
Abstract
Background
Epigenetic silencing of the MGMT gene by promoter methylation is associated with loss of MGMT expression, diminished DNA-repair activity and longer overall survival in patients with glioblastoma who, in addition to radiotherapy, received alkylating chemotherapy with carmustine or temozolomide. We describe and validate a rapid methylation sensitive quantitative PCR assay (MS-qLNAPCR) using Locked Nucleic Acid (LNA) modified primers and an imprinted gene as a reference.
Methods
An analysis was made of a database of 159 GBM patients followed between April 2004 and October 2008. After bisulfite treatment, methylated and unmethylated CpGs were recognized by LNA primers and molecular beacon probes. The SNURF promoter of an imprinted gene mapped on 15q12, was used as a reference. This approach was used because imprinted genes have a balanced copy number of methylated and unmethylated alleles, and this feature allows an easy and a precise normalization.
Results
Concordance between already described nested MS-PCR and MS-qLNAPCR was found in 158 of 159 samples (99.4%). The MS-qLNAPCR assay showed a PCR efficiency of 102% and a sensitivity of 0.01% for LNA modified primers, while unmodified primers revealed lower efficiency (69%) and lower sensitivity (0.1%). MGMT promoter was found to be methylated using MS-qLNAPCR in 70 patients (44.02%), and completely unmethylated in 89 samples (55.97%). Median overall survival was of 24 months, being 20 months and 36 months, in patients with MGMT unmethylated and methylated, respectively. Considering MGMT methylation data provided by MS-qLNAPCR as a binary variable, overall survival was different between patients with GBM samples harboring MGMT promoter unmethylated and other patients with any percentage of MGMT methylation (p = 0.003). This difference was retained using other cut off values for MGMT methylation rate (i.e. 10% and 20% of methylated allele), while the difference was lost when 50% of MGMT methylated allele was used as cut-off.
Conclusions
We report and clinically validate an accurate, robust, and cost effective MS-qLNAPCR protocol for the detection and quantification of methylated MGMT alleles in GBM samples. Using MS-qLNAPCR we demonstrate that even low levels of MGMT promoter methylation have to be taken into account to predict response to temozolomide-chemotherapy.
Similar content being viewed by others


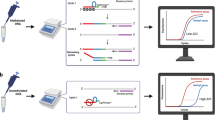
Background
Transcriptional inactivation by cytosine methylation at promoter CpG islands of tumour suppressor genes is thought to be an important mechanism in human carcinogenesis. A number of tumour suppressor genes, including CDKN2A, MGMT, MLH1, etc, are silenced by promoter methylation in a variety of tumors [1]. In the course of tumor development, gene silencing by DNA methylation is an early and important mechanism by which tumor-suppressor genes are inactivated [2, 3].
Epigenetic silencing of the MGMT gene by promoter methylation is associated with loss of MGMT expression [4–6], diminished DNA-repair activity and longer overall survival in patients with glioblastoma (GBM) who, in addition to radiotherapy, received alkylating chemotherapy with temozolomide [7]. The MGMT gene is located on chromosome 10q26 and encodes a DNA-repair protein that removes alkyl groups from the O6position of guanine, an important site of DNA alkylation. The restoration of the DNA consumes the MGMT protein, which the cell must replenish. Left unrepaired, chemotherapy-induced lesions, especially O6-methylguanine, trigger cytotoxicity and apoptosis [8, 9]. High levels of MGMT activity in cancer cells create a resistant phenotype by blunting the therapeutic effect of alkylating agents and may be an important determinant of treatment failure [3, 8–13]. Patients with glioblastoma containing a methylated MGMT promoter showed a major benefit from temozolomide [14].
Given the key roles of cytosine methylation, there has been a wide interest in the development of procedures for DNA methylation analyses [2, 3, 6, 15–26].
Vlassenbroeck I et al. [27] described a Real Time by SYBRGreen method to detect MGMT methylation status. The copy number of the methylated MGMT promoter, normalized to the ACTB gene, provides a quantitative test result. Woidacz TK et al. showed that MGMT methylation could be detected at levels as low as 0.1%. by high resolution melting analysis [28].
Here we present a novel methylation sensitive quantitative real time PCR assay (MS-qLNAPCR) which permits high throughput quantification of the methylation status of the MGMT promoter in an accurate, very sensitive and cost-effective manner. High specificity was achieved recognizing methylated and unmethylated CpGs by 3'-locked nucleic acid (LNA) primers and molecular beacon probes [29]. The CpG islands of SNURF were selected as a reference locus. SNURF belongs to the 15q imprinted center mapped on 15q12. The maternal allele is usually methylated, while the paternal one is unmethylated [30]. In theory in a homogeneous population of cells of the same individual the methylated maternal alleles should be balanced with the unmethylated paternal alleles if the tumor cells did not acquire any deletion for this locus or aberrant methylation of the paternal allele. This feature allows an easy and precise calculation of the ratio between the methylated and unmethylated alleles of MGMT following the method described by Ginzinger et al. [31].
Methods
Tumor Samples
For this study, samples were retrieved from the Pathology Section of the University of Bologna at Bellaria Hospital (Bologna). Tumors were classified and graded according to 2007 WHO [32] criteria. The use of brain tumor tissue after completing histopathological diagnosis for research purposes was approved by the regional ethics committee.
Following patient charts review, a retrospective analysis was made of a database of GBM patients followed prospectively between April 2004 and October 2008. We evaluated only patients who met the following inclusion criteria: age ≥ 18 years; Performance Status (PS) at diagnosis, 0-2; histological diagnosis of newly diagnosed GBM; MGMT methylation status assessed by methylation specific nested PCR; postoperative treatment consisting of radiotherapy (RT) followed by adjuvant temozolomide (TMZ) (14 patients) or TMZ concurrent with and adjuvant to RT (145 patients) [7]. Twenty-four blood samples from healthy donors were used as controls.
DNA isolation
Tissue blocks were selected for DNA extraction after careful examination on hematoxylin and eosin staining of corresponding sections to exclude contaminating necrotic debris. Molecular genetic analyses were performed on samples showing an estimated tumor cell content of at least 90% from five sections of 10 μm from paraffin embedded tissue (FFPE) blocks. Tumor area was macrodissected manually by a sterile blade or were microdissected using the laser assisted SL μcut Microtest (MMI GmbH, Glattbrugg, Switzerland) as previously described[33]. Two incubations with xylene at 60°C and two incubations with absolute ethanol at room temperature for ten minutes each were used to eliminate paraffin. The tissues were then incubated with NaSCN 1 M for one hour at 37°C and lysed with proteinase K at 55°C overnight. Genomic DNA was extracted using the GENTRA Puregene tissue kit (Qiagen, Hilden, Germany) in accordance to the manufacturer's instructions. The pellet was then eluted in 35 μl of TE buffer and total DNA was quantified by Quant-iT™ dsDNA BR kit (Invitrogen, Carlsbad, California). At least 200 ng of DNA was then treated with bisulfite using the EpiTect Bisulfite kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
In VitroMethylation Assay and Standards
To test for sensitivity and specificity of MS-qLNAPCR, titration experiments were performed using normal pooled genomic DNA (DNA Female pool, Cod. G1521, Promega, Madison, Wisconsin) which was methylated in vitro using SssI (New England Biolabs, Ipswich, MA). In brief, 1.5 μg was treated with SssI to methylate all CpG sites (near complete methylation and no loss of DNA was assumed) for two hours at 37°C following the instruction of the provider. Mixtures of SssI-treated DNA: untreated DNA (100%, 50%, 10%, 1%, 0.1% 0.01%) were prepared in duplicate (each containing 1.5 μg of template DNA). 50% SssI-treated DNA: untreated DNA served as calibrator for MGMT MSP-qPCR, while the same DNA amount of untreated DNA pool (Promega, Madison, Wisconsin) served as calibrator for SNURF MSP-qPCR to confirm equal amounts of maternal methylated allele and paternal unmethylated allele. The same DNA pool was used to test for specificity in terms of absence of amplicons for the MGMT methylated allele for single runs.
MS-nested PCR and MS-qLNAPCR
Nested MS-PCR was performed as previously described [34, 35] with minor modifications: a total of 26 cycles for the flanking primers and a total of 30 cycles for the methylation specific primers were performed. Amplicons were detected by SeaKem LE agarose gel (3%, Lonza, Basel, Switzerland) by the use of GelStar (Lonza, Basel, Switzerland) as intercalator. Real Time PCR analysis was performed using an SDS-ABI Prism 7000 (Applied Biosystems, Foster City, CA). 3'-locked nucleic acid (LNA) primers (see Table 1) were synthesized by SIGMA-Proligo (SIGMA-Proligo, Paris, France). Reactions were performed in a final volume of 25 μl, adjusted to 4 mM of MgCl2 and containing 1 Unit of FastStartTaq DNA Polymerase (Roche, Mannheim, Germany), 1× related Buffer and 5 μl of 5× GC rich solution, 200 μM of dNTPs, 0.5 μl of ROX 50× (Invitrogen, Carlsbad, California), 500 nM of each primers, 250 nM of beacon probe and 3 μl of bisulfite treated DNA. The real time qPCR cycling conditions were as follows: 95°C for 4 min, 60°C for 2 min, 72°C for 2 min, followed by 40 cycles for 20 s at 95°C, 45 s at 60°C with fluorescence measurement, 30 s at 72°C.
Relative Quantification of the Methylated Allele
The number of PCR cycles (Ct) required for the FAM intensities to exceed a threshold just above background was calculated for the test (MGMT) and reference (SNURF) reactions as described previously [31] with some modifications: Ct values were determined for each sample and subtracted to obtain:

(mMGMT: methylated allele; uMGMT: unmethylated allele; mSNURF: methylated allele; uSNURF: unmethylated allele).
ΔCt values were measured for each unknown sample [ΔCt (test DNA)] and for calibrator [ΔCt (equal amount of SssI-treated DNA mixed with the same amount of untreated DNA)].
Relative copy number at each locus in the test sample was then calculated as:
Relative Methylated allele copy number = (1+E)-ΔΔCt where:
ΔΔCt = ΔCt (test DNA) - ΔCt (calibrator DNA), and E = PCR efficiency. The efficiency of PCR was calculated from the slope of the line, E = 10-1/slope - 1. We used primers with PCR efficiencies of >95% and We calculated the relative DNA copy number ratio respect the unmethylated allele following the formula = 2-ΔΔCtas previously described [31].
A sample of 24 healthy controls was used to estimate the normality range of the differences between methylated and unmethylated SNURF alleles; the normality range was estimated as the mean value ± 1.96 * standard deviation.
Results were considered invalid if criteria of DNA quantity and quality were not met. These were based on minimal starting amplifiable DNA of approximately 6.25 ng (equivalent of ~1000 copies of a diploid human genome) available for amplification after bisulfite treatment and calculated by ABI 7000 SDS software using the standard curve for uMGMT. For this purpose, serial dilutions of normal female DNA (DNA Female pool, Cod. G1521, Promega, Milan) were tested by uMGMT primers to create a standard curve and calculate the starting amount of each DNA specimen. Positive signals from uMGMT, mSNURF and uSNURF were considered as internal controls for PCR reaction in order to exclude the presence of PCR inhibitors. Cases below a threshold of approximately 6.25 ng were repeated starting from a higher amount of tumor tissue. Specimens showing a threshold below this limit were considered invalid and were excluded from the study. The investigators who performed the MS-qLNAPCR tests were blinded to the clinical data and patient outcome.
Primers and molecular beacon probes design
Previously described primers for methylated and unmethylated MGMT promoter [14, 19, 35] were modified only to insert LNA nucleotides at the 3' end of their sequence (See Table 1). Primers for SNURF were designed using MethPrimer http://www.urogene.org/methprimer/index1.html[36] and verified by methBlast http://medgen.ugent.be/methBLAST (see Fig. 1 and Table 1).

CpG island of SNURF promoter. The upper lane identifies the exact sequence before bisulfite modification. The bottom lane represents the bisulfite treated methylated forward sequence where all C's are changed to T's, except for those followed by a G. Primer sequences are underlined by >. The internal probe covers two consecutive CpGs (marked as +).
Molecular beacon probes (see Table 1) were designed using the Primer3 software http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi including two CpG sites each. Flanked molecular beacon arms were designed using the OLIGO 6.0 software reaching a temperature between 57°C and 61°C in the stem loop conformation. LNA nucleotide were added to primers as described previously [37] using a Tm prediction tool available at exiqon web site http://www.exiqon.com. Amplicons were tested by MFOLD http://www.bioinfo.rpi.edu/applications/mfold/old/dna/ in order to avoid secondary structures within primer and probe positions.
Direct Sequencing
PCR primers to amplify the CpG island of SNURF promoter were designed by MethPrimer software on line http://www.urogene.org/methprimer/, which was also used to predict CpG islands and CpG sites in the sequence (see Table 1). The amplification of bisulfited-modified DNA was performed using FastStartTaq™ polymerase (Roche, Milan), with the following conditions: 95°C for 4 min, followed by 40 three steps cycles at 95°C for 30 sec, 60°C for 30 sec and at 72°C for 30 sec. The PCR products were separated on 3% SeaKem® LE agarose gels and purified (Agencourt, Beverly, MA), followed by sequencing by CEQ2000XL automatic DNA sequencer (Beckman Coulter, Fullerton, CA). The presence of a cytosine residue after bisulfite treatment shows that the cytosine residue was protected by methylation from bisulfite modification (Fig. 2).

Direct Sequencing of SNURF promoter. Direct sequencing after bisulfite modification of two CpGs of SNURF for case BF215. The arrows indicates the simultaneous presence of C/T (C: black peak; T: blue peak) due to a balanced amount of methylated and unmethylated cytosine in the CpGs amplified with our primer set.
Results
159 patients (M:F 99:60, median age: 57 years, range 25-77 years) met the inclusion criteria. TMZ concurrent with and adjuvant to RT was administered in 145 patients (91.2%). MGMT promoter was found methylated by nested MS-PCR in 69 samples (43.39%), and unmethylated in 90 samples (56.60%), and was found methylated by MS-qLNAPCR in 70 patients (44.02%), and unmethylated in 89 samples (55.97%). Concordance between the two assays was found in 158 of 159 samples (99.4%). The only patient discordant was found unmethylated by nested MS-PCR and showed methylation in less than 1% of cells by MS-qLNAPCR (ratio between mMGMT/uMGMT = 0.0008).
We tested in parallel two sets of primers specific for mMGMT and uMGMT with and without LNA modifications for PCR efficiency in the MS-qLNAPCR assay. LNA modified primers for mMGMT showed higher PCR efficiency (slope: -3.271; efficiency: 102%, see Fig. 3) than unmodified primers (slope: -4.339; efficiency: 69% see Fig. 4). The analytical detection limit of 0.01% was reached only for LNA modified primers, while conventional primers showed a detection limit of 0.1%. The analytical detection limit (sensitivity limit) of the MS-qLNAPCR assay was determined using the ABI7000™ instrument using serial dilution mixtures of SssI-treated DNA: untreated DNA (100%, 50%, 10%, 1%, 0.1% 0.01% each containing 1.5 μg of template DNA).

Standard curves for MS-qLNAPCR with LNA primers. MS-qLNAPCR standard curves with serial dilution mixtures of SssI-treated DNA:untreated DNA (100%, 50%, 10%, 1%, 0.1%, 0.01%, 0.001%; each containing 1.5 μg of template DNA) for LNA primers specific for mMGMT. LNA modified primers for mMGMT show higher PCR efficiency (slope: -3.271; efficiency: 102%) than conventional primers (slope: -4.339; efficiency: 69%, see Fig.4). The analytical detection limit of 0.01% is reached only with LNA primers.

Standard curves for MS-qPCR with conventional primers. MS-qPCR standard curves with serial dilution mixtures of SssI-treated DNA:untreated DNA (100%, 50%, 10%, 1%, 0.1%, 0.01%, 0.001%; each containing 1.5 μg of template DNA) for conventional primers[35] specific for mMGMT. Conventional primers for mMGMT show lower PCR efficiency (slope: -4.339; efficiency: 69%) than LNA modified primers (see Fig.3). The analytical detection limit with the use of conventional primers was 0.1%.
1.5 μg of SssI treated DNA (hypothetically fully methylated DNA) maintained a small amount of unmethylated DNA for MGMT promoter (0.00079%). By this protocol a sensitivity of 0.01% methylated alleles was detected and confirmed by the 24 replicates with a 95% cut-off value, according to common diagnostic practices [38].
The specificity of the assays was assessed by purification and analysis of a series of DNA from whole blood of 24 healthy donors without detecting any methylated allele for MS-qLNAPCR. On the contrary we detected two methylated cases by nested MS-PCR.
The total failure rate of unmethylated allele PCR was 0%. Inhibitors were not observed due to the high efficiency of DNA extraction and bisulfite treated DNA purification.
During the evaluation tests, 12 replicates of dilution mixture of 0.01% SssI-treated DNA in a background of 1.5 μg of untreated DNA were evaluated with no false negative results.
Evaluating SNURF data from 24 healthy blood donors, we found a mean difference between methylated and unmethylated SNURF alleles of 0.001 (SD = 0.81); the normality range was therefore estimated as [-1.57, +1.59]. Values outside the SNURF normality range were detected in 14 out of 159 GBM (8.8%; see the Additional file 1 for details). In these cases we could not calculate the methylated/unmethylated MGMT allele ratio because the requirement for using SNURF as a reference is that methylated and unmethylated SNURF alleles are at a ratio of 1:1.
The SNURF promoter of the 14 cases with Ct ratio [(mSNURF - uSNURF)] outside the normality range was sequenced to verify whether they showed the simultaneous presence of both methylated and unmethylated cytosine in each of the CpGs interrogated by the primers and beacon probes. Every specimen showed the presence of both methylated and unmethylated cytosine in the CpGs (see Fig. 1 and Fig. 2), demonstrating that the Ct ratios outside normalcy are not due to primer mismatch and different PCR efficiency for the methylated and unmethylated alleles. Of the 14 GBMs with Ct ratio outside the normality range only five among methylated cases had MGMT methylation and the ΔCt of SNURF had some impact on the final calculation for the mMGMT/uMGMT ratio in only one of 70 cases (1.42%, sample BF215). The SNURF promoter of BF215 was sequenced showing both methylated and unmehtylated cytosines for each of the CpGs tested (see Fig. 2). This indicates that the BF215 ratio of Ct [(mSNURF - uSNURF)] is not due to primer/probe mismatch but may be the result of a partial loss of imprinting (LOI) of the maternal allele (see Additional file 1 for details).
With our MS-qLNAPCR protocol we identified 70 of 159 MGMT methylated cases (44.0%). In addition, MS-qLNAPCR allows a fine discrimination of the relative amounts of methylated vs. unmethylated MGMT in a given case (see Fig. 5). Quantitative analysis showed a bimodal distribution of ratio values between methylated and unmethylated MGMT alleles, with two prevalent groups with ratios between 0.001-0.33 and 0.67-1, respectively (see Fig. 6). This bimodal distribution is similar to that found by Vlassenbroeck et al. [27].

A representative MS-qLNAPCR plot. A representative MS-qLNAPCR plot (GBM case BF240) showing an MGMT methylated ratio value of 0.46.

Bimodal distribution of methylated and unmethylated alleles. Normalized ratio value groups between methylated and unmethylated alleles of the 70 MGMT methylated GBMs identified with our MS-qLNAPCR protocol. There is a bimodal distribution of cases with two prevalent groups showing ratio values between 0.001-0.33 and 0.67-1, respectively.
Survival
Median overall survival of the 159 patients was of 24 months (95%CI: 21 - 27), being 20 months and 36 (95%CI: 17.7 - 54.3) months, in patients with MGMT unmethylated and methylated tumors by nested MS-PCR, respectively (log-rank test, p = 0.004). Considering MGMT methylation data provided by our MS-qLNAPCR protocol as a binary variable, overall survival was statistically different between patients with GBM samples harboring MGMT promoter unmethylated and other patients with any percentage of MGMT methylation (log-rank test, p = 0.003, Fig. 7). This difference was retained using other cut off values for MGMT methylation rate (i.e. 10% and 20% of methylated cells), while the difference was lost when 50% of MGMT methylated cells was used as cut-off. Overall survival significantly correlated with MGMT promoter methylation status determined either by nested MS-PCR (p = 0.021) or MS-qLNAPCR (p = 0.025) even after the 14 patients that received RT followed by adjuvant TMZ were excluded.

Kaplan-Meier survival curves. Survival curves of GBM patients with methylated MGMT promoter (thick line) and unmethylated MGMT promoter (thin line) determined by MS-qLNAPCR. Patients with tumors with unmethylated MGMT promoter have lower overall survival (Log rank test p = 0.003).
Discussion
Given the key roles of cytosine methylation, there has been a wide interest in the development of procedures for DNA methylation analyses [15]. The prognostic significance of MGMT promoter methylation has been shown in two chemoradiotherapy clinical trials; first in a phase II study testing concomitant and adjuvant temozolomide and radiation [39] and subsequently in EORTC 26981/22981 & NCIC CE.3 [14]. In the later study, MGMT promoter methylation was an independent favourable prognostic factor and patients whose tumour contained a methylated MGMT promoter had median survival of 21.7 months and 2-year survival of 46%, when treated with temozolomide and radiotherapy. These studies suggest that determination of MGMT methylation status maybe an important factor in determining which glioblastoma patients should receive chemoradiotherapy [40], but its prognostic significance in the routine clinical setting is not clearly established. Methylation-specific PCR (MSP) is widely used to test MGMT promoter methylation; however, in EORTC 26981/22981 & NCIC CE.3 Hegi et al. [14] only achieved methylation data for 67% of samples analysed, representing 36% of cases.
Sensitive, accurate, high-throughput and cost-effective methods should give a better definition of the role and control of this epigenetic modification, especially in cancer. Many different experimental approaches have been developed to allow either global large-scale or specific analyses [2, 3, 6, 15–23]. The most popular approaches rely on bisulfite treatment of DNA [41]. This treatment is performed in such a way that, while cytosine is quantitatively deaminated to uracil, 5-methyl cytosine remains unmodified, thus allowing identification of the cytosine methylation status following PCR amplification. There are many possible variations in the subsequent DNA sequence detection methods used, which achieve diverse levels of performance and adequacy. The quantitative analysis of the cytosine methylation levels at several independent proximal positions can be achieved using Pyrosequencing analysis of PCR products amplified in a methylation-independent manner [18, 41]. This procedure is not particularly sensitive and does not perform well with neoplastic populations that represents <10% of the sample, which limits its usefulness in the analysis of heterogeneous pathological samples and of early events in epigenetic reprogramming. MS-PCR [42] is the most widely used assay for the detection of hypermethylation in CpG islands. It relies on the selective PCR amplification of sequences corresponding to either unmethylated or methylated DNA using primers that anneal specifically with either one of the DNA species. For methylation-specific annealing, each primer must contain sequences corresponding to at least two CpG dinucleotides. This method allows sensitive detection of particular methylation patterns and is currently used for the analysis of pathological samples.
MS-PCR has been adapted to fluorescence-based real-time PCR technology, thus allowing for both quantitative and high-throughput sample analysis [17, 18, 27, 28, 42, 43].
Eads CA et al. introduced quantitative methylation sensitive PCR based on TaqMan® chemistry [24], determining the relative amounts of a particular methylation pattern with quantitative accuracy. Hattermann K et al. [44] described a method combining methylation specific and SYBR green based quantitative PCR allowing the quantification of fully methylated and fully unmethylated MGMT DNA species. Values were related to standard curves, corrected for DNA input by an internal calibrator, and calculated in relation to methylated and unmethylated control DNAs as a percentage share [44]. Smith E et al. [26] described an elegant and cost effective quantitative methylation sensitive PCR based on melting curve analysis obtained after amplification of bisulfite modified DNA in a real-time thermocycler. Vlassenbroeck I et al. [27] validated a direct quantitative methylation sensitive PCR assay based on SYBRGreen detection chemistry, which quantifies the copy number of the methylated MGMT promoter after normalization of Ct values with the ACTB reference gene. To the best of our knowledge, we are the first to describe and validate a method to quantitate DNA methylation using LNA modified primers and an imprinted gene as a reference, instead of a methylation independent calibrator such as ACTB [27]. In our opinion ACTB does not represent the best reference gene for normalization because it is located at 7p15-p12, a chromosomal site subject to copy number variations in gliomas [45, 46], and because it is close EGFR (located at 7p12) that is amplified in about 40% of GBMs [47]. We use of an imprinted gene (SNURF) as an internal control, because it may check the efficiency of the assay from DNA purification, through bisulfite treatment to PCR. Additionally it should be used as a reference because mSNURF and uSNURF mimic the biallelic MGMT status. In fact, in normal cells the maternal allele of SNURF is methylated at the promoter locus, while the paternal allele of the same gene is usually unmethylated and expressed [30]. This condition is thus similar to a tumor population of cells in which the MGMT is methylated at one of the two alleles. In order to consider SNURF as an ideal reference, the ratio between methylated and unmethylated SNURF alleles might not be disturbed by copy number changes, or by loss of imprinting, both of which are common in cancer. However, several references demonstrate that SNURF, which has been mapped at 15q12, is hardly ever altered in gliomas [45, 48–51]. These data were confirmed by our study because the methylated and unmethylated SNURF ΔCt of the most part of cases (91.2%) was nearly always very close to 0. The SNURF methylation values outside the normality range in 14 of the 159 GBMs (8.8%) may be due to a distinct CpG methylation pattern among tumor cell population, to partial loss of one allele (LOI: loss of imprinting), or to a methylation machinery disorder that methylates the paternal allele (GOI: gain of imprinting). The requirement for using SNURF as a reference is that methylated and unmethylated SNURF alleles are at a ratio of 1:1, and in our series in only one of these 14 cases did the ΔCt of SNURF have a negative impact on the final calculation for the mMGMT/uMGMT ratio. In these circumstances we avoid using SNURF as a reference loosing relative quantification data.
By sequencing the SNURF promoter of this abnormal case (BF215, Fig. 2), we found a balanced ratio between methylated and unmethylated cytosine in CpG interrogated loci. These data demonstrate for this case that the ΔCt found to be outside the normality range is not due to primer/probe mismatch but may be the result of a partial LOI of the maternal allele.
LNA based PCR has been widely used to achieve high specificity for the detection of single nucleotide polymorphism (SNP). LNA primers show very accurate mismatch discrimination in comparison with conventional DNA primers at all 3'-terminal positions [37]. Moreover, they offer greater sensitivity with respect to TaqMan based SNP detection due to the fact that in allele specific PCR for SNP detection two PCR amplify selectively each allele. On the other hand, in case of TaqMan technology a single PCR with flanking primers will amplify in parallel both alleles. If one allele is poorly represented (e.g. in samples with low density of tumor cells in a background of normal cells) the TaqMan probe may fail to detect or underestimate the less represented allele.
Because of these reasons we have taken advantage of 3'-LNA modified primers and molecular beacon detection chemistry [52, 53] to developed a direct methylation sensitive quantitative PCR (MS-qLNAPCR) assay. Although allele specific assays provide an elegant method to discriminate between alleles, quantification of the variants is not attainable because of the intrinsic endpoint detection by conventional PCR. This limitation is fully addressed using real-time qPCR methods [54], whereby PCR product accumulation is monitored at each PCR cycle by means of fluorescent detection using molecular beacon chemistry. Unlike previously described methylation sensitive quantitative PCR protocols [27, 28], our new MS-qLNAPCR approach is based on two different detection beacon probes, one recognizing the methylated allele and the other one recognizing the unmethylated allele.
These probes were designed to span two CpGs in order to obtain high specificity compared with previous MS-PCR or MS-qPCR methods with SYBRGreen detection, with high discriminatory power between the methylated and unmethylated MGMT alleles even with DNA extracted from FFPE samples. Variation of standard curves run in parallel with tumor samples in each batch of cases were small and acceptable, indicating optimal reproducibility. We found our MS-qLNAPCR protocol particularly reliable when dealing with large amounts of input DNA since it avoids false positive results [37].
To limit experimental variation we chose to utilize the same primers used by Hegi et al. which have been clinically validated on a large number of samples [14, 19, 35]. We modified the primers only to insert the LNA nucleotides to improve the assay and demonstrated that our LNA modified primers show optimal PCR efficiency and high sensitivity, better than that of large clinical studies [14]. These have so far largely used nested MS-PCR. MS-qLNAPCR offers many advantages compared with nested MS-PCR, including ease of interpretation compared with gel-based methods where weak bands are often difficult to interpreted, quantitative results, a one step procedure that saves time and reduces contamination risk, a broad dynamic range with reproducible results with both high and very low input DNA amounts. Low DNA amounts could still be processed also because the use of the EpiTect Bisulfite kit reduced the intrinsic DNA fragmentation during the bisulfite treatment. The advantages of our MS-qLNAPCR protocol make it thus ideal for high throughput analysis, when large numbers of clinical samples need to studied.
Vlassenbroeck I et al. [27] using MS-qPCR have shown that the amount of MGMT promoter methylation in GBMs is variable, with a bimodal distribution of cases. With MS-qLNAPCR we have fully confirmed their data. In addition we demonstrate that epigenetic silencing of MGMT is associated with response to temozolomide-chemotherapy even when the amount of the methylated MGMT allele is low. This is very important for the clinical implications of a molecular diagnosis of MGMT promoter methylation in GBMs. Since low levels of MGMT promoter methylation retain a predictive value, even weak methylation signals should be reported. The biological explanation of the phenomenon is unclear, but it should be kept in mind that, next to MGMT, other factors may be involved in temozolomide response (e.g. the high activity of poly-ADP ribose polymerase and base excision repair machinery [55, 56]).
Conclusions
We report and validate clinically, a novel, accurate, robust, and cost effective MS-qLNAPCR protocol for the detection and quantification of methylated MGMT alleles. This protocol is the only quantitative method validated to date from both technical and clinical standpoints for GBM samples. Using MS-qLNAPCR we demonstrate that even low levels of MGMT promoter methylation have to be taken into account to predict response to temozolomide-chemotherapy.
Abbreviations
- CDKN2A:
-
cyclin-dependent kinase inhibitor 2A
- MGMT:
-
O6-Methylguanine-DNA methyltransferase
- MLH1:
-
mutL homolog 1
- GBM:
-
Glioblastoma
- LNA:
-
locked nucleic acid
- RT:
-
Radiotherapy
- TMZ:
-
temozolomide
- FFPE:
-
paraffin embedded and formalin fixed
- MS-PCR:
-
methylation sensitive PCR
- MS-qLNAPCR:
-
methylation sensitive quantitative locked nucleic acid real time PCR
- AS-PCR:
-
allelic specific PCR
- LOI:
-
loss of imprinting
- GOI:
-
gain of imprinting.
References
Laird PW: Cancer epigenetics. Hum Mol Genet. 2005, 14 (Spec No 1): R65-76. 10.1093/hmg/ddi113.
Herman JG, Baylin SB: Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003, 349 (21): 2042-2054. 10.1056/NEJMra023075.
Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T: Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol. 2003, 13 (2): 176-184.
Qian XC, Brent TP: Methylation hot spots in the 5' flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997, 57 (17): 3672-3677.
Watts GS, Pieper RO, Costello JF, Peng YM, Dalton WS, Futscher BW: Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol Cell Biol. 1997, 17 (9): 5612-5619.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG: Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59 (4): 793-797.
Stupp R, Mason WP, Bent van den MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005, 352 (10): 987-996. 10.1056/NEJMoa043330.
Liu L, Markowitz S, Gerson SL: Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res. 1996, 56 (23): 5375-5379.
Gerson SL: MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004, 4 (4): 296-307. 10.1038/nrc1319.
Hotta T, Saito Y, Fujita H, Mikami T, Kurisu K, Kiya K, Uozumi T, Isowa G, Ishizaki K, Ikenaga M: O6-alkylguanine-DNA alkyltransferase activity of human malignant glioma and its clinical implications. J Neurooncol. 1994, 21 (2): 135-140. 10.1007/BF01052897.
Belanich M, Pastor M, Randall T, Guerra D, Kibitel J, Alas L, Li B, Citron M, Wasserman P, White A, et al: Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996, 56 (4): 783-788.
Jaeckle KA, Eyre HJ, Townsend JJ, Schulman S, Knudson HM, Belanich M, Yarosh DB, Bearman SI, Giroux DJ, Schold SC: Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998, 16 (10): 3310-3315.
Friedman HS, McLendon RE, Kerby T, Dugan M, Bigner SH, Henry AJ, Ashley DM, Krischer J, Lovell S, Rasheed K, et al: DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol. 1998, 16 (12): 3851-3857.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al: MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005, 352 (10): 997-1003. 10.1056/NEJMoa043331.
Kress C, Thomassin H, Grange T: Local DNA demethylation in vertebrates: how could it be performed and targeted?. FEBS Lett. 2001, 494 (3): 135-140. 10.1016/S0014-5793(01)02328-6.
Liu ZJ, Maekawa M: Polymerase chain reaction-based methods of DNA methylation analysis. Anal Biochem. 2003, 317 (2): 259-265. 10.1016/S0003-2697(03)00169-6.
Cottrell SE, Distler J, Goodman NS, Mooney SH, Kluth A, Olek A, Schwope I, Tetzner R, Ziebarth H, Berlin K: A real-time PCR assay for DNA-methylation using methylation-specific blockers. Nucleic Acids Res. 2004, 32 (1): e10-10.1093/nar/gnh008.
Uhlmann K, Brinckmann A, Toliat MR, Ritter H, Nurnberg P: Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. Electrophoresis. 2002, 23 (24): 4072-4079. 10.1002/elps.200290023.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG: Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000, 343 (19): 1350-1354. 10.1056/NEJM200011093431901. Erratum, N Engl J Med 2000;1343: 1740
Jeuken JW, Cornelissen SJ, Vriezen M, Dekkers MM, Errami A, Sijben A, Boots-Sprenger SH, Wesseling P: MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest. 2007, 87 (10): 1055-1065. 10.1038/labinvest.3700664.
Cankovic M, Mikkelsen T, Rosenblum ML, Zarbo RJ: A simplified laboratory validated assay for MGMT promoter hypermethylation analysis of glioma specimens from formalin-fixed paraffin-embedded tissue. Lab Invest. 2007, 87 (4): 392-397.
Thomassin H, Oakeley EJ, Grange T: Identification of 5-methylcytosine in complex genomes. Methods. 1999, 19 (3): 465-475. 10.1006/meth.1999.0883.
Fraga MF, Esteller M: DNA methylation: a profile of methods and applications. Biotechniques. 2002, 33 (3): 636-649.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW: MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28 (8): E32-10.1093/nar/28.8.e32.
Lorente A, Mueller W, Undargarin E, Lázcoz P, vonDeimling A, Castresana JS: Detection of methylation in promoter sequences by melting curve analysis-based semiquantitative real time PCR. BMC Cancer. 2008, 8: 61-10.1186/1471-2407-8-61.
Smith E, Jones ME, Drew PA: Quantitation of DNA methylation by melt curve analysis. BMC Cancer. 2009, 9: 123-10.1186/1471-2407-9-123.
Vlassenbroeck I, Califice S, Diserens AC, Migliavacca E, Straub J, Di Stefano I, Moreau F, Hamou MF, Renard I, Delorenzi M, et al: Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008, 10 (4): 332-337. 10.2353/jmoldx.2008.070169.
Wojdacz TK, Dobrovic A: Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007, 35 (6): e41-10.1093/nar/gkm013.
Gustafson KS: Locked nucleic acids can enhance the analytical performance of quantitative methylation-specific polymerase chain reaction. J Mol Diagn. 2008, 10 (1): 33-42. 10.2353/jmoldx.2008.070076.
El-Maarri O, Buiting K, Peery EG, Kroisel PM, Balaban B, Wagner K, Urman B, Heyd J, Lich C, Brannan CI, et al: Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet. 2001, 27 (3): 341-344. 10.1038/85927.
Ginzinger DG, Godfrey TE, Nigro J, Moore DH, Suzuki S, Pallavicini MG, Gray JW, Jensen RH: Measurement of DNA copy number at microsatellite loci using quantitative PCR analysis. Cancer Res. 2000, 60 (19): 5405-5409.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK: World Health Organization Classification of Tumours of the Central Nervous System. 2007, IARC Lyon
Morandi L, Marucci G, Foschini MP, Cattani MG, Pession A, Riva C, Eusebi V: Genetic similarities and differences between lobular in situ neoplasia (LN) and invasive lobular carcinoma of the breast. Virchows Arch. 2006, 449 (1): 14-23. 10.1007/s00428-006-0192-7.
Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA: Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000, 60 (21): 5954-5958.
van Engeland M, Weijenberg MP, Roemen GM, Brink M, de Bruine AP, Goldbohm RA, Brandt van den PA, Baylin SB, de Goeij AF, Herman JG: Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res. 2003, 63 (12): 3133-3137.
Li LC, Dahiya R: MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002, 18 (11): 1427-1431. 10.1093/bioinformatics/18.11.1427.
Latorra D, Campbell K, Wolter A, Hurley JM: Enhanced allele-specific PCR discrimination in SNP genotyping using 3' locked nucleic acid (LNA) primers. Hum Mutat. 2003, 22 (1): 79-85. 10.1002/humu.10228.
Liikanen E: Common technical specification for in vitro diagnostic medical devices. Official Journal of European Community. 2002, L131: 17-30.
Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, Otten P, VanMelle G, deTribolet N, Stupp R: Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004, 10 (6): 1871-1874. 10.1158/1078-0432.CCR-03-0384.
Gorlia T, Bent van den MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, et al: Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008, 9 (1): 29-38. 10.1016/S1470-2045(07)70384-4.
Tost J, Dunker J, Gut IG: Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques. 2003, 35 (1): 152-156.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996, 93 (18): 9821-9826. 10.1073/pnas.93.18.9821.
Thomassin H, Kress C, Grange T: MethylQuant: a sensitive method for quantifying methylation of specific cytosines within the genome. Nucleic Acids Res. 2004, 32 (21): e168-10.1093/nar/gnh166.
Hattermann K, Mehdorn HM, Mentlein R, Schultka S, Held-Feindt J: A methylation-specific and SYBR-green-based quantitative polymerase chain reaction technique for O6-methylguanine DNA methyltransferase promoter methylation analysis. Anal Biochem. 2008, 377 (1): 62-71. 10.1016/j.ab.2008.03.014.
Roversi G, Pfundt R, Moroni RF, Magnani I, van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers EF: Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006, 25 (10): 1571-1583. 10.1038/sj.onc.1209177.
Yin D, Ogawa S, Kawamata N, Tunici P, Finocchiaro G, Eoli M, Ruckert C, Huynh T, Liu G, Kato M, et al: High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009, 7 (5): 665-677. 10.1158/1541-7786.MCR-08-0270.
von Deimling A, Louis DN, von Ammon K, Petersen I, Hoell TCRY, Martuza RL, Schoenfeld DA, Yaşargil MG, Wiestler OD: Association of epidermal growth factor receptor gene amplification with loss of chromosome 10 in human glioblastoma multiforme. J Neurosurg. 1992, 77: 295-301. 10.3171/jns.1992.77.2.0295.
Freire P, Vilela M, Deus H, Kim YW, Koul D, Colman H, Aldape KD, Bogler O, Yung WK, Coombes K, et al: Exploratory analysis of the copy number alterations in glioblastoma multiforme. PLoS One. 2008, 3 (12): e4076-10.1371/journal.pone.0004076.
Gardina PJ, Lo KC, Lee W, Cowell JK, Turpaz Y: Ploidy status and copy number aberrations in primary glioblastomas defined by integrated analysis of allelic ratios, signal ratios and loss of heterozygosity using 500K SNP Mapping Arrays. BMC Genomics. 2008, 9: 489-10.1186/1471-2164-9-489.
Lo KC, Bailey D, Burkhardt T, Gardina P, Turpaz Y, Cowell JK: Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization. Genes Chromosomes Cancer. 2008, 47 (3): 221-237. 10.1002/gcc.20524.
Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, Hariono S, Silber J, Feiler HS, Gray JW, et al: Comparative analyses of gene copy number and mRNA expression in GBM tumors and GBM xenografts. Neuro Oncol. 2009, 11 (5): 477-87. 10.1215/15228517-2008-113.
Bottema CD, Sommer SS: PCR amplification of specific alleles: rapid detection of known mutations and polymorphisms. Mutat Res. 1993, 288 (1): 93-102.
Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF: Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17 (7): 2503-2516. 10.1093/nar/17.7.2503.
Maertens O, Legius E, Speleman F, Messiaen L, Vandesompele J: Real-time quantitative allele discrimination assay using 3' locked nucleic acid primers for detection of low-percentage mosaic mutations. Anal Biochem. 2006, 359 (1): 144-146. 10.1016/j.ab.2006.07.039.
Curtin NJ, Wang LZ, Yiakouvaki A, Kyle S, Arris CA, Canan-Koch S, Webber SE, Durkacz BW, Calvert HA, Hostomsky Z, et al: Novel poly(ADP-ribose) polymerase-1 inhibitor, AG1 restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res. 4361, 10 (3): 881-889. 10.1158/1078-0432.CCR-1144-3.
Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW: The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005, 65 (14): 6394-6400. 10.1158/0008-5472.CAN-05-0715.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/10/48/prepub
Acknowledgements
This study was supported in part by a regional grant (1897, 2008) given by Friuli Venezia Giulia to Alphagenics Diaco Biotechnologies S.r.l., Area Science Park, Basovizza, Trieste Italy grant to L.M and in part by a R. F. O. (ex MURST 40%) grant to G.T.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Conception and Design: LM; Provision of study materials or patients: AB, EF, AT; Collection and assembly of data: LM, DB, GM, AP, GT; Data analysis and interpretation: LM, EF, AT, AP, GT, ME, GM; Manuscript writing: LM, GT. Final approval of manuscript: LM, EF, GM, DB, AT, AP, GT, AB, ME
Annalisa Pession, Giovanni Tallini and Alba Brandes contributed equally to this work.
Electronic supplementary material
12885_2009_1847_MOESM1_ESM.XLS
Additional file 1: MS-qLNAPCR and nested PCR data comparison. this file includes MS-qLNAPCR and nested PCR data for each of the 159 cases. GBM = glioblastoma; Nested-PCR: methylation status obtained by nested PCR; MS-qLNAPCR: methylation status obtained by MS-qLNAPCR. Met/Unmet Ratio: relative DNA copy number ratio between methylated and UnMethylated allele calculated from the formula described in Materials and methods; Ct [(mSNURF - uSNURF)]: delta Ct between SNURF methylated allele and SNURF UnMethylated allele. (XLS 53 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Morandi, L., Franceschi, E., de Biase, D. et al. Promoter methylation analysis of O6-methylguanine-DNA methyltransferase in glioblastoma: detection by locked nucleic acid based quantitative PCR using an imprinted gene (SNURF) as a reference. BMC Cancer 10, 48 (2010). https://doi.org/10.1186/1471-2407-10-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-10-48