
Abstract
Background
Chemotherapy with trastuzumab is widely used for patients with human epidermal growth factor receptor 2-positive (HER2+) breast cancer, but a significant number of patients with the tumor fail to respond, or relapse. The mechanisms of recurrence and biomarkers that indicate the response to the chemotherapy and outcome are not fully investigated.
Methods
Genomic alterations were analyzed using single-nucleotide polymorphism arrays in 46 HER2 immunohistochemistry (IHC) 3+ or 2+/fluorescent in situ hybridization (FISH)+ breast cancers that were treated with neoadjuvant chemotherapy with paclitaxel, cyclophosphamid, epirubicin, fluorouracil, and trastuzumab. Patients were classified into two groups based on presence or absence of alterations of 65 cancer-associated genes, and the two groups were further classified into four groups based on genomic HER2 copy numbers or hormone receptor status (HR+/−). Pathological complete response (pCR) and relapse-free survival (RFS) rates were compared between any two of the groups.
Results and discussion
The pCR rate was 54% in 37 patients, and the RFS rate at 3 years was 72% (95% CI, 0.55-0.89) in 42 patients. The analysis disclosed 8 tumors with nonamplified HER2 and 38 tumors with HER2 amplification, indicating the presence of discordance in tumors diagnosed using current HER2 testing. The 8 patients showed more difficulty in achieving pCR (P=0.019), more frequent relapse (P=0.018), and more frequent alterations of genes in the PI3K pathway (P=0.009) than the patients with HER2 amplification. The alterations of the PI3K and estrogen receptor (ER) pathway genes generally indicated worse RFS rates. The prognostic significance of the alterations was shown in patients with a HR+ tumor, but not in patients with a HR- tumor when divided. Alterations of the PI3K and ER pathway genes found in patients with a HR+ tumor with poor outcome suggested that crosstalk between the two pathways may be involved in resistance to the current chemotherapy with trastuzumab.
Conclusions
We recommend FISH analysis as a primary HER2 testing because patients with IHC 2+/3+ and nonamplified HER2 had poor outcome. We also support concurrent use of trastuzumab, lapatinib, and cytotoxic and anti-hormonal agents for patients having HR+ tumors with alterations of the PI3K and ER pathway genes.
Similar content being viewed by others

Background
Patients with human epidermal growth factor receptor 2 (HER2)-positive (HER2+) breast cancer were known to have a poor prognosis in the era when trastuzumab was not available [1–3]. After the introduction of trastuzumab, the outcome of HER2+ operable patients changed significantly, and many patients who achieved a pathological complete response (pCR) were expected to have been cured of the disease [2, 3]. However, pCR rates are 30-60%, and the 3-year relapse-free survival (RFS) is 71-78% in patients with operable breast cancer, indicating that a substantial number of patients who undergo surgical resection after the chemotherapy have recurrence [2, 3]. Patients with HER2+ breast cancer are usually treated with a combination of trastuzumab and taxanes with or without other chemotherapeutic agents [1–3], but predictors that indicate the response to the chemotherapy and outcome are not fully investigated.
Alterations in the HER2-PI3K-AKT pathway, which include expression of an extracellular domain-truncated HER2 (p95HER2), mutation and amplification of PIK3CA, loss of PTEN or INPPB4, and mutation of AKT1, are known to result in a poor response to chemotherapy with trastuzumab or poor outcome for breast cancer patients [4, 5]. In addition, there are two types of HER2+ breast cancer; namely, hormone receptor (HR)-positive (HR+) and HR-negative (HR-), and some investigators have reported different biological characteristics including pathological responses between the two [6, 7]. Crosstalk between the estrogen receptor (ER) pathway and the PI3K or ERK/MAPK pathway is thought to be involved in the resistance to trastuzumab-containing chemotherapy in HER2+/HR+ breast cancer [8]. However, there have been few studies aiming to resolve the mechanism of chemotherapy resistance or to identify biomarkers that indicate pCR and relapse using clinical samples.
It has been reported that 0.9% - 18.5% of HER2 immunohistochemistry (IHC) 3+ tumors had a single copy of HER2[8]. The technical shortcomings of IHC that can result in false-positive and false-negative results may be one of the reasons for the discordance between IHC grades and HER2 copy numbers [9, 10], however, there may be true single-gene overexpressers although the incidence may be low. Although metastatic breast cancer patients with the discordance between IHC and HER2 copy numbers seemed to show a low probability of responding to HER2-targeted therapy [11], there has been no study to clarify that single-gene overexpressers with operable breast cancer will respond to trastuzumab, and the mechanisms for the possible resistance to the trastuzumab-containing chemotherapy.
An alternatively spliced form of the human HER2 gene, Δ16HER2, containing an in-frame deletion was found in human breast cancer [12]. Mitra et al. showed that ectopic expression of the Δ16HER2 transcript, but not wild-type HER2 transcript, promotes receptor dimerization, cell invasion, and trastuzumab resistance in NIH3T3 and MCF7 tumor cells [13]. More recently, it was reported that Δ16HER2-expressing transgenic mice, but not wild-type HER2-expressing mice, developed multiple mammary adenocarcinomas [14]. However, the clinical significance of Δ16HER2 has not been fully examined in human breast cancer.
We hypothesized that genomic alterations detectable by single-nucleotide polymorphism (SNP) arrays and HER2 copy numbers and levels of HER2 transcripts would suggest mechanisms of resistance and prognostic factors for patients treated with trastuzumab-containing chemotherapy. Thus, we studied SNP array patterns of 143 breast cancer samples, including 46 HER2+ tumors, obtained at the time of diagnosis. We found that alterations of genes involved in the estrogen-receptor (ER) and PI3K pathways indicated worse RFS rates in patients with a HR+ but not HR- tumor, who were treated with chemotherapy with trastuzumab, followed by adjuvant trastuzumab (plus endocrine therapy for patients with a HR+ tumor). We also found that patients with a tumor showing a single HER2 copy number had more difficulty in achieving pCR, and tended to have worse RFS rates than those having a tumor with HER2 amplification. These findings may help to clarify the mechanisms for resistance to the chemotherapy with trastuzumab, and improve the efficacy of chemotherapy in HER2+ breast cancer.
Methods
Patients and samples
One hundred and fifty four tumor tissue and peripheral blood samples were obtained from 152 Japanese women, including two with bilateral tumors, who underwent a diagnostic core-needle biopsy between April 2005 and August 2011. The first specimen was used for the pathological diagnosis with H&E staining, the determination of ER, progesterone receptor (PgR), and HER2 status using IHC, and fluorescent in situ hybridization (FISH) [15]. The second and third specimens, which were directly frozen in liquid nitrogen, were used for DNA analysis including SNP assays and for RNA and definitive FISH analyses, respectively. Eleven specimens were excluded after evaluation of the content of tumor area, which was less than 30% of the whole specimen. Thus, 143 specimens from 141 patients were used for the present study. All patients included in the analysis provided consent to participate in the study and to publish the results. The study design was approved by the ethics committee of Saitama Cancer Center.
Histological examination and immunohistochemistry
The core-needle specimens were evaluated microscopically by pathologists, and classified according to the system proposed by Elston and Ellis [16]. Positive rates (%) for the ER and PgR were determined as a ratio of positive cells to total cancer cells, and a value of 10% or higher was defined as positive [17]. HER2 expression was defined as 0 to 3+ based on positive cell rates and the intensity of IHC staining (HercepTest, DAKO, Japan). Tumors showing moderate expression (2+) of HER2 were also tested by FISH to clarify amplification of the HER2 gene in paraffin specimens with the use of PathVysion (Abbott, IL); positive FISH was defined as a ratio of HER2 signals to centromere 17 signals of >2.2. Thus, a HER2-positive reaction was defined as either 3+ for IHC or 2+ for IHC with positive routine FISH results.
P-cadherin (monoclonal mouse anti-human P-cadherin clone 56, BD Transduction Lab.) was subjected to immunohistochemical staining using an avidin-biotin complex for the validation of genomic alterations identified by the SNP array in breast cancers.
Neoadjuvant and adjuvant chemotherapy and adjuvant hormone therapy
Of 141 patients, 46 were determined as having a HER2+ tumor. Of these 46 patients, 37 received neoadjuvant chemotherapy consisting of 12 weekly cycles of paclitaxel with trastuzumab, and four cycles of cyclophosphamide, epirubicin, and fluorouracil with concurrent trastuzumab throughout the chemotherapy [1, 18], and then underwent surgery. Of the remaining 9 patients, 4 chose immediate surgery, one received neoadjuvant chemotherapy without trastuzumab, which was added after surgery, and three with metastatic cancers at diagnosis and one with bilateral tumors who chose treatment with trastuzumab, exemestane, and radiation did not undergo surgery. All 5 patients who underwent surgery, received essentially the same trastuzumab-containing chemotherapy, and were included in the RFS analysis. Thus, pathological response after neoadjuvant chemotherapy was evaluated in 37 tumors, and RFS was evaluated for 42 patients. After surgery, weekly trastuzumab therapy was given to HR- patients for 6 to 12 months [3], and the same therapy plus hormone therapy; tamoxifen for premenopausal patients and an aromatase inhibitor for postmenopausal patients, was given to patients with a HR+ tumor.
Pathological response
Pathological response was assessed by a pathologist (M. K.) according to the “histopathological criteria for assessment of therapeutic response in breast cancer” proposed by the Japanese Breast Cancer Society [19]. The extent of responses is classified as grade 0, 1, 2, and 3, which represents no response, slight response, marked response, and complete response (CR), respectively. The grade 1 is further classified as 1a and 1b, which represents mild response indicating mild changes in cancer cells regardless of the area, or marked changes in less than one third of cancer cells, and moderate response indicating marked changes in one third or more but less than two thirds of tumor cells, respectively. Grade 2 indicates marked changes in two thirds or more of tumor cells. Grade 3 indicates necrosis or disappearance of all tumor cells.
Copy number and loss of heterozygosity (LOH) analysis using SNP arrays
Affymetrix Mapping 250K-Nsp arrays (Affymetrix, Santa Clara, CA) were used to analyze the chromosomal copy number and LOH status in 143 tumors as described previously [20]. Partial uniparental disomy (UPD) was defined as a region of copy number-neutral LOH spanning over 3 Mb. Copy numbers and LOH were calculated using CNAG and AsCNAR programs with paired references as controls [21, 22]. Amplification, gain, and loss are defined as copy number ratios of >2.5, 1.2-2.5, and <0.8, respectively.
Sixty-five genes were chosen for analyzing genomic alterations (gain, amplification, loss, and UPD) because of previous studies reporting the involvement of these genes in the neoplastic process of breast and other cancers. They were GPSM2, GSTM1, ATR, PIK3CA, MUC4, INPP4B, TERT, MAP3K1, CCNB1, GCCR, FOXC1, DEK, ID4, E2F3, NOTCH4, VEGFA, ESR1, HOXA9, CHIP, MET, EGR3, FGFR1, MYC, CDKN2A, BAG1, CTSL2, GATA3, PTEN, FGFR2, MKI67, SCUBE2, CCND1, EMSY, HBXAP, GAB2, PGR, BIRC2, HER3, MDM2, BRCA2, RB1, SPRY2, FOXA1, MTA1, RAD51, PTPN9, IGFR1, CDH3, CDH1, CD68, TP53, AURKB, ERBB2/HER2, GRB7, BECN1, BRCA1, MME1, TRIM25, BCAS3, BIRC5, BCL2, MYBL2, AIB1, AURKA, and MMP11.
Quantitative PCR (QPCR) analysis of HER2copy numbers
The QPCR analysis of HER2 copy numbers was carried out with a Light Cycler (Roche Diagnostics, Indianapolis, IN) and TaqMan probe (Roche Diagnostics). We designed two regions of HER2 [HER2 5´ region, 5´-GACAGCCGCAGTAGCTTCTTA-3´ and 5´-CAAAATGGAGCGCAGGTT-3´ (UPL#34); HER2 3´ region, 5´- GAGAACCCCGAGTACTTGACAC-3´ and 5´- CCAGTAATAGAGGTTGTCGAAGG-3´ (UPL#63)] to quantify the copy number of HER2. MOCS2 at 5q11.2 and SCN7A at 2q24.3, where the normal copy was identified by a SNP array-based analysis, were used as reference genes.
Quantitative reverse-transcription PCR (QRT-PCR) analysis of wild-type and variant HER2transcripts
First strand cDNA was synthesized as described previously and the quantification of ACTB mRNA was performed as a control to confirm the series of procedures [20]. QRT-PCR using StepOne Plus and MGB probes (Applied Biosystems, Foster City, CA) was used to quantify the wild-type and variant HER2 mRNA, ΔHER2, containing an in-frame deletion. Primers used were; wild-type HER2 5´-TCCTGTGTGGACCTGGATGA-3´ and 5´-GACCAGCAGAATGCCAACCA-3´, probe 5´-AAGGGCTGCCCCGC-3´; Δ HER, 5´-CAACTGCACCCACTCCCC-3´, 5´-CTTGATGAGGATCCCAAAGACC-3´, probe 5´-CATCATCTCTGCGGTGGT-3´. The copy number for wild-type HER2 or ΔHER2 was calculated in absolute units by comparing the signal generated by the test samples to that generated by a set of external plasmid standards containing the sequence of wild-type HER2 or ΔHER2[23, 24]. The stock plasmid standard was created by ligating a PCR product containing the wild-type HER2 or ΔHER2 sequence into a plasmid vector system, pGEM-T Easy Vector System I (Promega, Madison, WI), according to the manufacturerΔs protocol. The amount of plasmid DNA was determined by spectrophotometric analyses of the insert-containing plasmid DNA at A 260 (1 optical density = 50 μg/ml plasmid DNA), and the copy number per milliliter was determined based on molecular weight. Dilutions of the plasmid ranged from 10 to 300,000 copies per reaction, and quantitative determination for clinical samples was carried out by reading from this standard curve.
Analysis of TP53 and PIK3CAmutations
To detect specific point mutations, genomic DNA from tumor samples was examined using PCR primers to cover exons 2–10 of TP53, and to cover exons 9 and 20 of PIK3CA. PCR products were directly sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Fluorescence in situ hybridization (FISH)
Some tumors showed discordance between HER2 genomic status determined with SNP arrays or HER2 copy numbers examined by QPCR and IHC (HercepTest, DAKO) with or without FISH using paraffin specimens (PathVysion, Abbott, Japan). Some of these tumors were subsequently analyzed by FISH, using defrosted tumor specimens stamped on slide glasses. The chromosome 17 alpha satellite DNA was generated by PCR [25]. A BAC clone, RP11-62N23, was used for detection of the HER2 region.
Statistical analysis
Patients were classified into two groups based on presence or absence of alterations of 65 cancer-associated genes, and the two groups were further classified into four groups based on HER2 genomic copy numbers, hormone receptor status (HR+/−), or the response to neoadjuvant chemotherapy (pCR vs. no pCR). Significance of differences in clinical and genetic characteristics between patientΔs groups was examined using the chi-square or FisherΔs exact test and StudentΔs t-test. RFS for each group of patients classified on the basis of clinical and genetic characteristics was estimated using the Kaplan-Meier method, and compared using the log-rank test. Time to failure was defined as the interval between diagnosis and the time of first recurrence or last follow-up. We also assessed the association between HER2 genomic copy numbers and wild-type HER2 or Δ16HER2 mRNA levels by determining the Spearman rank correlation coefficient and associated P-value.
Results
On the basis of routine methods, 77 tumors were classified as the HER2-/HR+ type, 28 as the HER2+/HR+ type, 18 as the HER2+/HR- type, and 20 as the HER2-/HR- type. The 46 HER2+/HR+ and HER2+/HR- tumors are the subjects of this paper. The numbers of four chromosome aberrations, including gain, loss, amplification and UPD, were examined in each tumor. Gain and amplification of oncogenes were individually described in Additional file 1: Table S1, however, they were combined and are referred to as gain in the following analyses. All the 46 tumors showed at least some chromosome aberrations.
Discordance ratios between the results of the routine and definitive analyses on HER2 status
The routine method identified 46 tumors with the HER2+/HR+/− type. Of the 46 tumors, 39 were classified as HER2 IHC 3+ and 7 as HER2 IHC 2+ with positive routine FISH results. SNP array patterns for the HER2 locus disclosed gain in 38, normal in 6, and UPD in two of the 46 tumors. Genomic HER2 copy numbers were successfully examined in 45 of the 46 tumors by QPCR; 37 tumors showed HER2 copy numbers with >2.0 indicating HER2 gain, and 8 showed genome copy numbers between 0.5 and 2.0, indicating no gain of HER2. FISH analyses using defrosted tissue specimens were carried out in 11 tumors for the validation of the results of SNP array and QPCR analyses; SNP array patterns were normal or showed UPD in 7, and gain in four. The data obtained by the definitive FISH analysis were consistent with those obtained by SNP array and QPCR analyses (Additional file 1: Table S1). Accordingly, one tumor (No. 28), which was not examined by QPCR and identified to have HER2 gain by SNP array, was included in the tumors with HER2 copy numbers >2.0 for further analysis. Thus, 4 HER2 IHC 3+ (8.7%) and 4 HER2 IHC 2+ (8.7%) tumors of the 46 tumors showed HER2 copy numbers ≤ 2.0 by QPCR, and nonamplified HER2 by definitive FISH analysis.
The relationship between HER2 genomic copy numbers and clinical and genetic factors or levels of HER2mRNA
Patients having tumors with lower HER2 copy numbers (≤ 2.0) showed more difficulty in achieving pCR than patients having tumors with higher HER2 copy numbers (> 2.0) (P=0.019) (Table 1). Tumors with the lower HER2 copy numbers had lower levels of wild-type HER2 mRNA (<400) than tumors with the higher HER2 copy numbers (P=0.035), and showed higher incidences of PIK3CA mutation (P=0.024), mutations and gain of PIK3CA (P=0.005), loss of PTEN (P=0.008), or a combined alteration of HER2 downstream genes, PIK3CA, PTEN, and INPP4B (P=0.009) than tumors with the higher HER2 copy numbers with respective genetic alterations.
HER2 transcripts were successfully examined in 41 tumors, in all of which genomic copy numbers were also examined by QPCR. The Spearman rank correlation coefficient analysis showed that HER2 genome copy numbers tended to be correlated with the levels of wild-type HER2 mRNA (rS=0.280, P=0.076) and Δ16HER2 mRNA (rS=0.297, P=0.059), but not with the percentages of Δ16HER2 mRNA (rs=−0.200, P=0.202). The expression levels of wild-type HER2 mRNA were correlated with the expression levels of Δ16HER2 mRNA (rS=0.901, P=1.05E-15).
Genetic aberrations and HER2copy numbers and transcripts detected in HER2+/HR+ and HER2+/HR- tumors
Of 28 HR+ tumors, two were ER-negative/PgR-positive, 6 were ER-positive/PgR-negative, and 20 were ER-positive/PgR-positive. There were no significant differences in the frequency of three aberrations (gain, loss, and UPD) between 28 HER2+/HR+ tumors and 18 HER2+/HR- tumors. HR+/HER2+ tumors had lower levels of wild-type HER2 mRNA (P=0.05) and Δ16HER2 mRNA (P<0.001) and a lower incidence of BIRC5 gain (P=0.018) than HER2+/HR- tumors (Table 2). In addition, HER2+/HR+ tumors tended to have higher incidences of PTEN loss (P=0.058) and CDH3 gain (P=0.058) than HER2+/HR- tumors. If we excluded the 8 patients with nonamplified HER2, the same genes showed different incidences between the two types of patients (Table 2, numbers in parentheses).
Response to neoadjuvant chemotherapy and RFS between two groups of patients classified by clinical and genetic characteristics in tumors
The response to neoadjuvant chemotherapy and RFS were evaluated in 37 and 42, respectively, of 46 patients (Table 3). Clinical factors, including age and clinical T and N stages, did not affect the response to neoadjuvant chemotherapy or RFS (data not shown). HR+ tumors showed more difficulty in achieving pCR than HR- tumors (P=0.009). Tumors with higher copy numbers of HER2 and higher levels of Δ16HER2 mRNA entered pCR more frequently than their counterparts (P=0.019 and P=0.020). Tumors with gain of FGFR1 or MYBL2 had more difficulty in achieving pCR than the tumors without (P=0.035 and P=0.028). Tumors with loss of PTEN had more difficulty entering pCR than the tumors without (P=0.009). Tumors with a combination of mutations and gain of PIK3CA, or combined aberrations of the PI3K pathway genes, PIK3CA, PTEN and INPP4B tended to have more difficulty entering pCR than tumors without (P=0.08 and P=0.086).
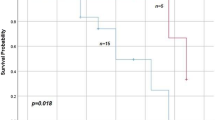
The median follow-up time of the 42 patients was 41.2 months ranging from 19.3 to 85.3 months. There was no difference in RFS between patients who achieved pCR and those who did not (P=0.764) (Figure 1A) (Table 3). Patients with lower HER2 genome copy numbers tended to have worse RFS rates than those with higher HER2 genome copy numbers (P=0.095) (Figure 1B). In regard to HER2 transcripts, patients with lower levels of the wild-type HER2 mRNA (< 400) in tumors showed worse RFS rates than those with higher levels of the wild-type mRNA (≥ 400) (P=0.022) (Figure 1C), and patients with higher percentages of the Δ16HER2 transcript (≥ 2.4%) in tumors showed worse RFS rates than those with lower percentages (< 2.4%) (P=0.039) (Figure 1D). Patients with a combination of mutations and gain of PIK3CA (P=0.041), gain of DEK (P=0.006), CCND1 (P=0.043)), FOXA1 (P=0.012) (Figure 1E), CDH3 (P=0.009) (Figure 1F), BIRC5 (P=0.005) (Figure 2A), or AIB1 (P=0.017) (Figure 2B) in tumors had worse RFS rates than patients without.

Relapse-free survival curves for patients classified by clinical and genetic characteristics. (A) Patients who achieved pCR and those who did not, (B) patients with HER2 copy numbers ≤ 2.0 and those with HER2 copy numbers >2.0 in the tumors, (C) patients with wild-type HER2 mRNA < 400 and those with wild-type HER2 mRNA ≥ 400 in the tumors, (D) patients with Δ16HER2 mRNA < 2.4% and those with Δ16HER2 mRNA ≥ 2.4% in the tumors, (E) patients with gain of FOXA1 and those with a normal copy or UPD of FOXA1 in the tumors, (F) patients with gain of CDH3 and those with a normal copy, loss, or UPD of CDH3 in the tumors.

Relapse-free survival curves for patients classified by genetic characteristics. (A) Patients with gain of BIRC5 and those with a normal copy, loss, or UPD of BIRC5 in the tumors, (B) patients with gain of AIB1 and those with a normal copy or loss of AIB1 in the tumors, (C) 20 patients with HR+ or HR- tumors with combined aberrations of PIK3CA, PTEN, and INPP4B, and 22 patients with HR+ or HR- tumors with no aberrations of PIK3CA, PTEN, and INPP4B, (D) 13 patients with HR+ tumors with combined aberrations of PIK3CA, PTEN, and INPP4B, and 12 patients with HR+ tumors with no aberrations of PIK3CA, PTEN, and INPP4B, (E) 13 patients with HR+ or HR- tumors with combined aberrations of PIK3CA, PTEN, and INPP4B, and 21 patients with HR+ or HR- tumors with no aberrations of PIK3CA, PTEN, and INPP4B, and (F) 7 patients with HR+ tumors with combined aberrations of PIK3CA, PTEN, and INPP4B, and 12 patients with HR+ tumors with no aberrations of PIK3CA, PTEN, and INPP4B.
Next, we evaluated the pCR and RFS rates in 30 and 34 patients, respectively, excluding 7 and 8 patients with HER2 copy numbers ≤ 2.0 from the 37 and 42 patients, respectively, because data on patients only with a HER2-amplified tumor may be important to show outcome of the therapy given to a specified group of patients (Additional file 2 Table S2). The positive HR status (P=0.034), loss of PTEN (P=0.054), and gain of FGFR1 (P=0.077), MYBL2 (P=0.088) or AIB1 (P=0.088) indicated or tended to indicate more difficulty in achieving pCR than the respective counterparts. In contrast, predictive significance of Δ16HER2 < 4.5 for difficulty in achieving pCR disappeared (P=0.132), and that of DEK gain for more likely to achieve pCR newly appeared (P=0.037). In regard to RFS, patients with wild-type HER2 mRNA < 400 (P=0.066), mutation and gain of PIK3CA (P=0.06), gain of CDH3 (P<0.001), BIRC5 (P=0.007), MYBL2 (P=0.013), or AIB1 (P=0.013) had or tended to have worse RFS rates than those without. In contrast, the prognostic significance of Δ16HER2 ≥ 2.4% (P=0.1) and gain of DEK (P=0.108), CCND1 (P=0.247), or FOXA1 (P=0.217) found in the 42 patients disappeared in the 34 patients. Thus, the studies including or excluding the 8 patients showed or suggested prognostic significance of certain genetic alterations, especially those involved in the PI3K and ER pathways.
Genetic characteristics that show significant differences in RFS between HR+ tumors and HR- tumors
Patients having a HR+ tumor with mutations of PIK3CA (P=0.006), a combination of mutations and gain of PIK3CA (P=0.001), a combined aberration of PIK3CA, PTEN and INPP4B (P=0.002), and gain of FOXA1 (P=0.002), CDH3 (P=0.007), BIRC5 (P=0.016), MYBL2 (P=0.015), and AIB1 (P=0.006) had worse RFS rates than patients having a HR+ tumor without (Table 4). However, no such significance of the genetic aberrations was found in patients with a HR- tumor. It is noteworthy that while there was no significant difference in a RFS rate between 20 patients having a HR+ or HR- tumor with aberrations of the PI3K pathway genes and 22 patients having a HR+ or HR- tumor with no such aberrations (Table 3 and Figure 2C, P=0.195), 13 patients having a HR+ tumor with the same aberrations had a worse RFS rate than 12 patients having a HR+ tumor with no such aberrations (Table 4, Figure 2D, P=0.002). When we excluded the 8 patients with HER2 copy numbers ≤ 2.0 in tumors, the prognostic significance of the genetic alterations was also demonstrated in 19 patients with a HR+ tumor (Additional file 3: Table S3, Figure 2F, P=0.03), but not in the 34 patients with a HR+ or HR- tumor (Additional file 2: Table S2, Figure 2E, P=0.248).
Clinical and genetic characteristics that predict pCR or RFS, or both
Patients with higher HER2 copy numbers, a combination of mutations and gain of PIK3CA, and gain of MYBL2 and AIB1 had or tended to have worse pCR and RFS rates than the respective counterparts (Table 3). In contrast, patients with positive HR status, lower levels of Δ16HER2 transcript, loss of PTEN, and gain of FGFR1 had more difficulty in achieving pCR, but not worse RFS rates than the respective counterparts, whereas patients with lower levels of wild-type HER2 mRNA, higher percentages of Δ16HER2 mRNA, and gain of DEK, CCND1, FOXA1, CDH3, or BIRC5 had worse RFS rates, but not more difficulty in achieving pCR than the respective counterparts. Gain of MYC, EMSY, and AURKA, TP53 mutations, and other genetic aberrations did not affect pCR or RFS (data not shown). Thus, some genetic aberrations indicated either the response to the chemotherapy or the RFS, and others indicated both. These findings also indicated that genetic aberrations that correlate with difficulty in achieving pCR and those that correlate with worse RFS rates do not always overlap.
Correlation of immunohistochemical findings of CDH3with hormone receptor status, genetic alterations, and RFS
All 5 tumors with gain of CDH3 belonged to the HR+ type, and patients having a tumor with gain of CDH3 had worse RFS rates than patients without. To validate the findings shown by the SNP array, we performed immunohistochemical staining of P-cadherin encoded by CDH3. While 5 tumors with CDH3 gain showed 1+, 2+, or 3+ P-cadherin expression, 8 of 33 tumors with CDH3 loss or a normal CDH3 genetic status showed negative P-cadherin expression. While all 18 HR- tumors showed 1+, 2+, or 3+ P-cadherin expression, 8 of 22 HR+ tumors showed negative P-cadherin expression (P<0.01). Patients with P-cadherin 3+ expression/HR+ tumors had worse RFS rates than patients with P-cadherin negative or 1+ expression/HR+ tumors (P=0.032). Thus, while all HR- tumors showed P-cadherin expression, some HR+ tumors were negative for P-cadherin expression, indicating that the staining patterns and their prognostic implications differed between the two types of tumors.
Discussion
In the present SNP array-based study of 46 HER2+ breast cancers, we evaluated clinical and genetic factors that indicate pCR and RFS in patients who were treated with trastuzumab-containing neoadjuvant chemotherapy. SNP array analysis has a merit to detect whole genomic aberrations at once, and therefore could find a predictive or prognostic impact of combined genomic aberrations. Such an attempt seems not to have been made before. The 46 tumors were selected based on a routine HER2 study using IHC with or without FISH in deparaffinized tissue samples. We found discordance between results of the routine analysis and the present SNP and QPCR analysis with FISH in defrosted tissue samples in 17.4% (8/46) of tumors; 4 IHC 3+ (8.7%) and 4 IHC 2+ (8.7%) tumors. Previous studies reported the IHC3+, FISH negative type in 0.9%-18.5% of tumors examined [9], and the percentage of 8.7% shown in the present study may be comparable to or a little higher than the previous results. Another discordance of 4 patients with the IHC 2+ and nonamplified HER2 type may be caused by the difficulty of FISH analysis using needle biopsied tissues embedded in paraffin. It is difficult to compare this percentage of 8.7% with those of other series, because there have been no comparable studies reported.
Overexpression of protein occurs not only by mRNA overexpression, but also by post-transcriptional, translational and protein degradation regulation [26]. HER2 is efficiently ubiquitinated and downregulated by the chaperone binding ubiquitin ligase CHIP/STUB1 [27]. Recently, Jan et al. studied the expression and correlations among TID1, CHIP, and HER2 in a total of 183 breast cancer histology sections using IHC and immunoblotting assay, and found that the immunohistochemical expression of TID1 and CHIP were positively correlated with each other but were both inversely correlated to that of HER2 [28]. These findings suggest that down-regulation of CHIP could cause overexpression of HER2 by preventing its degradation. In a study of metastatic breast cancer with the HER2 IHC2+/3+ and nonamplified HER2 type, patients treated with trastuzumab plus chemotherapy had better progression-free survival than those treated with chemotherapy alone (P=0.03), suggesting the presence of the true single-gene overexpressers in the study population [11]. Thus, some HER2-single-gene overexpressers were not the results of technical shortcomings of HER2 IHC, but could be the results of disruption of HER2 protein degradation occurring in the tumor cells. Overexpression of MYCN caused by the disruption of the protein degradation but not by the amplification indicated poor outcome in patients with neuroblastoma [29].
The present study indicated that the patients with nonamplified HER2 showed more difficulty in achieving pCR and more frequent relapse than those with HER2 amplification. Unexpectedly, we found that tumors with the lower HER2 copy numbers had more frequent alterations in the PI3K pathway genes than those with the higher copy numbers (Table 1). Alterations of the PI3K pathway genes were reported to be associated with poor response to HER2-targeted therapy in patients with HER2+ tumors [30], and this finding concurs with the present finding indicating the poor outcome of patients having a tumor with HER2 IHC 2+/3+ and nonamplified HER2. These findings support the statement that FISH analysis should be the primary HER2 testing for patients who are candidates for HER2-targeted therapies [9].
In regard to the relationship between clinical and genetic factors and RFS, patients with various genetic changes in tumors identified by SNP array had or tended to have worse RFS rates than those without (Table 3). Previous studies reported that overexpression of these genes identified by various methods was associated with a poor outcome in patients with breast cancer [31–37]. Thus, the present study indicated that the results of the SNP array analysis detecting gain of specific genes and those of other analyses detecting overexpression of the gene products were mostly consistent.
When the prognostic implications of the genetic aberrations were separately examined in HR+ and HR- tumors, a combination of mutations and gain of PIK3CA, a combined aberration of the PI3K pathway genes, and gain of FOXA1, CDH3, BIRC5, MYBL2, and AIB1 indicated worse RFS rates only in patients with a HR+ tumor (Table 4). It is noteworthy that the incidences of the most genetic changes did not differ between the two types of tumors (Table 2). We confirmed the result of a study reporting that increased PI3K pathway activity was associated with poor outcome for patients treated with HER2-targeting therapy [30], and newly found that the prognostic significance of the alterations in the PI3K and ER pathway genes applied only to the patients with a HR+ tumor (Table 4). FOXA1, a forkhead family transcription factor, is essential for optimum expression of ER and estrogen responsive genes. Although one study reported that FOXA1 expression was correlated with the luminal A subtype and a favorable outcome [38], others depicted a complicated picture of FOXA1 as a participant in multiple signaling pathways in breast cancer, which are both oncogenic and tumor suppressive [33]. Actually, a recent study reported co-expression of ER and FOXA1 in metastatic breast cancer samples, indicating oncogenic activities of FOXA1[39]. AIB1 is a nuclear receptor coactivator that interacts with ERs in a ligand-dependent fashion and enhances estrogen-dependent transcription. Harigopal et al. reported AIB1 activation to be mediated by crosstalk with other signaling kinases such as growth factor receptors including MAPK, which can be activated by HER2 in a ligand-independent fashion [40]. AIB1 is considered as a key factor in the regulation of tumor growth and carcinogenesis. In addition, patients with AIB1-positive tumors identified by IHC showed worse RFS rates than those with AIB1-negative tumors [40]. These findings indicate that crosstalk between the PI3K and ER pathways is causally involved in resistance to the chemotherapy with trastuzumab, and thus, that hormone receptor status impacted the prognostic significance of genomic alterations in HER2+ tumors.
The present study also showed that gain of DEK and BIRC5 genes, which seems not to be involved in the PI3K or ER pathway, had clear prognostic significance in patients with breast cancer (Table 3), suggesting presence of other pathways exhibiting crosstalk with the PI3K or ER pathway. Lu et al. reported that HER2-mediated up-regulation of survivin expression contributed to Taxol resistance through a survivin-mediated faster exit from mitosis [41]. Xia et al. showed that acquired resistance to lapatinib in the BT474 cell line was associated with a switch in the regulation of survivin from HER2 to ER [42]. Thus, various roles of survivin in chemotherapy resistance may contribute to the prognostic significance of BIRC5 gain in the present series of patients with HR+ and HR- breast cancers.
A splice variant of the human HER2 transcript lacking exon 16 (Δ16HER2) has been detected in human breast cancers [12]. Mitra and colleagues showed that ectopic expression of Δ16HER2, but not wild-type HER2 mRNA, promoted receptor dimerization, cell invasion, and trastuzumab resistance in the NIH3T3 and MCF7 cell lines [13]. Recently, a mouse line that transgenically expresses the Δ16HER2 transcript has been generated and all the transgenic females developed multifocal mammary tumors with a rapid onset [14]. This oncogenic isoform has been associated with trastuzumab resistance in various in vitro and transgenic mouse studies [12, 14]. The present study showed that levels of the wild-type HER2 transcript and those of the Δ16HER2 transcript were correlated, and that there was no difference in the RFS rate between patients with higher levels of Δ16HER2 mRNA and those without. In addition, we also found that patients with higher percentages of the Δ16HER2 mRNA (≥ 2.4%) and lower levels of the wild-type HER2 mRNA (< 400) were likely to have recurrence. The decreased RFS rates for patients with lower levels of wild-type HER2 mRNA may be explained by the decreased susceptibility to trastuzumab in patients with such tumors. The patients with the higher percentage of Δ16HER2 mRNA also had the lower level of wild-type HER2 mRNA, and this lower level might have resulted in a lower denominator and hence the higher percentages of Δ16HER2 mRNA. Thus, the poor prognosis of patients with the higher percentage of Δ16HER2 mRNA may be related to the lower levels of the wild-type HER2 mRNA, or to presently unknown mechanisms.
The present study showed no difference in RFS between patients who attained pCR and those who did not. Previous studies indicated better overall survivals for patients with pCR than those with no pCR [2, 3]. von Minckwits and colleagues studied pCR and its prognostic impact on survival in intrinsic breast cancer subtypes and concluded that pCR is a suitable surrogate end point for patients with HER2+ (nonluminal) but not luminal B/HER2+ breast cancer [43]. More recently, results of a meta-analysis showed that patients with a HR-/HER2+ or HR+/HER2+ tumor who achieved pCR had greater event-free survival than those with the respective subtype of tumor who did not [44]. The contradictory finding of the present study might be caused by the small sample size and inclusion of HR+/HER2+ and HR-/HER2+ breast cancer samples.
Several clinical trials have been carried out to improve the outcome of patients with HER2+ tumors. Two trials reported that the combination of an aromatase inhibitor with trastuzumab or lapatinib improved outcome for patients with HER2+/HR+ metastatic breast cancer compared with an aromatase inhibitor alone [45, 46]. Both trials showed better progression-free survival for the patients treated with the combination. These studies were undertaken because the crosstalk between the PI3K and ER pathways could cause resistance to trastuzumab and anti-hormone agents. In addition, one phase 2 and another phase 3 clinical trials were carried out using a combination of lapatinib and trastuzumab with or without cytotoxic chemotherapy for patients with a HER2+/HR+ or HER2+/HR- early stage tumor, and both studies showed improved pCR rates [47, 48]. These studies were undertaken because primary and acquired resistance to both agents could be overcome, their partly non-overlapping mechanisms of action, and the well-characterized synergistic interaction between them could be expected in patients with HER2+ breast cancer. The present study showed that the PI3K and ER pathway genes were specifically altered in HER2+/HR+ tumors of patients who subsequently relapsed after receiving neoadjuvant chemotherapy with trastuzumab. Lapatinib targets the intracellular ATP domain of HER2, preventing self-phosphorylation and subsequent activation of the PI3K and MAPK signal pathways [5]. It may be reasonable to add lapatinib to the present trastuzumab-containing chemotherapy for patients with a HER2+/HR+ tumor to overcome the resistance due to the altered PI3K pathway genes and to obtain a better outcome.
Conclusion
The present study showed that 17.4% of tumors with positive HER2 testing had nonamplified HER2, and patients with this type of tumor showed difficulty in achieving pCR and frequent relapses, supporting FISH analysis as a primary HER2 testing. The study also disclosed that alterations of the PI3K and ER pathway genes indicated worse RFS rates in patients with a HR+ but not with a HR- tumor who were treated with neoadjuvant chemotherapy with trastuzumab. It may be reasonable to add lapatinib to the present trastuzumab-containing chemotherapy for patients with a HER2+/HR+ tumor to overcome the resistance due to the altered PI3K pathway genes and to obtain a better outcome.
AuthorsΔ contributions
MT, TH, KT, MH, and JW participated in data collection, interpretation and molecular analysis. FK and MK carried out molecular cytogenetic and pathological studies. ASO and OS were responsible for SNP array analysis. MT, HT, KI, MM, and YK contributed to concept design and drafted the manuscript. All authors have read and approved the manuscript.
Abbreviations
- ER:
-
Estrogen receptor
- FISH:
-
Fluorescence in situ hybridization
- H & E:
-
Haematoxylin and eosin
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hormone receptor
- IHC:
-
Immunohistochemistry
- pCR:
-
Pathological complete response
- RFS:
-
Relapse-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase
- SNP:
-
Single nucleotide polymorphisms
References
Buzdar AU, Valero V, Ibrahim NK, Francis D, Broglio KR, Theriault RL, Pusztai L, Green MC, Singletary SE, Hunt KK, Sahin AA, Esteva F, Symmans WF, Ewer MS, Buchholz TA, Hortobagyi GN: Neoadjuvant therapy with paclitaxel followed by 5-fluorouracil, epirubicin, and cyclophosphamide chemotherapy and concurrent trastuzumab in human epidermal growth factor receptor 2-positive operable breast cancer: an update of the initial randomized study. Clin Cancer Res. 2007, 13: 228-233. 10.1158/1078-0432.CCR-06-1345.
Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, Margolese RG, Hoehn JL, Vogel VG, Dakhil SR, Tamkus D, King KM, Pajon ER, Wright MJ, Robert J, Paik S, Mamounas EP, Wolmark N: Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J Clin Oncol. 2008, 26: 778-785. 10.1200/JCO.2007.15.0235.
Untch M, Fasching PA, Konecny GE, Hasmüller S, Lebeau A, Kreienberg R, Camara O, Müller V, du Bois A, Kühn T, Stickeler E, Harbeck N, Höss C, Kahlert S, Beck T, Fett W, Mehta KM, von Minckwitz G, Loibl S: Pathologic complete response after neoadjuvant chemotherapy plus trastuzumab predicts favorable survival in human epidermal growth factor receptor 2-overexpressing breast cancer: Results From the TECHNO Trial of the AGO and GBG Study Groups. J Clin Oncol. 2011, 29: 3351-3357. 10.1200/JCO.2010.31.4930.
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R: A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007, 12: 395-402. 10.1016/j.ccr.2007.08.030.
OΔBrien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, OΔDonovan N, Slamon DJ: Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010, 6: 1489-1502.
Bhargava R, Dabbs DJ, Beriwal S, Yildiz IA, Badve P, Soran A, Johnson RR, Brufsky AM, Lembersky BC, McGuire KP, Ahrendt GM: Semiquantitative hormone receptor level influences response to trastuzumab-containing neoadjuvant chemotherapy in HER2-positive breast cancer. Mod Pathol. 2011, 24: 367-374. 10.1038/modpathol.2010.209.
Vaz-Luis I, Ottesen RA, Hughes ME, Marcom PK, Moy B, Rugo HS, Theriault RL, Wilson J, Niland JC, Weeks JC, Lin NU: Impact of hormone receptor status on patterns of recurrence and clinical outcomes among patients with human epidermal growth factor-2-positive breast cancer in the National Comprehensive Cancer Network: a prospective cohort study. Breast Cancer Res. 2012, 14: R129-10.1186/bcr3324.
Arpino G, Wiechmann L, Osborne CK, Schiff R: Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008, 29: 217-233.
Sauter G, Lee J, Bartlett JM, Slamon DJ, Press MF: Guidelines for human epidermal growth factor receptor 2 testing: biologic and methodologic considerations. J Clin Oncol. 2009, 27: 1323-1333. 10.1200/JCO.2007.14.8197.
Pauletti G, Godolphin W, Press MF, Slamon DJ: Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene. 1996, 13: 63-72.
Mass RD, Press MF, Anderson S, Cobleigh MA, Vogel CL, Dybdal N, Leiberman G, Slamon DJ: Evaluation of clinical outcomes according to HER2 detection by fluorescence in situ hybridization in women with metastatic breast cancer treated with trastuzumab. Clin Breast Cancer. 2005, 6: 240-246. 10.3816/CBC.2005.n.026.
Siegel PM, Ryan ED, Cardiff RD, Muller WJ: Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999, 18: 2149-2164. 10.1093/emboj/18.8.2149.
Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE: An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009, 8: 2152-2162. 10.1158/1535-7163.MCT-09-0295.
Marchini C, Gabrielli F, Iezzi M, Zenobi S, Montani M, Pietrella L, Kalogris C, Rossini A, Ciravolo V, Castagnoli L, Tagliabue E, Pupa SM, Musiani P, Monaci P, Menard S, Amici A: The human splice variant Δ16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS One. 2011, 6: e18727-10.1371/journal.pone.0018727.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, Van de Vijver MJ, Wheeler TM, Hayes DF: American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007, 25: 118-145.
Elston CW, Ellis IO: Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991, 19: 403-410. 10.1111/j.1365-2559.1991.tb00229.x.
Harvey JM, Clark GM, Osborne K, Allred DC: Estrogen receptor status by IHC is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999, 17: 1474-1481.
Inoue K, Nagai S, Kaneko S, Uchida S, Hayashi Y, Higuchi T, Yoshida T, Takei H, Tabei T: Efficacy of oxycodone against anti-cancer agent-induced pain in breast cancer patients during adjuvant treatment before and after surgery. Jpn J Cancer Chemother. 2011, 38: 1137-1142.
Kurosumi M, Akiyama F, Iwase T, Motomura K, Okazaki M, Tsuda H: Histopathological criteria for assessment of therapeutic response in breast cancer. Breast Cancer. 2001, 8: 1-2.
Haruta M, Arai Y, Sugawara W, Watanabe N, Honda S, Ohshima J, Soejima H, Nakadate H, Okita H, Hata J, Fukuzawa M, Kaneko Y: Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer. 2008, 47: 712-727. 10.1002/gcc.20572.
Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S: A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005, 65: 6071-6079. 10.1158/0008-5472.CAN-05-0465.
Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, Koeffler HP, Ogawa S: Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of Affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007, 81: 114-126. 10.1086/518809.
Hammond E, Shaw K, Carnley B, PΔng S, James I, Herrmann R: Quantitative determination of JAK2 V617F by TaqMan: An absolute measure of averaged copies per cell that may be associated with the different types of myeloproliferative disorders. J Mol Diagn. 2007, 9: 242-248. 10.2353/jmoldx.2007.060125.
Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Ménard S: Role of exon-16-deleted HER2 in breast carcinomas. Endocri Relat Cancer. 2006, 13: 221-232. 10.1677/erc.1.01047.
Warburton PE, Greig GM, Haaf T, Willard HF: PCR amplification of chromosome-specific alpha satellite DNA. Genomics. 1991, 11: 324-333. 10.1016/0888-7543(91)90139-6.
Vogel C, Marcotte EM: Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012, 13: 227-232.
Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L: Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA. 2002, 99: 12847-12852. 10.1073/pnas.202365899.
Jan CI, Yu CC, Hung MC, Harn HJ, Nieh S, Lee HS, Lou MA, Wu YC, Chen CY, Huang CY, Chen FN, Lo JF: Tid1, CHIP and ErbB2 interactions and their prognostic implications for breast cancer patients. J Pathol. 2011, 225: 424-437. 10.1002/path.2921.
Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, Molenaar JJ, van Nes J, Versteeg R: Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci USA. 2012, 109: 19190-19195. 10.1073/pnas.1208215109.
Jensen JD, Knoop A, Laenkholm AV, Grauslund M, Jensen MB, Santoni-Rugiu E, Andersson M, Ewertz M: PIK3CA mutations, PTEN, and pHER2 expression and impact on outcome in HER2-positive early-stage breast cancer patients treated with adjuvant chemotherapy and trastuzumab. Ann Oncol. 2000, 23: 2034-2042.
Abba MC, Sun H, Hawkins KA, Drake JA, Hu Y, Nunez MI, Gaddis S, Shi T, Horvath S, Sahin A, Aldaz CM: Breast cancer molecular signatures as determined by SAGE: correlation with lymph node status. Mol Cancer Res. 2007, 5: 881-890. 10.1158/1541-7786.MCR-07-0055.
Lundgren K, Brown M, Pineda S, Cuzick J, Salter J, Zabaglo L, Howell A, Dowsett M, Landberg G, TransATAC investigators: Effects of cyclin D1 gene amplification and protein expression on time to recurrence in postmenopausal breast cancer patients treated with anastrozole or tamoxifen: a TransATAC study. Breast Cancer Res. 2012, 14: R57-
Bernardo GM, Keri RA: FOXA1: a transcription factor with parallel functions in development and cancer. Biosci Rep. 2012, 32: 113-130. 10.1042/BSR20110046.
Paredes J, Albergaria A, Oliveira JT, Jerónimo C, Milanezi F, Schmitt FC: P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res. 2005, 11: 5869-5877. 10.1158/1078-0432.CCR-05-0059.
Lv YG, Yu F, Yao Q, Chen JH, Wang L: The role of survivin in diagnosis, prognosis and treatment of breast cancer. J Thoracic Dis. 2010, 2: 100-110.
Sala A: B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur J Cancer. 2005, 41: 2479-2484. 10.1016/j.ejca.2005.08.004.
Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS: Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008, 456: 663-666. 10.1038/nature07483.
Badve S, Turbin D, Thorat MA, Morimiya A, Nielsen TO, Perou CM, Dunn S, Huntsman DG, Nakshatri H: FOXA1 expression in breast cancer–correlation with luminal subtype A and survival. Clin Cancer Res. 2007, 13: 4415-4421. 10.1158/1078-0432.CCR-07-0122.
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS: Differential estrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012, 481: 389-393.
Harigopal M, Heymann J, Ghosh S, Anagnostou V, Camp RL, Rimm DL: Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Res Treat. 2009, 115: 77-85. 10.1007/s10549-008-0063-9.
Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkammongkol W, Andreeff M, Symmans F, Yu D: Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance. Clin Cancer Res. 2009, 15: 1326-1334. 10.1158/1078-0432.CCR-08-0954.
Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J, Harris J, Spector NL: A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA. 2006, 103: 7795-7800. 10.1073/pnas.0602468103.
von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich J, Huober J, Jackisch C, Kaufmann M, Konecny GE, Denkert C, Nekljudova V, Mehta K, Loibl S: Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012, 30: 1796-1804. 10.1200/JCO.2011.38.8595.
Cortazar P, Zhang L, Untch M, Mehta K, Costantino J, Wolmark N, Bonnefoi H, Cameron D, Gianni L, Valagussa P, Zujewski JA, Justice R, Loibl S, Wickerham L, Bogaerts J, Baselga J, Perou C, Blumenthal G, Blohmer J, Mamounas E, Bergh J, Semiglazov V, Prowell T, Eidtmann H, Paik S, Piccart M, Sridhara R, Fasching P, Swain SM, Slaets L: Meta-analysis Results from the Collaborative Trials in Neoadjuvant Breast Cancer (CTNeoBC) (Abstract). Cancer Res. 2012, 72 (24 Suppl): 93S-
Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, Tjulandin S, Jahn M, Lehle M, Feyereislova A, Révil C, Jones A: Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2–positive, hormone receptor–positive metastatic breast cancer: Results from the randomized phase III TAnDEM Study. J Clin Oncol. 2009, 27: 5529-5537. 10.1200/JCO.2008.20.6847.
Johnston S, Pippen J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, Press MF, Maltzman J, Florance A, OΔRourke L, Oliva C, Stein S, Pegram M: Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor–positive metastatic breast cancer. J Clin Oncol. 2009, 27: 5538-5546. 10.1200/JCO.2009.23.3734.
Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gómez H, Dinh P, Fauria K, Van Dooren V, Aktan G, Goldhirsch A, Chang TW, Horváth Z, Coccia-Portugal M, Domont J, Tseng LM, Kunz G, Sohn JH, Semiglazov V, Lerzo G, Palacova M, Probachai V, Pusztai L, Untch M, Gelber RD, Piccart-Gebhart M, NeoALTTO Study Team: Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012, 376: 633-640.
Rimawi MF, Mayer IA, Forero A, Nanda R, Goetz MP, Rodriguez AA, Pavlick AC, Wang T, Hilsenbeck SG, Gutierrez C, Schiff R, Osborne CK, Chang JC: Multicenter phase II study of neoadjuvant lapatinib and trastuzumab with hormonal therapy and without chemotherapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer: TBCRC 006. J Clin Oncol. 2013, 31: 1726-1731. 10.1200/JCO.2012.44.8027.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/13/241/prepub
Acknowledgements
This study was supported in part by the Ministry of Health, Labor and Welfare, Japan for Third-term Comprehensive Control Research for Cancer, and the City Area Program of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
12885_2013_3877_MOESM1_ESM.docx
Additional file 1: Table S1: Clinical and genetic characteristics of 28 patients with a breast cancer of HER2+/HR+ type and 18 with a breast cancer of HER2+/HR- type. (DOCX 36 KB)
12885_2013_3877_MOESM2_ESM.doc
Additional file 2: Table S2: RFS and pCR rates for 34 patients with a HR+ or HR- tumor classified by clinical and genetic characteristics; 8 patients with HER2 copy numbers ≤ 2.0 are excluded. (DOC 58 KB)
12885_2013_3877_MOESM3_ESM.doc
Additional file 3: Table S3: RFS in patients with a HR+ or HR- tumor classified by clinical and genetic characteristics; 8 patients with HER2 copy numbers ≤ 2.0 are excluded. (DOC 64 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Takada, M., Higuchi, T., Tozuka, K. et al. Alterations of the genes involved in the PI3K and estrogen-receptor pathways influence outcome in human epidermal growth factor receptor 2-positive and hormone receptor-positive breast cancer patients treated with trastuzumab-containing neoadjuvant chemotherapy. BMC Cancer 13, 241 (2013). https://doi.org/10.1186/1471-2407-13-241
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-13-241