
Abstract
Background
The identification of a BRCA1 or BRCA2 mutation in familial breast cancer kindreds allows genetic testing of at risk relatives. However, considerable controversy exists regarding the cancer risks in women who test positive for the family mutation.
Methods
We reviewed 385 unrelated families (223 with BRCA1 and 162 with BRCA2 mutations) ascertained through two regional cancer genetics services. We estimated the penetrance for both breast and ovarian cancer in female mutation carriers (904 proven mutation carriers – 1442 females in total assumed to carry the mutation) and also assessed the effect on penetrance of mutation position and birth cohort.
Results
Breast cancer penetrance to 70 and to 80 years was 68% (95%CI 64.7–71.3%) and 79.5% (95%CI 75.5–83.5%) respectively for BRCA1 and 75% (95%CI 71.7–78.3%) and 88% (95%CI 85.3–91.7%) for BRCA2. Ovarian cancer risk to 70 and to 80 years was 60% (95%CI 65–71%) and 65% (95%CI 75–84%) for BRCA1 and 30% (95%CI 25.5–34.5%) and 37% (95%CI 31.5–42.5%) for BRCA2. These risks were borne out by a prospective study of cancer in the families and genetic testing of unaffected relatives. We also found evidence of a strong cohort effect with women born after 1940 having a cumulative risk of 22% for breast cancer by 40 years of age compared to 8% in women born before 1930 (p = 0.0005).
Conclusion
In high-risk families, selected in a genetics service setting, women who test positive for the familial BRCA1/BRCA2 mutation are likely to have cumulative breast cancer risks in keeping with the estimates obtained originally from large families. This is particularly true for women born after 1940.
Similar content being viewed by others

Background
Since the identification of the BRCA1 [1] and BRCA2 [2] genes a great deal of debate has focussed on the issue of breast and ovarian cancer risk associated with mutations in these genes. It is clear that calculated cancer risks are dependent on the method of ascertainment of the families studied. Thus, breast cancer risks in large familial breast cancer kindreds with BRCA1/BRCA2 mutations are substantially higher than risks derived from population based studies [3, 7, 8]. In the high-risk families that recruited to the Breast Cancer Linkage Consortium (BCLC) cohort, BRCA1 and BRCA2 mutations were estimated to cause a cumulative lifetime risk of breast cancer at age 70 years of 85–87% and 77–84% respectively [3, 7, 8]. However, estimates of breast cancer risks to age 70 years of age derived from previous population based studies to date are much lower at 28–60% [4–6] for BRCA1, and lower still for BRCA2. It has been suggested that even these studies may overestimate the effect of the BRCA1/2 mutation alone [9]. Whilst there is some evidence of variation of cancer risk by position of mutation within each gene [10–12], more variation occurs between families with the same mutation. Therefore it is likely that a substantial proportion of the breast cancer risk in strong familial clusters with a BRCA1/2 mutation (the group of families that are usually seen by a Cancer Genetics Service), might be contributed to by modifier genes [13]. Optimum clinical practice requires, that the cancer risks provided to families undergoing genetic testing are appropriate to the setting in which the mutation was detected. To determine the most appropriate risks for women attending clinical cancer genetics services we determined the cumulative risks of breast and ovarian cancer for 385 families with pathogenic BRCA1/2 mutations identified in North West and Central England covering a population of 10 million.
Methods
Index cases and relatives
Breast and ovarian cancer families have been tested for BRCA1/2 mutations (using a whole gene analysis including a test for large deletions) since 1996 in the overlapping regions of Manchester and Birmingham in mid-north England. All genetic testing is undertaken with informed consent and consent is also taken to confirm cancer diagnosis. The study was carried out with Local Ethical committee approval. Women who attend the specialist genetic clinics in these regions with a family history of breast/ovarian cancer have a detailed family tree elicited with all first, second and if possible third degree relatives recorded. If a BRCA1/2 mutation is identified, further extensive attempts are made to ensure that all individuals at risk of inheriting the family mutation are represented on the pedigree. All cases of breast or abdominal cancers are confirmed by means of hospital/pathology records, from the Regional Cancer Registries (data available from 1960) or from death certification. Once a family specific pathogenic BRCA1/2 mutation is identified predictive testing is offered to all blood relatives. Where possible all affected women with breast/ovarian cancer are tested to establish the true extent of BRCA1/2 involvement in the family. In many large families it is possible to establish "obligate" gene carriers by testing for the same mutation in different branches of the family, thereby establishing that intervening relatives carry the same mutation.
All female BRCA1/2 mutation carriers identified were included in this study, and their details, those of all tested relatives and first-degree untested female relatives were entered onto a Filemaker Pro 5 database. The initial individual in which a mutation was identified was designated the "index" case, with all other individuals being classified as to their position in the pedigree compared to a proven mutation carrier. All women reaching 20 years were entered if untested for a mutation. The exception was mothers of a mutation carrier when it was clear that the mutation was paternally inherited. 385 index cases were studied and from these extended pedigrees information on a total of 2466 females was collected. Information was entered on date of birth, date of last follow up, breast cancer status, ovarian cancer status, dates of diagnoses and date of death (if applicable), gene mutation carried in the family, their relationship to a known mutation carrier and their mutation status and date at which testing took place.
The proportions of unaffected first-degree relatives (FDRs) testing positive or negative was derived for each age cohort. Figures from this were used to estimate the proportion of untested relatives that were likely to test positive in each age group. The proportion of untested FDRs with breast or ovarian cancer that were likely to test positive was similarly estimated from testing that had taken place in each family. Penetrance analysis was performed by including all mutation positive individuals and appropriate numbers of untested FDRs on a proportional basis. Kaplan Meier curves were derived for breast and ovarian cancer incidence for each gene and by dividing each gene into the previously identified ovarian cancer cluster region (OCCR): exon 11 (nucleotides 2401–4190) for BRCA1 and exon 11 (nucleotides 3035–6629) for BRCA2. For BRCA1 we used the nucleotide range identified by the BCLC [11], although this is not traditionally called an OCCR it is the region published as having the greatest proportional risk of ovarian cancer. Individuals were censored at age of death, age of last follow up, age at appropriate cancer or age at appropriate risk reducing surgery (oophorectomy for ovarian cancer, mastectomy and oophorectomy for breast cancer). The Manchester scoring system was used to assess the strength of the breast/ovarian cancer history [14]. This system was devised to assess the likelihood of a BRCA1/2 mutation and scores breast and ovarian cancers individually in the family, giving a higher score the younger the age at diagnosis [14]. A combined score of 20 reflects a 20% likelihood of identifying a BRCA1/2 mutation.
Results
The 385 families consisted of 223 apparently unrelated BRCA1 and 162 BRCA2 families. Mutations were spread throughout the BRCA1 and BRCA2 genes with the commonest mutation being the Jewish exon 2 185 DelAG (31 families). There were also 20 families with single or multiple exon deletions or duplications in BRCA1 and 6 in BRCA2. These families contained 904 proven female mutation carriers (526 in BRCA1; 378 in BRCA2). There were 992 female FDRs of unknown mutation status: 554 in BRCA1; 438 in BRCA2 kindreds. Of these 244 had been diagnosed with breast cancer, 88 with ovarian cancer and 14 with both. 21/206 (10%) FDRs with breast cancer tested negative for the family mutation, but only 1/101 FDRs with ovarian cancer. The age distribution of the breast cancer cases testing negative for the mutation was identical to those testing positive. We therefore assumed that every tenth untested FDR with breast cancer (only) was negative for the mutation in each gene. All 21 individuals testing negative for the family mutation were also negative for the 1100delC mutation in CHEK2. As 99% of the ovarian cancers tested were positive we assumed that all FDRs with ovarian cancer were positive. The results for predictive testing of unaffected females for each gene are shown in Table 1. We assumed that similar proportions of untested unaffected female relatives would test positive for each gene. We therefore stratified these relatives by age and excluded an increasing proportion of the relatives as indicated for each age group. For the age group of 60 years and over we assumed that 10% would be positive for BRCA1 and 20% for BRCA2.
The proportion testing positive for each gene with Manchester scores [14] above and below 20 and 23 are presented in Table 2. This shows a substantial effect of cancer burden for BRCA2 with high-risk families (scores above 20 points) having a much lower proportion of positive predictive tests after 50 years.
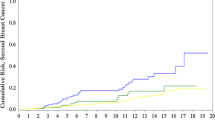
Overall, of the FDRs of unknown mutation status, 92/92 with ovarian cancer, 220/244 with breast cancer and 234/648 unaffected FDRs were included in the analysis. In total this amounted to 839 actual and presumed carriers for BRCA1 and 603 actual and presumed carriers for BRCA2. There were 243/839 (29%) BRCA1 individuals with ovarian cancer compared to 64/603 (11%) female BRCA2 carriers. 411/839 (49%) BRCA1 carriers and 355/603 (59%) BRCA2 carriers had developed breast cancer. Penetrance estimates for each gene are shown (Table 3; Figures 1, 2) for breast and ovarian cancer. The curves were remarkably similar for each gene, with breast cancer penetrance to 70 and 80 years of 68% (95%CI 65–71%) and 79.5% (95%CI 75–84%) for BRCA1 and 74% (95%CI 71–77%) and 88% (95%CI 85–91%) for BRCA2. Ovarian cancer risk to 70 and 80 years was 60% (95%CI 65–71%) and 65% (95%CI 75–84%) for BRCA1 and 30% (95%CI 25.5–34.5%) and 37% (95%CI 31.5–42.5%) for BRCA2. The penetrance for ovarian cancer was significantly higher for BRCA1 (p < 0.0001), but breast cancer incidence for BRCA2 was borderline significantly higher than for BRCA1 (p = 0.09). Indeed breast cancer penetrance estimates for BRCA2 after 60 years were significantly higher as was overall penetrance including the index case (p = 0.02). There was no significant effect of ovarian cancer cluster regions (OCCR, nucleotides 2401–4190 BRCA1 and nucleotides 3035–6629 in BRCA2) for either gene with lifetime ovarian cancer risks (to 80 years) of 65% for 573 BRCA1 carriers outside the OCCR and 70% for 266 women with mutations within the OCCR (p = 0.18). Similarly there was no effect of position for BRCA2 with lifetime risks of 37% for 373 BRCA2 carriers outside the OCCR and 41% for 230 BRCA2 women with OCCR mutations (p = 0.17). There was a 10% higher cumulative incidence at most ages for breast cancer in those outside the BRCA2 OCCR, although lifetime risk was little different at 90% and statistical significance was not reached (p = 0.07). No such difference was seen for BRCA1 with virtually identical incidence curves (p = 0.25). DCIS was included as breast cancer. However, this only amounts to 1% of BRCA1 breast cancers and 2% for BRCA2. It is likely that nearly all of these would have become invasive as only 1/16 occurred after 60 years of age. Tamoxifen is not licensed in the UK for prevention. Only 23 mutation carriers took tamoxifen as part of the IBIS1 prevention trial and this is unlikely to have materially changed the penetrance estimates.

Breast cancer cumulative incidence by gene ( BRCA1 or BRCA2 ).

Ovarian cancer cumulative incidence by gene ( BRCA1 or BRCA2 ).
An estimate for breast cancer penetrance was also made for each 10–20 year birth cohort. A highly significant difference was identified with those born after 1960 having a breast cancer risk to 40 years of age of 40% compared to only 7.5% for those with a year of birth between 1900 and 1920 (Figure 3: p < 0.00001). However, after exclusion of the index case the cumulative risk to 40 years dropped to between 21–23% for the birth cohorts after 1940. This was, nonetheless still a highly significant trend (p = 0.0005). After exclusion of the index case there was no significant birth cohort effect observed for ovarian cancer (p = 0.086). To assess the earlier age at breast cancer diagnosis on life expectancy we carried out a Kaplan-Meier survival analysis on the birth cohorts, again excluding the index case. There was no significant difference in survival from birth (Table 4; log rank df 6, p = 0.07), although there was a trend to better survival in the earlier birth cohorts. Indeed if the index case was included 21/83 (25%) index cases post birth year 1960 had died by 45 years of age, equivalent to a cumulative mortality of 35% to that age.

Cumulative risk of breast cancer by age cohort for BRCA1 and BRCA2 combined after exclusion of the index case. Risk to 40 years: Group 1 (birth year <1900; n = 45) 4%; Group 2 (1900–1920; n = 154) 8%; Group 3 (1920–1930; n = 154) 10%; Group 4 (1930–1940; n = 124) 17%; Group 5 (1940–1950; n = 162) 21%; Group 6 (1950–1960; n = 265) 23%; Group 7 (1960+; n = 276) 22%. Log rank (df 6) 153; p = 0.0005.
Breast cancer incidence was also assessed after family ascertainment. Incidence figures for breast/ovarian cancer are shown in Table 5. These reflect the incidence in unaffected women at the time of family ascertainment and follow up was censored at the time of risk reducing surgery (oophorectomy/mastectomy). As the index case was used to identify the mutation usually on surveillance the incidence rates for these cases are artificially high. Excluding the index cases there was an incidence of 2.5–2.7 per thousand for breast cancer in proven carriers. Even including 40% of the follow up time and 80% of the breast cancers from the FDR unknown category (Tables 1 and 2), this still gave an annual incidence of breast cancer of 1.98% for both BRCA1/2 mutation carriers (38/1917; 35/1763.6). An annual rate of 2% averaged over the risk period of 30–79 years would if anything indicate a higher risk than those indicated by the Kaplan-Meier analysis.
Discussion
We present data on a large cohort of women identified as carriers or presumed carriers of BRCA1 and BRCA2 mutations in a large proportion of the UK population. The penetrance estimates derived from these women are very similar to those derived from the BCLC cohort of high-risk families with lifetime risks of breast cancer of close to 85% for both genes [3, 7, 8]. The estimate of ovarian cancer was also very similar with risks to 70 years of 60% for BRCA1 carriers and 33% as opposed to 27% [3] for BRCA2 carriers. It is possible that the higher overall breast cancer estimates for BRCA2 were related to competing mortality from ovarian cancer. Many risk factors for breast and ovarian cancer are similar (early menarche, late menopause, nulliparity) and women with these may have died from ovarian cancer before they developed breast cancer. This effect would be more prominent for BRCA1 and would potentially explain the higher breast cancer penetrance for BRCA2. The ratio of those testing positive:negative for the BRCA mutation whilst still unaffected also gives support to high penetrance. Of those women without an affected daughter, <10% of those aged over 60 years, tested positive for BRCA1 and <20% for BRCA2. The figures over 60 years are, nonetheless based on small numbers. The earlier drop in positive:negative ratio for BRCA1 almost certainly represents a higher combined risk of both breast and ovarian cancer to 50 and 60 years. Another supportive feature is shown in Table 2. The typical families tested in our centre have a Manchester score of 20+ reflecting multiple early onset breast and/or ovarian cancer in the family. The less "high" risk clusters as evidenced by lower Manchester scores had a higher proportion testing positive >50 years. This suggests that Manchester score could be used as a bench-mark to predict penetrance particularly in BRCA2 families. Whilst all attempts to assess penetrance have their inherent biases and assumptions this cannot be said of the results of presymptomatic testing. The only potential bias would be if women had an inkling that they would test positive or negative prior to coming forward. This is not borne out by our results particularly accounting for Manchester score.
The previously reported positional effect of mutations for both BRCA1 and BRCA2 is not borne out by our analysis. No substantial effect of increased risk of ovarian cancer was seen in the respective ovarian cluster regions of each gene and only a borderline significant reduction of breast cancer risk was seen for BRCA2. Much of the OCCR association has been based on ratios of breast to ovarian cancer [10] or on the presence or not of ovarian cancer in the family [11]. Even this reliance on the presence of ovarian cancer for BRCA2 has been questioned by the report of 58% of BRCA2 related ovarian cancer families having mutations outside the OCCR [12]. Although the BCLC study on BRCA1 positional effect [10] included 356 families compared to our 223 families no absolute estimate of penetrance was made. Whilst the breast cancer incidence was lower in the central portion of the gene (nucleotides 2401–4190) (RR 0.71) in their analysis it was not possible to derive absolute risk figures for each portion of the gene. Additionally it is likely that our more extensive testing of unaffected relatives may provide a more accurate overall picture as reported here. Accurate estimates of cancer risk are essential for families and individuals undertaking genetic testing. Based on our analysis, it is questionable whether any account should be taken of the OCCR in each gene or indeed any substantial positional effect in genetic counselling.
It is also clear that for individuals undertaking predictive genetic testing in the context of families ascertained from cancer genetic clinics as opposed to population testing that risk figures similar to those derived in our study or the BCLC is quoted in our own clinics and we recommend that penetrance estimates are derived for the population being counselled. Our data are nonetheless at variance to a similar analysis carried out in North America [15]. A series of 1948 families were tested for mutations in BRCA1/2 in eight centres. 283 families with BRCA1 mutations were identified and 143 in BRCA2. The authors used statistical modelling to arrive at penetrance figures by 70 years of 46% (95%CI 39–54%) for BRCA1 and 43% (95%CI 36–51%) for BRCA2. The authors did not appear to take advantage of any further testing of relatives in the family. Whilst they corrected for potential ascertainment bias, they did not allow for the effects of modifier genes in these families and purely looked at attributable risk from BRCA1 and BRCA2 mutations alone. This was based on the apparent lack of heterogeneity in another study of Jewish families from North America [16]. What is particularly concerning is the risk attributed to "non mutation carriers" to 70 years. A figure of 5% as a general population risk for breast cancer may have been correct 20–30 years ago, but is certainly not the risk faced by women in the US or the UK today. Breast cancer risk to age 70 is 7.6% in the UK [17] and nearer 8% in the US. A correction for this difference might give penetrance figures of nearer 74% for BRCA1 and 69% for BRCA2. The decision not to include any adjustment in these families for the effects of modifier genes is questionable. The difference in penetrance obtained from the BCLC and from population studies strongly suggests the presence of additional genetic factors in high-risk families. We have recently reported that those testing negative for a family BRCA mutation are still at 3-fold relative risk of breast cancer [13]. This phenocopy effect was also seen in the Iceland data for their founder BRCA2 mutation, although to a lesser extent given the strong population based element of their analysis [18]. However, it is possible that modifier genes are more prevalent in some populations and that penetrance in North America is less affected by modifier genes than in the UK. The presence of these modifier alleles is now indisputable from recent genome wide association studies [19–21].
A potential criticism of our study is that we have not taken enough account of ascertainment bias and that additional adjustment maybe necessary beyond excluding the index case. An analysis using these adjustments was carried out in the North American study [15] and recent reports from the Cambridge group [22]. These studies did not take into account the widespread testing of relatives and as explained above the American study deliberately excluded any effect other than of the BRCA1/2 mutation. Whilst it is clearly interesting to know the effect of BRCA1/2 alone, women undergoing testing will want to know what their own specific risk of breast and ovarian cancer are, including that contributed by other potential "modifier" genes in their family. We must also acknowledge that confidence intervals in table 3 should also be wider due to forcing the data on unknown FDRs into a known category.
The high-risk women testing positive is also supported by the prospective part of our study. The 2–2.7% annual risk demonstrated is equivalent to the highest risk in a 10-year period (23% BRCA1; 30% BRCA2-Table 3). Although most of the breast cancers were detected by screening, only one was detected at a prevalence mammogram. These follow up risks are also supported by a similar follow up study in the Netherlands where 8 breast cancers occurred in 63 mutation carriers with a calculated annual risk of 2.5% [23].
Our own study and recent analyses from North America and Iceland demonstrate that women in the most recent birth cohort have a substantially higher risk of developing breast cancer than past cohorts [16, 18]. The incidence of breast cancer in BRCA2 carriers has risen 4 fold in 80 years in Iceland (as has breast cancer in the general population) and we have observed a similar increase from <10% risk by 40 years in those born before 1930 to a 40% risk on those born after 1960, although this was less significant after allowing for ascertainment bias. It is, therefore, inappropriate to quote risks as low as 43–46% (based on population studies) for lifetime breast cancer risk to women in their twenties or early thirties if they test positive for a mutation in a high-risk family. Another potential effect of earlier breast cancer might be a reduction in life expectancy. With increasing survival from birth in the general population and improved survival from diagnosis of breast cancer we might have expected to see improved life expectancy. However, it would appear that these elements almost completely cancel each other out and there is no evidence for improved survival from birth in modern BRCA birth cohorts.
When discussing the higher risks of breast cancer in recent generations, it is nonetheless important to couch any discussion on risk in terms of future prospects for risk reduction by preventive measures. Increasing numbers of women are opting for risk reducing surgery particularly early RRO, which will substantially reduce the risk of both breast and ovarian cancer [24]. It is also likely that new treatments or substantial changes from the Western lifestyle may have a sufficient effect to help in risk management in the future.
Conclusion
We believe our results show that when counselling women on their risks of breast and ovarian cancer if they carry a family BRCA1/2 mutation the risks should reflect the context of cancer in their family and not just an average risk from possibly over-corrected penetrance estimates from population studies. Indeed a recent review in a prestige journal quoted "headline" risks for BRCA2 of only 40% and 8% for breast and ovarian cancer to 80 years [25]. Understandably many clinicians and counsellors may quote these risks. The use of family cancer burden in adjusting risks to carriers is already used in the BOADICEA programme [26] and the Manchester score could also be used as a bench mark of where in the range of 40–90% breast cancer risk a women should be steered, especially for BRCA2.
References
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994, 266 (5182): 66-71. 10.1126/science.7545954.
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al: Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995, 378 (6559): 789-792. 10.1038/378789a0.
Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al: Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998, 62 (3): 676-689. 10.1086/301749.
Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al: The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997, 336 (20): 1401-1408. 10.1056/NEJM199705153362001.
Warner E, Foulkes W, Goodwin P, Meschino W, Blondal J, Paterson C, et al: Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst. 1999, 91 (14): 1241-1247. 10.1093/jnci/91.14.1241.
Hopper JL, Southey MC, Dite GS, Jolley DJ, Giles GG, McCredie MR, et al: Population-based estimate of the average age-specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Australian Breast Cancer Family Study. Cancer Epidemiol Biomarkers Prev. 1999, 8 (9): 741-747.
Cancer risks in BRCA2 mutation carriers: The Breast Cancer Linkage Consortium. J Natl Cancer Inst. 1999, 91 (15): 1310-1316. 10.1093/jnci/91.15.1310.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE: Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994, 343 (8899): 692-695. 10.1016/S0140-6736(94)91578-4.
Begg CB: On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst. 2002, 94 (16): 1221-1226.
Lubinski J, Phelan CM, Ghadirian P, Lynch HT, Garber J, Weber B, Tung N, Horsman D, Isaacs C, Monteiro AN, Sun P, Narod SA: Cancer variation associated with the position of the mutation in the BRCA2 gene. Fam Cancer. 2004, 3 (1): 1-10. 10.1023/B:FAME.0000026816.32400.45.
Thompson D, Easton D, Breast Cancer Linkage Consortium: Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol Biomarkers Prev. 2002, 11 (4): 329-36.
Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R: BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005, 104 (12): 2807-16. 10.1002/cncr.21536.
Smith A, Moran A, Boyd MC, Bulman M, Shenton A, Smith L, Iddenden I, Woodward E, Lalloo F, Rahman N, Maher ER, Evans DGR: The trouble with phenocopies: are those testing negative for a family BRCA1/2 mutation really at population risk?. J Med Genet. 2007, 44 (1): 10-15. 10.1136/jmg.2006.043091.
Evans DGR, Eccles DM, N Rahman, Young K, Bulman M, Amir E, Shenton A, Howell A, Lalloo F: A new scoring system for the chances of identifying a BRCA1/2 mutation, outperforms existing models including BRCAPRO. J Med Genet. 2004, 41 (6): 474-480. 10.1136/jmg.2003.017996.
Chen S, Iversen ES, Friebel T, Finkelstein D, Weber BL, Eisen A, Peterson LE, Schildkraut JM, Isaacs C, Peshkin BN, Corio C, Leondaridis L, Tomlinson G, Dutson D, Kerber R, Amos CI, Strong LC, Berry DA, Euhus DM, Parmigiani G: Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol. 24 (6): 863-71. 10.1200/JCO.2005.03.6772. 2006 Feb 20
King MC, Marks JH, Mandell JB: Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003, 302: 643-6. 10.1126/science.1088759.
McIntosh A, Shaw C, Evans G, Turnbull N, Bahar N, Barclay M, Easton D, Emery J, Gray J, Halpin J, Hopwood P, McKay J, Sheppard C, Sibbering M, Watson W, Wailoo A, Hutchinson A: Clinical Guidelines and Evidence Review for The Classification and Care of Women at Risk of Familial Breast Cancer. 2004, London: National Collaborating Center for Primary Care/University of Sheffield, NICE guideline CG014, [http://www.nice.org.uk]
Tryggvadottir L, Sigvaldason H, Olafsdottir GH, Jonasson JG, Jonsson T, Tulinius H, Eyfjord JE: Population-based study of changing breast cancer risk in Icelandic BRCA2 mutation carriers, 1920–2000. J Natl Cancer Inst. 2006, 98 (2): 116-22.
Easton DF, Pharoah PDP, Dunning AM, Pooley K, Cox DR, Ballinger D, Thompson D, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, the Search collaborators2, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Rahman N, Stratton MR, Peto J, Fletcher O, Ponder BAJ: A genome-wide association study identifies multiple novel breast cancer susceptibility loci. Nature. 2007, 447 (7148): 1087-93. 10.1038/nature05887.
Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Hoover RN, Thomas G, Chanock SJ: A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007, 39 (7): 870-4. 10.1038/ng2075.
Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE, CIMBA: An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007, 9 (2): 104-10.1186/bcr1670.
Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper J, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles D, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallionemi O-P, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF: Breast and ovarian cancer risks to carriers of the BRCA1 5382insC and 185delAG and BRCA2 6174delT mutations: a combined analysis of 22 population based studies. J Med Genet. 2005, 42 (7): 602-3. 10.1136/jmg.2004.024133.
Meijers-Heijboer EJ, van Geel B, van Putten WLJ, et al: Breast cancer after prophylactic bilateral mastectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2001, 345: 159-164. 10.1056/NEJM200107193450301.
Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van 't Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E, Olopade OI, Weber BL: Reduction in Cancer Risk After Bilateral Prophylactic Oophorectomy in BRCA1 and BRCA2 Mutation Carriers. N Engl J Med. 2002, 346: 1616-1622. 10.1056/NEJMoa012158.
Robson M, Offit K: Management of an inherited predisposition to breast cancer. N Engl J Med. 2007, 357: 154-162. 10.1056/NEJMcp071286.
Antoniou AC, Durocher F, Smith P, Simard J, Easton DF, INHERIT BRCAs program members: BRCA1 and BRCA2 mutation predictions using the BOADICEA and BRCAPRO models and penetrance estimation in high-risk French-Canadian families. Breast Cancer Res. 2006, 8 (1): R3-10.1186/bcr1365.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/155/prepub
Acknowledgements
We would like to thank The Genesis Appeal and The Breast Cancer Research Trust (BCRT) for their financial support for this work. We also dedicate this paper to Andrew Shenton who died tragically young on February 19th 2008.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DGE: Conception. DGE, AS, EW, FL: Data collection. DGE and AS: Data analysis. DGE, AS, FL, AH, ERM: Manuscript writing. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Evans, D.G., Shenton, A., Woodward, E. et al. Penetrance estimates for BRCA1 and BRCA2based on genetic testing in a Clinical Cancer Genetics service setting: Risks of breast/ovarian cancer quoted should reflect the cancer burden in the family. BMC Cancer 8, 155 (2008). https://doi.org/10.1186/1471-2407-8-155
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-155