HJ, Jang KL: Natural variants of hepatitis B virus X protein have differential effects on the expression of cyclin-dependent kinase inhibitor p21 gene. Nucleic Acids Res. 2004, 32: 2202-2213. 10.1093/nar/gkh553.
Sheahan S, Bellamy CO, Dunbar DR, Harrison DJ, Prost S: Deficiency of G1 regulators P53, P21CIP and/or pRb decreases hepatocyte sensitivity to TGFbeta cell cycle arrest. BMC Cancer. 2007, 7: 215-225. 10.1186/1471-2407-7-215.
Sheahan S, Bellamy CO, Treanor L, Harrison DJ, Prost S: Additive effect of p53, p21 and Rb deletion in triple knockout primary hepatocytes. Oncogene. 2004, 23: 1489-1497. 10.1038/sj.onc.1207280.
Chen YW, Klimstra DS, Mongeau ME, Tatem JL, Boyartchuk V, Lewis BC: Loss of p53 and Ink4a/Arf cooperate in a cell autonomous fashion to induce metastasis of hepatocellular carcinoma cells. Cancer Res. 2007, 67: 7589-7596. 10.1158/0008-5472.CAN-07-0381.
Vooijs M, te RH, van , Berns A: Tumor formation in mice with somatic inactivation of the retinoblastoma gene in interphotoreceptor retinol binding protein-expressing cells. Oncogene. 2002, 21: 4635-4645. 10.1038/sj.onc.1205575.
Purdie CA, Harrison DJ, Peter A, Dobbie L, White S, Howie SE, Salter DM, Bird CC, Wyllie AH, Hooper ML, .: Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994, 9: 603-609.
Renton KW, Deloria LB, Mannering GJ: Effects of Polyriboinosinic Acid, Polyribocytidylic Acid and A Mouse Interferon Preparation on Cytochrome P-450-Dependent Mono-Oxygenase Systems in Cultures of Primary Mouse Hepatocytes. Molecular Pharmacology. 1978, 14: 672-681.
Kreamer BL, Staecker JL, Sawada N, Sattler GL, Hsia MTS, Pitot HC: Use of A Low-Speed, Iso-Density Percoll Centrifugation Method to Increase the Viability of Isolated Rat Hepatocyte Preparations. In Vitro Cellular & Developmental Biology. 1986, 22: 201-211. 10.1007/BF02623304.
Bellamy CO, Prost S, Wyllie AH, Harrison DJ: UV but not gamma-irradiation induces specific transcriptional activity of p53 in primary hepatocytes. J Pathol. 1997, 183: 177-181. 10.1002/(SICI)1096-9896(199710)183:2<177::AID-PATH909>3.0.CO;2-E.
Bellamy C: PhD thesis. The roles of P53 in hepatocytes. 1997, PhD Thesis.Edinburgh University
Prost S, Sheahan S, Rannie D, Harrison DJ: Adenovirus-mediated Cre deletion of floxed sequences in primary mouse cells is an efficient alternative for studies of gene deletion. Nucleic Acids Res. 2001, 29: E80-10.1093/nar/29.16.e80.
Prost S, Bellamy CO, Cunningham DS, Harrison DJ: Altered DNA repair and dysregulation of p53 in IRF-1 null hepatocytes. FASEB J. 1998, 12: 181-188.
Kerr JF, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972, 26: 239-257.
Rotello RJ, Lieberman RC, Purchio AF, Gerschenson LE: Coordinated regulation of apoptosis and cell proliferation by transforming growth factor beta 1 in cultured uterine epithelial cells. Proc Natl Acad Sci U S A. 1991, 88: 3412-3415. 10.1073/pnas.88.8.3412.
Oberhammer F, Bursch W, Parzefall W, Breit P, Erber E, Stadler M, Schulte-Hermann R: Effect of transforming growth factor beta on cell death of cultured rat hepatocytes. Cancer Res. 1991, 51: 2478-2485.
Harbour JW, Dean DC: Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000, 2: E65-E67. 10.1038/35008695.
Fridman JS, Lowe SW: Control of apoptosis by p53. Oncogene. 2003, 22: 9030-9040. 10.1038/sj.onc.1207116.
Roberts AB, McCune BK, Sporn MB: TGF-beta: regulation of extracellular matrix. Kidney Int. 1992, 41: 557-559. 10.1038/ki.1992.81.
Iwig M, Czeslick E, Muller A, Gruner M, Spindler M, Glaesser D: Growth regulation by cell shape alteration and organization of the cytoskeleton. Eur J Cell Biol. 1995, 67: 145-157.
Assoian RK: Control of the G1 phase cyclin-dependent kinases by mitogenic growth factors and the extracellular matrix. Cytokine Growth Factor Rev. 1997, 8: 165-170. 10.1016/S1359-6101(97)00011-7.
Thiery JP, Sleeman JP: Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006, 7: 131-142. 10.1038/nrm1835.
Ahmed S, Nawshad A: Complexity in interpretation of embryonic epithelial-mesenchymal transition in response to transforming growth factor-beta signaling. Cells Tissues Organs. 2007, 185: 131-145. 10.1159/000101314.
Assoian RK, Zhu X: Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr Opin Cell Biol. 1997, 9: 93-98. 10.1016/S0955-0674(97)80157-3.
Hay ED: An overview of epithelio-mesenchymal transformation. Acta Anatomica. 1995, 154: 8-20.
Hay ED, Zuk A: Transformations Between Epithelium and Mesenchyme - Normal, Pathological, and Experimentally-Induced. American Journal of Kidney Diseases. 1995, 26: 678-690. 10.1016/0272-6386(95)90610-X.
Zeisberg M, Chandraker A, Sayegh MH, Kalluri R: Involvement of epithelial-to-mesenchymal transition (EMT) in chronic renal allograft dysfunction. American Journal of Transplantation. 2004, 4: 458-458. 10.1111/j.1600-6143.2004.00387.x.
Zavadil J, Bottinger EP: TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005, 24: 5764-5774. 10.1038/sj.onc.1208927.
Grunert S, Jechlinger M, Beug H: Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nature Reviews Molecular Cell Biology. 2003, 4: 657-665. 10.1038/nrm1175.
Nagaki M, Sugiyama A, Naiki T, Ohsawa Y, Moriwaki H: Control of cyclins, cyclin-dependent kinase inhibitors, p21 and p27, and cell cycle progression in rat hepatocytes by extracellular matrix. J Hepatol. 2000, 32: 488-496. 10.1016/S0168-8278(00)80401-8.
Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP: Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci U S A. 2001, 98: 6686-6691. 10.1073/pnas.111614398.
Fan G, Ma X, Kren BT, Steer CJ: The retinoblastoma gene product inhibits TGF-beta1 induced apoptosis in primary rat hepatocytes and human HuH-7 hepatoma cells. Oncogene. 1996, 12: 1909-1919.
Fan G, Ma X, Wong PY, Rodrigues CM, Steer CJ: p53 dephosphorylation and p21(Cip1/Waf1) translocation correlate with caspase-3 activation in TGF-beta1-induced apoptosis of HuH-7 cells. Apoptosis. 2004, 9: 211-221. 10.1023/B:APPT.0000018803.11260.02.
Valdes F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernandez M, Benito M, Nieto MA, Fabregat I: The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res. 2002, 1: 68-78.
Caja L, Ortiz C, Bertran E, Murillo MM, Miro-Obradors MJ, Palacios E, Fabregat I: Differential intracellular signalling induced by TGF-beta in rat adult hepatocytes and hepatoma cells: Implications in liver carcinogenesis. Cell Signal. 2006
Ju W, Ogawa A, Heyer J, Nierhof D, Yu L, Kucherlapati R, Shafritz DA, Bottinger EP: Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol. 2006, 26: 654-667. 10.1128/MCB.26.2.654-667.2006.
Guicciardi ME, Gores GJ: Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005, 54: 1024-1033. 10.1136/gut.2004.053850.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/191/prepub
Acknowledgements
This work was supported by grants from the Melville Trust for the Care and Cure of Cancer to SP and SS. The p21 Cip1-/- mice were kind gift from Philip Leder (Harvard Medical School, Boston, MA) and the Rb-floxed (Rb lox/lox) mice from Anton Berns (Netherlands Cancer Institute, Amsterdam), to whom we are very grateful. We wish to thanks Helen Caldwell for technical assistance, Professor Sarah Howie (Centre for Inflammation, QMRI, Edinburgh) for kindly donating Snail primers, Rebecca Aucott (Centre for Inflammation, QMRI, Edinburgh) for providing N-cadherin antibody and Helen Wilson (Centre for Reproductive Biology, QMRI, Edinburgh) for providing the cleaved-caspase 3 antibody.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SS initiated the study and carried out some of the experiments, COB contributed to the experimental design, interpretation of data, supervision and gave critical review of the manuscript, SNH carried out some of the experiments, DJH contributed to the experimental design and gave general supervision and funding support, SP made substantial contribution to the conception & design, acquisition and interpretation of data and wrote the manuscript.
Electronic supplementary material
12885_2008_1129_MOESM1_ESM.pdf
Additional file 1: Representative photos of primary hepatocytes necrosis and apoptosis. Our method of quantifying apoptosis and necrosis is based on the morphological characteristics of the cells. Those have been well described, by Kerr JF, Wyllie AH, and Currie AR who first described apoptosis in 1972 (Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics.Br J Cancer 1972; 26: 239–257). Enclosed are photos of normal, apoptotic and necrotic hepatocytes, showing how easy these different cells can be recognised after Feulgen staining and light green counterstain. The colours on the photos are real and have not been modified; the cytoplasm is green and the chromatin pink. Normal hepatocytes are flat, with big pale pink nuclei and green cytoplasm. Necrotic hepatocytes have shrunken and distorted nuclei darker pink and no condensation of the chromatin. Apoptotic cells have condensed, uniformly refractile chromatin with retracted condensed cytoplasm (dark green) often with blebbing. Typically apoptotic bodies are best seen at a different plane of focus (above) normal cells. 1 & 2: photos of the same field showing some apoptotic cells in a different plan of focus compared with normal and necrotic cells. 3: In this photo, showing apoptotic, necrotic and normal hepatocytes, the apoptotic cells in the black rectangles have been taken on a different plan of focus to show the apoptotic bodies. Note that necrotic nuclei is "detaching" from cytoplasm area that is not condensed, whereas apoptotic cells have highly condensed chromatin and cytoplasm. (PDF 56 KB)
12885_2008_1129_MOESM2_ESM.pdf
Additional file 2: Proliferation does not dictate the level of apoptosis. A: The graph represents the index of proliferation versus the percentage of apoptosis in TGFβ-treated hepatocytes of indicated genotypes. The percentage of proliferation after TGFβ-treatment was calculated by integration of the number of cells incorporating BrDU between 72 and 108 hours after plating (48 and 84 hours of TGFβ) [22]. Using videomicroscopy, we have observed that, in our culture conditions, the apoptotic primary hepatocytes remain attached for many days to the plate. The number of apoptotic cells counted at a given time is therefore a good estimation of the number of cells undergoing apoptosis until that time. The graph therefore shows the % apoptotic cells at 108 hours. A similar analysis was performed with proliferation and apoptosis values for 120 hours with similar results. Note the absence of relationship between proliferation and apoptosis. B: As above with arrows showing the effect of p53 (red), Rb (purple) or p21 Cip1 (blue) deficiencies on both apoptosis levels and proliferation for the various genotypes. (PDF 31 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sheahan, S., Bellamy, C.O., Hartland, S.N. et al. TGFbeta induces apoptosis and EMT in primary mouse hepatocytes independently of p53, p21 Cip1 or Rbstatus. BMC Cancer 8, 191 (2008). https://doi.org/10.1186/1471-2407-8-191
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-191