
Abstract
Background
Promoter methylation of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) is an acknowledged predictive epigenetic marker in glioblastoma multiforme and anaplastic astrocytoma. Patients with methylated CpGs in the MGMT promoter benefit from treatment with alkylating agents, such as temozolomide, and show an improved overall survival and progression-free interval. A precise determination of MGMT promoter methylation is of importance for diagnostic decisions. We experienced that different methods show partially divergent results in a daily routine. For an integrated neuropathological diagnosis of malignant gliomas, we therefore currently apply a combination of methylation-specific PCR assays and pyrosequencing.
Results
To better rationalize the variation across assays, we compared these standard techniques and assays to deep bisulfite sequencing results in a cohort of 80 malignant astrocytomas. Our deep analysis covers 49 CpG sites of the expanded MGMT promoter, including exon 1, parts of intron 1 and a region upstream of the transcription start site (TSS). We observed that deep sequencing data are in general in agreement with CpG-specific pyrosequencing, while the most widely used MSP assays published by Esteller et al. (N Engl J Med 343(19):1350–1354, 2000. https://doi.org/10.1056/NEJM200011093431901) and Felsberg et al. (Clin Cancer Res 15(21):6683–6693, 2009. https://doi.org/10.1158/1078-0432.CCR-08-2801) resulted in partially discordant results in 22 tumors (27.5%). Local deep bisulfite sequencing (LDBS) revealed that CpGs located in exon 1 are suited best to discriminate methylated from unmethylated samples. Based on LDBS data, we propose an optimized MSP primer pair with 83% and 85% concordance to pyrosequencing and LDBS data. A hitherto neglected region upstream of the TSS, with an overall higher methylation compared to exon 1 and intron 1 of MGMT, is also able to discriminate the methylation status.
Conclusion
Our integrated analysis allows to evaluate and redefine co-methylation domains within the MGMT promoter and to rationalize the practical impact on assays used in daily routine diagnostics.
Similar content being viewed by others


Background
Glioblastoma multiforme (GBM) are the most frequent human brain tumors in adults and represent the most aggressive form of tumors deriving from astroglia. With their accelerated, infiltrative, often multiple growth, and characteristic hallmarks such as necrosis, endothelial proliferation and high mitotic activity, they are graded as WHO grade IV tumors with an average survival of 15 months upon optimized standard treatment [1,2,3,4]. Anaplastic astrocytomas (grade III) are histologically slightly less aggressive lacking necrotic areas and endothelial proliferations, but patients are subjected to the same kind of treatment. Besides surgery and radiotherapy, the therapeutic decision mainly depends on the promoter DNA methylation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase (MGMT). Methylation-induced downregulation or silencing of MGMT inhibits its DNA repair mechanism of alkyl group removal, thereby making tumor cells sensitive to cytotoxic alkylating agents like temozolomide which improves patients’ overall survival [5, 6]. Another genetic alteration in astrocytic tumors that confers a significant survival benefit is a mutation of isocitrate dehydrogenase (IDH). Point mutations in the IDH1 or IDH2 gene are correlated with a better overall survival and while they occur frequently in lower-grade astrocytomas (WHO grade II and III) and secondary glioblastomas, they are rare (< 10%) in primary glioblastomas [7]. This genetic influence on prognosis needs to be considered when therapies in astrocytic tumors are compared or a predictive parameter like the MGMT promoter methylation is determined.
Currently, there is no direct method to determine the enzymatic activity of MGMT in tumor samples, which is the reason why the MGMT promoter methylation serves as an indirect tool. Methylation-specific PCR (MSP) [8] and pyrosequencing [9] are commonly used techniques for routine testing in molecular diagnostics of glioblastoma. While the latter is based on quantitative measurements of a handful of CpGs, MSP returns a binary decision with uncertainties in the determination of technical cutoffs [10,11,12]. MSP, however, is cost-effective and easy when compared to other methods, since it does not ask for special equipment beyond that present in any laboratory doing PCRs.
The positioning of PCR and sequencing primers in CpG-rich regions such as the MGMT promoter is challenging. Thus, it is unclear whether MSP or pyrosequencing assays, like the MGMT Pyro Kit (QIAGEN), perform in an unbiased manner. Illumina 850 K (EPIC) BeadChip microarrays are increasingly used for clinical diagnostic and prognostic estimations as they allow a genome-wide methylation profiling [13,14,15]. However, BeadChip arrays only cover few CpGs in promoter regions such as MGMT and detection is highly dependent on probe position and primer extension effects [16]. In contrast to all these selective methods, profiling by local deep bisulfite sequencing (LDBS) offers a direct and (countable) quantitative single CpG methylation readout covering regions of up to 500 bp [17]. By cautious primer design, the potential biases in LDBS are reduced to a minimum. We therefore generated LDBS data across the MGMT promoter region as a “gold standard” in 69 glioblastoma and 11 anaplastic astrocytoma samples. We then compared these results with the routinely obtained data from pyrosequencing (therascreen MGMT Pyro Kit, Q24, MDx) and two different MSPs [18, 19], and discuss the results with regard to their practical implementation in everyday diagnostic procedures [7].
Results
Bisulfite profiling of the exon 1 and intron 1 region of the MGMT promoter
In a glioblastoma/anaplastic astrocytoma cohort of 80 tumors, standard molecular diagnostic procedures were conducted, including MGMT promoter methylation analysis using two methylation-specific PCRs (MSP) [8, 18, 19] and pyrosequencing [20,21,22]. Following the cutoff values for the therascreen MGMT Pyro Kit as suggested by Reifenberger and colleagues, we regard an average methylation percentage larger than 8% as methylated for diagnosis [23]. For both “methylated” MSPs, any visible PCR product will signify a methylation of the MGMT promoter. Comparing MSP results using primer pairs published by Esteller et al. (2000) and primer pairs published by Felsberg et al. (2009), we observed concordance, i.e., presence or absence of PCR products in both MSPs, for 58 tumors, while discordant results, i.e., absence of PCR product in one of the MSPs, were obtained for 22 tumors (Additional file 1: Table S1) [18, 19]. Out of 22 discordant samples, 13 were detected as methylated using the Esteller primer pair, while 9 samples were found to be methylated by the Felsberg primer pair only. Quantitative pyrosequencing of four CpG positions in MGMT exon 1 revealed average methylation values between 2.5 and 70.3% for the MSP-concordant methylated tumors, between 1.5 and 16.3% for the MSP-concordant unmethylated tumors and between 1.2% and 23.5% for the discordant tumors. Among the MSP-discordant cases, five of the 13 “Esteller-methylated/Felsberg-unmethylated” (38.5%) and three of the nine “Felsberg-methylated/Esteller-unmethylated” (30%) samples were considered methylated by pyrosequencing, i.e., above a cutoff of 8% average methylation. In three of the 26 cases with concordant MSP-unmethylated results (11.5%), quantitative pyrosequencing also produced an average methylation value above 8%.
To obtain a detailed picture of the CpG methylation levels covered by MSP primers and pyrosequencing, we performed local deep bisulfite sequencing (LDBS) using oligos that amplify the exon 1/intron 1 region in a methylation-independent manner (Fig. 1) [17]. Out of 80 tumors (82 samples), we managed to obtain 72 (exon 1) and 70 (intron 1) amplicons that were subjected to LDBS, respectively. As shown in Fig. 2, sample-wise averaged MGMT exon 1 (MGMTe1) methylation levels showed an almost bimodal distribution, with unmethylated tumor samples clearly separated from methylated ones in the hierarchical clustering in exon 1.

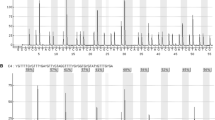
Schematic overview on the MGMT promoter region with positioning of exon 1 (ENSG00000170430), promoter-associated CpG island (CGIp) and sequenced amplicon regions; hg19 genomic position is given in bp; relative position of CGIp and amplicons subjected to local deep bisulfite sequencing (MGMTup, MGMTe1 and MGMTi1) are shown as grey bars; MGMTup and MGMTe1 forward primer sequences are highlighted in turquoise, MGMTup and MGMTi1 reverse primer sequences are highlighted in pink; the sequence used for MGMTi1 forward and MGMTe1 reverse primers is highlighted in green; arrows indicate the directionality of primers; exon1 sequence is highlighted in yellow; grey: ambiguously called nucleotides (taken from dbSNP153); red: CpG sites, numbered relative to the TSS

DNA methylation heatmap of all CpGs in the MGMTe1 (left) and MGMTi1 (right) amplicons in malignant astrocytoma samples; CpGs are numbered relative to the TSS; grey lines represent missing data for the respective sample; CpGs covered by pyrosequencing (PyroMark) and MSP (Felsberg and Esteller) or present on the 450 K/850 K (EPIC) BeadChip arrays are indicated at the top. Samples were clustered according to their methylation levels in MGMTe1 and put in the same order in MGMTi1 for clarity reasons
CpG-wise, MGMTe1 methylation appeared to be rather homogeneous across all CpGs with CpG + 6 and CpG + 12 showing lower DNA methylation compared to all other CpG sites in the MGMTe1 amplicon. In MGMT intron 1 (MGMTi1), sample- and CpG-wise methylation appeared to be more heterogeneous. The overall methylation decreased from MGMTe1 to MGMTi1 with single CpGs (CpG + 21, CpG + 32) showing an increase in methylation and some (CpG + 22, CpG + 28 to + 31) showing a decrease across all samples.
Taking all CpGs in MGMTe1 and MGMTi1 into account, we grouped the CpGs into five distinct methylation domains showing distinct patterns across the samples (Fig. 3A, see “Methods”). Interestingly, CpGs + 10, + 11, + 12 and CpG + 13, which are covered by the PyroMark assay, grouped into different methylation domains. Overall, the PyroMark results highly correlated with the local deep sequencing data (Pearson correlation r ≥ 0.89). Outlier samples were partially consistent between the single CpGs (Fig. 3B). The discordant MSP results (22 out of 80) showed an average methylation of 10.9% and 10% with bisulfite profiling for exon 1 (MGMTe1 and MGMTi1) for the solely “Esteller primer-methylated” (n = 13) and “Felsberg primer-methylated” (n = 9) samples, respectively. Looking into deep sequencing data of those CpGs within the MSP primer binding sites, we found at each CpG highly variable methylation values with an average between 4 and 12% for 13 out of 14 CpGs (Fig. 4A). CpG + 21 at the 3′-end of the Felsberg reverse primer showed an extraordinary high average methylation level of 19% and is grouped into a different methylation domain (Fig. 3A) making this oligo potentially the most unreliable one in the currently used oligo sets for routine testing.

A Grouping of CpGs in MGMTe1 (CpG + 6 to CpG + 17) and MGMTi1 (CpG + 18 to CpG + 32) into methylation domains; CpG sites covered by MSP primers are indicated by arrows, CpGs analyzed with pyrosequencing (PyroMark) are indicated as a box at the bottom. B Correlation plots of absolute methylation data obtained by local deep bisulfite sequencing (MiSeq) and pyrosequencing (PyroMark) for CpGs + 10, + 11, + 12 and + 13; each dot represents the methylation level of the indicated CpG from a single sample; Pearson correlation coefficient is indicated together with trend line, confidence intervals and IDs of consistent outlier samples

A Average DNA methylation of CpGs covered by MSP primers in samples discordant for MSP-based methylation detection; F, Felsberg primer set; E, Esteller primer set; f, forward primer; r, reverse primer. B Proposed localization of primers for an optimized MGMT promoter MSP assay; yellow: exon 1; turquoise: neighboring CpGs discriminating between unmethylated and methylated states with lowest logistic regression model p values; pink: forward and reverse primer binding sites, arrows indicate the directionality of the primers; grey: ambiguously called nucleotides (taken from dbSNP153); red: CpG sites, numbered relative to the TSS
Due to the classification into distinct methylation domains, we were able to systematically search for CpGs that reliably differentiate between methylated and unmethylated samples. Using logistic regression models, we computed p values for different combinations of CpGs and used this value as an indication for potentially new MSP primer designs (Fig. 4B). Lowest p values were obtained when forward primers were placed on CpG + 7 to CpG + 9 (p = 0.0016822, Additional file 2: Table S2, e1i1_domain2) and CpG + 15 to CpG + 17 (p = 0.0104365, Additional file 2: Table S2, e1i1_domain3). CpG + 10 and CpG + 11 together provided one of the lowest p values (p = 0.000128); however, the combined CpG + 7 to CpG + 11 (p = 0.003051) sequence stretch performed worse in comparison with CpG + 7 to CpG + 9 alone. The most significant CpGs for the binding of reverse primers could be found encompassing CpG + 18 to CpG + 20 (p = 0.006266, Additional file 2: Table S2, e1i1_domain4) and CpG + 26 to CpG + 29 (p = 0.0002321, Additional file 2: Table S2, e1i1_domain5). Because of an annotated common SNP (minor allele frequency > 1%) between CpG + 28 and CpG + 29, we recommend CpG + 26 to CpG + 28 as potential primer binding sites (p = 0.000777, Additional file 2: Table S2, e1i1_domain5).
When testing different MSP primer pairs, the best performance was obtained with the forward primer covering CpG + 15 to CpG + 17 and the reverse primer covering CpG + 26 to CpG + 28 (Fig. 4B). In fact, MSP on unmethylated peripheral blood (PBL) DNA and methylated EpiTect Control DNA (Qiagen) revealed the discriminative power of the proposed primer pair (Additional file 3: Fig. S1). Because of limited sample material, only 23 GBMs could be analyzed with the new primers resulting in 20/24 and 17/20 (83% and 85%) concordant results with pyrosequencing and LDBS, respectively (Additional file 1: Table S1 “Tierling MSP”).
Investigation of DNA methylation upstream of the MGMT promoter
Since the prominent promoter CpG island (CGIp, Fig. 1) extends 5′ beyond the so far analyzed region, we extended our analysis to a sequence section − 605 to − 353 bp relative to the transcriptional start site (TSS), called MGMTup, covering the 5′-portion of the CGIp (Fig. 1). Because of high CpG density and homopolymer stretches, the region between − 353 bp and the TSS was not amplifiable. In MGMTup, CpG-wise and sample-wise methylation were very heterogeneous. With the exception of samples #02, #07, #33 and #60, DNA methylation increased from a mosaic pattern (CpG − 47 to CpG − 61) towards highly methylated CpGs (CpG − 68 to CpG − 62) with increasing distance to the TSS (Additional file 4: Fig. S2a). Grouping MGMTup into different methylation domains similar to MGMTe1 and MGMTi1 revealed five different domains with two domains consisting of single CpGs (CpG − 67 and CpG − 68) (Additional file 4: Fig. S2a). CpGs with the highest discriminative power between lowly methylated (59) and highly methylated (13) samples were found in domain 5 (p < 7.74 × 10–5), namely CpG − 48 (10.07% ± 17% vs. 64.29% ± 32%), CpG − 52 (13.44% ± 21% vs. 60.63% ± 28%), CpG − 56 (17.85% ± 23% vs. 65.95% ± 20%), CpG − 49 (12.76% ± 19% vs. 53.28% ± 29%) and CpG − 51 (11.3% ± 19% vs. 56.22% ± 29%). Because of the rather high methylation level in the 59 lowly methylated samples, a diagnostic MSP assay with a positive/negative readout seems not to be feasible. Alternatively, quantitative methods like primer extension, qPCR or pyrosequencing assays focusing on CpG − 48 to CpG − 56 could be used. Comparing sample methylation levels in MGMTe1 with the other regions showed that lowly (< 9%), intermediately (9–30%) and highly methylated (> 30%) samples were, in tendency, proportionally higher methylated in MGMTup in comparison with MGMTe1 and MGMTi1 (Additional file 4: Fig. S2b). Interestingly, we observed a slight increase in methylation in MGMTi1 for the lowly methylated sample group (defined on the MGMTe1 methylation state) and a decrease for the intermediately and highly methylated sample group. This observation points to transcriptional downregulation of MGMT in glioblastoma being the result of higher activity of DNA methyltransferases in MGMTup and MGMTe1 compared to MGMTi1.
Association of CpG-wise methylation calls with progression-free survival
It was described that CpG methylation in the MGMT promoter region can be predictive for therapy outcome coupled with overall survival of glioblastoma patients [5, 6, 24,25,26,27,28]. Based on local deep bisulfite sequencing data, we performed Cox regression analysis on 50 samples (GBM without IDH mutation) excluding #18 and #23 which we regard as outliers because of their high progression-free survival (PFS, Additional file 1: Table S1). We did not find any CpG in MGMTe1 or MGMTi1 that significantly (p < 0.05) or in tendency (p < 0.1) correlated with progression-free survival. In MGMTup, we found CpG − 48, CpG − 61 and CpG − 64 close to p = 0.01 (Additional file 5: Table S3).
Using the average of all three CpGs as input to the Cox regression model, we found patients with low MGMTup methylation with significantly improved progression-free survival (PFS) (p = 0.04379) (Additional file 6: Fig. S3a). Analyzing 450 K data of 93 samples from The Cancer Genome Atlas (TCGA, data set “glioblastoma multiforme” excluding IDH1 mutants), we could not confirm any prognostic value of CpG − 48 which is represented as cg01341123 (p = 0.9056) (Additional file 6: Fig. S3b, for location compare Additional file 4: Fig. S2a). CpG − 47, CpG − 49 and CpG − 50, present on the array as cg25946389, cg23998405 and cg02022136, did also not show any predictive power with respect to progression-free survival (p = 0.6964, p = 0.8063 and p = 0.3582), respectively (data not shown, for location compare Additional file 4: Fig. S2a). Finally, we took a closer look on the methylation of CpG + 18 (cg12981137, compared Fig. 2), which was previously found by a logistic regression model to be predictive (together with cg12434587, which corresponds to CpG − 36, not present within our amplicons) for treatment outcome and prognosis in a test cohort of 63 glioblastomas and 450 K data taken from TCGA [13]. Neither local deep sequencing data of the here presented cohort nor the 450 K data set downloaded from TCGA could confirm the predictive quality of cg12981137 alone (p = 0.8671 and p = 0.9025, respectively, Additional file 6: Fig. S3c, d). We cannot exclude that adding local deep sequencing data of cg12434587 would enhance prognostic significance but at least doubt the informative power of cg12981137 as exclusive methylation marker in routine testing. Additionally, Bady and colleagues correlated the methylation status with the overall survival, while we used, due to limited OS data availability, progression-free intervals for a logistic regression [13]. While these two parameters are often correlated [18, 19], they do not necessarily show the same trend [20, 29].
Discussion
In this study, we characterized methylation of single CpG sites of the MGMT promoter in tumor samples from first neurosurgical intervention in a cohort of 69 glioblastoma multiforme (WHO grade IV) and 11 anaplastic astrocytoma (WHO grade III) patients, who subsequently received radiotherapy and temozolomide treatment. The transition from astrocytoma WHO grade III to glioblastoma multiforme often occurs as a smooth transition, so the classification as a WHO grade III astrocytic tumor may in some cases be due to a sampling error. In contrast to astrocytoma WHO grade II, astrocytic tumors WHO grade III and IV have a similarly high mitotic activity.
By using local deep bisulfite sequencing, we showed that for MGMTe1 DNA methylation distributed almost binary among the samples, a phenomenon already observed previously [30]. In MGMTi1 and MGMTup, methylation distribution was much more heterogeneous across samples and CpG sites. Overall, single CpG methylation levels tended to increase with distance to the TSS, an effect known for active genes and tumor suppressors in cancer [31, 32]. The detailed CpG-wise data collection enabled us to compare commonly used diagnostic assays such as pyrosequencing and MSP, with local deep bisulfite sequencing as the gold standard [16, 33], thus revealing that pyrosequencing data obtained with the PyroMark Q24 assay was highly correlated with local deep bisulfite sequencing results. This is in concordance with earlier studies confirming that pyrosequencing provides reliable quantitative and sensitive results in MGMT methylation detection [34,35,36,37,38]. However, this leaves the difficulty to decide on a cutoff value, i.e., for the average methylation of a limited number of CpGs in the MGMT promoter that should ideally reflect on the MGMT activity in the tumor, which is until now rather difficult to obtain. As clinically relevant cutoff, seven, eight and ten percent average methylation of CpGs analyzed by pyrosequencing have been suggested [39,40,41]. For the integrated neuropathological diagnosis, we usually follow the 8% cutoff recommended by Reifenberger et al. (2012) in concordance with the results obtained by both MSPs [41]. For local deep bisulfite sequencing, relevant cutoffs still need to be determined, which would require a larger study cohort. Yet, even with a suitable cutoff most neurosurgery/neuropathology laboratories, especially smaller ones, will probably not implement this method, due to a cost–benefit calculation and, even more importantly, the fact that collecting samples for one sequencing run would not comply with the delivery of results within a few workdays.
The question with any method to determine MGMT promoter methylation is whether the result really serves the patient, i.e., from what percentage of methylation on does the patient actually profit from a therapy with alkylating agents justifying the side effects. This applies especially in elderly patients and whenever an alternative treatment can be taken into consideration. Methylation-specific PCRs (MSPs) seem to produce a certain amount of unmethylated results that show methylation with quantitative methods (three cases in our cohort). One of our practical approaches to reduce false negatives, yet obtain a fast result during routine diagnostics, is the application of two MSP primer-sets on each sample. When both MSPs provide a methylated PCR product, we consider the sample as methylated. If one or both MSP primer pairs fail to produce a methylated PCR product, pyrosequencing immediately follows as the decisive factor for the MGMT promoter status. According to an international inter-laboratory study by Reifenberger et al. (2014), MSP was the most commonly used method for MGMT methylation status determination [41]. In our opinion, it still proves to be a cost-effective and comparatively easy method when being part of a diagnostic scheme with more than one method.
Comparing MSP results to local deep bisulfite sequencing revealed that primer design is crucial to reach sufficient sensitivity and specificity to reduce the number of false negative PCR results [42,43,44,45]. To avoid unreliable primer binding as suspected, e.g., the Felsberg reverse primer, we grouped CpG dinucleotides with similarly low or high methylation into methylation domains to select neighboring and discriminative CpGs within the same methylation domain. As a result, we proposed subsets of CpGs that could be part of optimized MSP primers in MGMTe1/i1 and for quantitative methylation assays like pyrosequencing, MethyLight [46] or single-nucleotide primer extension [47]. In addition, bisulfite profiling of MGMTup revealed discriminative CpG sites that could potentially be used in separate or supplementary quantitative assays to improve diagnostic and/or predictive power. Experimental testing of different primer combinations resulted in a new MSP primer pair with improved performance on a subset of our cohort. Application on larger independent cohorts will show if there is a benefit of new or adapted assays in diagnostic and/or predictive estimations.
Correlating patient’s clinical data to local deep bisulfite sequencing results showed that none of the CpGs in MGMTe1 and MGMTi1 was predictive for the duration of a progression-free survival in our sample cohort of primary glioblastoma without IDH mutations and treatment with radiotherapy and concomitant chemotherapy with temozolomide. This is in contrast to previous meta-analyses [28, 48, 49] and other recent studies [50,51,52,53]. However, studies with nonsignificant effect on survival data also exist [54, 55]. This may largely depend on how the progression-free survival is defined and detected. Routinely performed neuroimaging without neurological symptoms in set intervals may prepone the noted date of tumor remission in comparison with an onset of clinical aggravation. The actual location of the tumor in the brain, however, is crucial for the beginning of neurological symptoms. Massive tumor growth in an inconspicuous location may be overlooked for some time and artificially prolong the noted progression-free interval. In the present cohort, we had hardly any data on the overall survival of the patients. Also, the size of the cohort, immune cell composition, hot spot mutations or genomic aberrations within the group of glioblastoma multiforme may influence the outcome of survival analyses [56,57,58,59], possibly making them inconclusive.
We also found CpG + 18 methylation not to be meaningful towards prognosis in three different independent glioblastoma cohorts, which suggests that combinatorial analysis as shown by Bady and colleagues might improve survival analysis and biomarker detection significantly [13, 60]. In fact, for MGMTup we found three CpGs, which in combination showed significant correlation with PFS depending on their methylation level. We could not reveal a predictive effect of any of the single CpG methylation calls in our cohort. The predictive value of those CpGs needs to be evaluated in independent cohorts.
Conclusion
Taken together, our study shows that quantitative assays, like local deep bisulfite sequencing or pyrosequencing, provide reliable quantitative data of MGMT promoter methylation. We found a high correlation of bisulfite profiling with pyrosequencing. For pyrosequencing, cutoff values have been defined in other studies, while for local deep bisulfite sequencing relevant cutoffs still need to be determined using larger study cohorts. MSP data generation is fast and cost-effective, but CpGs, covered by the widely used Esteller and Felsberg MSP primers, group into different methylation domains, which results in significant numbers of negative MSP results that prove to be methylated with quantitative methods. In practice, a scheme involving more than one method and/or at least two primer pairs will improve the accuracy of the result. Together with bisulfite sequencing data of a newly identified region upstream of the MGMT promoter, MGMTup, we found neighboring CpGs within the same methylation domain being highly discriminative between unmethylated and methylated sequences, which were used to design a new MSP primer set with improved performance. Testing this primer set on larger sample cohorts will prove its suitability in routine diagnostics in the future.
Methods
Tumor sample cohort and DNA extraction
Tumor samples had been taken during the first surgical removal of the tumor (all but two in the department for Neurosurgery, Saarland University Medical Center in Homburg) and had been subsequently fixed in 4% buffered formaldehyde and embedded in paraffin for routine diagnostics. After the neuropathological diagnosis, we used the corresponding hematoxylin-/eosin-stained slide as reference to the 10-µm-thick FFPE sections for DNA preparation to choose tumor tissue parts without necrotic areas and isolated DNA using the QIAamp DNA Micro Kit (#56304, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. DNA yield and quality were determined using a NanoPhotometer® N60 (Implem GmBH, Munich, Germany). We admitted only patients with an astrocytic tumor grade III or IV that underwent the Stupp protocol [2], i.e., concomitant radiotherapy and temozolomide administration following surgery to our retrospective study. The study cohort includes 68 glioblastoma multiforme without proven IDH mutations, one glioblastoma with an R132C mutation and eleven anaplastic astrocytomas (grade III) with two having an IDH1 mutation (R132H and R132C) (see Additional file 1: Table S1). We determined the progression-free survival (PFS) by either neurological symptoms attributed to tumor remission or neuroimaging showing tumor recurrence. All relevant data regarding treatment and recrudescence were extracted from the patients’ records at the Saarland University Medical Center in Homburg (Saar), Germany. For subsequent analysis, we blinded all samples and kept only the mere necessary data such as progression-free survival (PFS), age at onset, gender and year of birth as shown in Additional file 1: Table S1 together with results for all applied methods. The project was approved by the ethics committee from the Aerztekammer des Saarlandes (No. 133/20).
Methylation-specific PCR (MSP) and Pyrosequencing
For MSP and pyrosequencing, we used 40 ng genomic DNA for bisulfite conversion using the EpiTect Fast Bisulfite Kit (#59824, QIAGEN, Hilden, Germany) according to manufacturer’s instructions. For each batch of samples, 20 ng of unmethylated human control DNA was also subjected to bisulfite conversion and the respective PCRs, in addition to bisulfite negative and positive bisulfite-treated controls (#59695 EpiTect PCR Control DNA Set, QIAGEN, Hilden) and non-template controls (Aqua bidest.).
To detect methylated and unmethylated bisulfite-converted DNA, we used two different primer sets. The first set, published by Esteller et al. (2000), amplifies an unmethylated fragment of 93 bp (primer sequences forward 5'-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3' and reverse 5'-AACTCCACACTCTTCCAAAAACAAAACA-3') and a methylated fragment of 89 bp (forward 5'-TTTCGACGTTCGTAGGTTTTCGC-3' and reverse 5´-GCACTCTTCCGAAAACGAAACG-3') [18]. The second set, published by Felsberg et al. (2009), amplifies an unmethylated fragment of 129 bp (primer sequences forward 5′-TGTGTTTTAGAATGTTTTGTGTTTTGAT-3′ and reverse 5′-CTACCACCATCCCAAAAAAAAACTCCA-3′) and a methylated fragment of 120 bp (primer sequences forward 5′-GTTTTTAGAACGTTTTGCGTTTCGAC-3′ and reverse 5′-CACCGTCCCGAAAAAAAACTCCG-3′) [19]. Our proposed MSP primer pair amplifies an unmethylated/methylated fragment of 126 bp (primer sequences forward 5′-GGTGC/TGTATC/TGTTTGC/TGATTTG-3′ and reverse 5′-CACCCG/AACG/AACG/AAAATAAAAAC-3′) using GoTaq Hot Start Green Master Mix (Promega) and 10 pmol of each primer in a 25 µl reaction. After an initial denaturation at 95° C for 2 min, 35 cycles of 95° C 45 s, 57° C 45 s and 72° C 30 s were applied with a final extension at 72° C for 5 min. The resulting PCR products were visualized with the LONZA FlashGel system using 2.2% gels (Flash Gel™ DNA cassettes, #57032, Biozym Scientific GmbH, Germany), a Flash Gel™ DNA marker (50–1.5 kb #57033) with the included camera for documentation. MSP results for the individual cases are documented in Additional file 1: Table S1 (0 = unmethylated; 1 = methylated).
For pyrosequencing, we applied the therascreen MGMT Pyro Kit (#971,061, QIAGEN, Hilden, Germany) using the PyroMark Q24 MDx according to manufacturer’s instructions and used the controls described above.
We determined isocitrate dehydrogenase (IDH) mutations for IDH1 (Arg 132) and IDH2 (Arg172) using the PyroMark Q24 MDx and a custom assay based on the publication by Thon et al. (2012) [61]. In brief, we used reagents from the PyroMark Gold Q24 Reagents (#970,802, QIAGEN, Hilden, Germany) according to manufacturer’s instructions with primers obtained from QIAGEN (IDH1 forward primer biotinylated 5′-biotin-GAAATCACCAAATGGCACCATAC-3′, reverse primer 5′-TTGCCAACATGACTTACTTGATCC-3′ and sequencing primer 5′-TGATCCCCATAAGCAT-3′; IDH2 forward primer biotinylated 5′-CATCCTGGGGGGGACTGT-3′, reverse primer 5′-ACCCTGGCCTACCTGGTCG-3′ and sequencing primer 5′-AGCCCATCACCATTG-3′).
Bisulfite amplicon preparation and sequencing
DNA obtained from the FFPE tissue sections was bisulfite-converted using the EpiTect Bisulfite Kit (QIAGEN, Hilden, Germany) or the EZ DNA Methylation Gold Kit (Zymo Res., Irvine, CA). Subsequently, three different PCRs were performed (4 µl bisulfite-treated DNA, 80 mM Tris–HCL, 20 mM (NH4)2SO4, 0.2% Tween-20, 2.5 mM MgCl2, 0.2 mM of each dNTP, 2.5U HotFirePol (Solis BioDyne, Tartu, Estonia)) using 250 pM of each primer (MGMTup: 5′-TTATTATAGGTTTTGGAGGTTGTT-3′, 5′-TACCTTTTCCTATCACAAAAATAAT-3′; MGMTe1: 5′-GGATATGTTGGGATAGTT-3′, 5′-ACCCAAACACTCACCAAAT-3′; MGMTi1: 5′-GATTTGGTGAGTGTTTGGGT-3′, 5′-AAACTAAACAACACCTAAA-3′) with Illumina universal adapter sequences attached at the 5′-end. To improve amplification efficiency, we added 0.2 µl Hot Start-IT Binding Protein (Thermo Scientific) to the MGMTup and MGMTi1 PCR reactions. PCRs were performed in a thermocycler starting with 15 min 95° C followed by 45 cycles 95° C 60 s, 54° C 75 s, 72° C 90 s and a 10-min final extension at 72° C. We purified amplicons using Agencourt AMPure XP beads (Beckman Coulter, Krefeld, Germany), and diluted, pooled and sequenced (v3 chemistry: 2 × 300 bp paired-end) them on the Illumina MiSeq following the manufacturer’s instructions.
Data evaluation and statistics
Sequencing reads were aligned to the reference sequence (Homo sapiens genome build GRCh37/hg19) and quality-filtered using the BiQ Analyzer HT program [62]. We conducted additional data analysis using custom R scripts (available at https://github.com/schmic05/MGMT_methylation) based on the aligned sequencing reads or the BiQ Analyzer HT output. Average DNA methylation levels for the CpGs were computed across the sequencing reads using BiQ Analyzer HT and used for visualization as methylation heatmaps. The samples were hierarchically clustered using the Euclidean distance and complete linkage on the CpG methylation values in exon 1.
We defined methylation domains in the amplicon sequencing data as adjacent CpGs that behave similarly across the samples within the amplicon. To that end, we first computed the mean and variance of all CpGs across the samples. Based on these values, we computed the difference between the mean and the variance between any two adjacent CpGs. According to the distribution of the differences in mean and variance of the methylation values, we defined the 90% quantiles as the threshold, where we set the border of each of the methylation domains for the three amplicons (MGMTup, MGMTe1, MGMTi1).
Based on the classification into these domains, we aimed to select those CpGs that reliably differentiate between the highly methylated and lowly methylated samples. First, we calculated the average DNA methylation value for all CpGs and samples within each of the domains. We termed those domains methylated that showed an average DNA methylation level of more than 50% and the others unmethylated. In the next step, we computed sample-wise average DNA methylation values for each of the domains individually and termed those samples as outliers per domain that showed an average DNA methylation more than two standard errors away from the mean. For the MGMTup region, we found higher differences across the samples and thus used four standard errors as the cutoff. For the unmethylated domains, methylated samples were termed outliers and vice versa. The information for each of the samples (outlier sample = 1, other samples = 0) was used as the output variable in a logistic regression model. We used the CpG-wise methylation values and all potential combinations of the CpG-wise methylation values as input variables in the regression models. Finally, we interpreted the p value of the individual logistic regression models (for each combination of CpGs) as an indicator of the discriminative power of these CpG-wise methylation states.
To determine CpGs that were associated with progression-free survival (PFS), we used the coxph() function from the survival R package [63] for all individual CpGs within the MGMTup, MGMTe1 and MGMTi1 amplicon. We used age and sex of the individuals as covariates.
Availability of data and materials
The data sets generated and/or analyzed during the current study and corresponding R scripts are available in the GitHub repository (https://github.com/schmic05/MGMT_methylation).
References
Kleihues P, Cavanee WK. Pathology and genetics of tumours of the nervous system. Lyon: IARC Press; 1998. https://doi.org/10.1002/1096-9896(200102)193:2<276::AID-PATH765>3.0.CO;2-Q
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. https://doi.org/10.1056/NEJMoa043330.
Nestler U, Lutz K, Pichlmeier U, Stummer W, Franz K, Reulen HJ, Bink A. Anatomic features of glioblastoma and their potential impact on survival. Acta Neurochir. 2015;157(2):179–86. https://doi.org/10.1007/s00701-014-2271-x.
Konar SK, Maiti TK, Bir SC, Kalakoti P, Bollam P, Nanda A. Predictive factors determining the overall outcome of primary spinal glioblastoma multiforme: an integrative survival analysis. World Neurosurg. 2016;86:341–348.e1–13. https://doi.org/10.1016/j.wneu.2015.08.078.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JEC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. https://doi.org/10.1056/NEJMoa043331.
Donson AM, Addo-Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer. 2007;48(4):403–7. https://doi.org/10.1002/pbc.20803.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20. https://doi.org/10.1007/s00401-016-1545-1.
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci. 1996;93(18):9821–6. https://doi.org/10.1073/pnas.93.18.9821.
Ronaghi M, Uhlén M, Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998;281(5375):363–5. https://doi.org/10.1126/science.281.5375.363.
Mansouri A, Hachem LD, Mansouri S, Nassiri F, Laperriere NJ, Xia D, Lindeman NI, Wen PY, Chakravarti A, Mehta MP, Hegi ME, Stupp R, Aldape KD, Zadeh G. MGMT promoter methylation status testing to guide therapy for glioblastoma: refining the approach based on emerging evidence and current challenges. Neuro Oncol. 2019;21(2):167–78. https://doi.org/10.1093/neuonc/noy132.
Hegi ME, Genbrugge E, Gorlia T, Stupp R, Gilbert MR, Chinot OL, Nabors LB, Jones G, van Criekinge W, Straub J, Weller M. MGMT promoter methylation cutoff with safety margin for selecting glioblastoma patients into trials omitting temozolomide: a pooled analysis of four clinical trials. Clin Cancer Res. 2019;25(6):1809–16. https://doi.org/10.1158/1078-0432.CCR-18-3181.
Pinson H, Hallaert G, van der Meulen J, Dedeurwaerdere F, Vanhauwaert D, van den Broecke C, van Dorpe J, van Roost D, Kalala JP, Boterberg T. Weak MGMT gene promoter methylation confers a clinically significant survival benefit in patients with newly diagnosed glioblastoma: a retrospective cohort study. J Neurooncol. 2020;146(1):55–62. https://doi.org/10.1007/s11060-019-03334-5.
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL, Mcdonald DR, Lacombe D, Stupp R, Delorenzi M, Hegi ME. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124(4):547–60. https://doi.org/10.1007/s00401-012-1016-2.
Bady P, Delorenzi M, Hegi ME. Sensitivity analysis of the MGMT-STP27 model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J Mol Diagn. 2016;18(3):350–61. https://doi.org/10.1016/j.jmoldx.2015.11.009.
Chai RC, Chang YZ, Wang QW, Zhang KN, Li JJ, Huang H, Wu F, Liu YQ, Wang YZ. A novel DNA methylation-based signature can predict the responses of MGMT promoter unmethylated glioblastomas to temozolomide. Front Genet. 2019;10:910. https://doi.org/10.3389/fgene.2019.00910.
Bock C, Halbritter F, Carmona FJ, Tierling S, Datlinger P, Assenov Y, Berdasco M, Bergmann AK, Booher K, Busato F, Campan M, Dahl C, Dahmcke CM, Diep D, Fernández AF, Gerhauser C, Haake A, Heilmann K, Holcomb T, The BLUEPRINT consortium. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34(7):726–37. https://doi.org/10.1038/nbt.3605.
Gries J, Schumacher D, Arand J, Lutsik P, Markelova MR, Fichtner I, Walter J, Sers C, Tierling S. Bi-PROF: bisulfite profiling of target regions using 454 GS FLX titanium technology. Epigenetics. 2013;8(7):765–71. https://doi.org/10.4161/epi.25242.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4. https://doi.org/10.1056/NEJM200011093431901.
Felsberg J, Rapp M, Loeser S, Fimmers R, Stummer W, Goeppert M, Steiger HJ, Friedensdorf B, Reifenberger G, Sabel MC. Prognostic significance of molecular markers and extent of resection in primary glioblastoma patients. Clin Cancer Res. 2009;15(21):6683–93. https://doi.org/10.1158/1078-0432.CCR-08-2801.
Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal PyrosequencingTM methylation analysis of CpG sites. Biotechniques. 2003;35(1):146–50. https://doi.org/10.2144/03351md01.
Håvik AB, Brandal P, Honne H, Dahlback HS, Scheie D, Hektoen M, Meling TR, Helseth E, Heim S, Lothe RA, Lind GE. MGMT promoter methylation in gliomas-assessment by pyrosequencing and quantitative methylation-specific PCR. J Transl Med. 2012;10:36. https://doi.org/10.1186/1479-5876-10-36.
Kim DC, Kim KU, Kim YZ. Prognostic role of methylation status of the MGMT promoter determined quantitatively by pyrosequencing in glioblastoma patients. J Korean Neurosurg Soc. 2016;59(1):26–36. https://doi.org/10.3340/jkns.2016.59.1.26.
Reifenberger G, Hentschel B, Felsberg J, Schackert G, Simon M, Schnell O, Westphal M, Wick W, Pietsch T, Loeffler M, Weller M. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer. 2012;131(6):1342–50. https://doi.org/10.1002/ijc.27385.
Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, Crooks D, Husband D, Shenoy A, Brodbelt A, Wong H, Liloglou T, Haylock B, Walker C. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer. 2009;101(1):124–31. https://doi.org/10.1038/sj.bjc.6605127.
Brandes AA, Tosoni A, Franceschi E, Sotti G, Frezza G, Amistà P, Morandi L, Spagnolli F, Ermani M. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation with MGMT promoter methylation status. J Clin Oncol. 2009;27(8):1275–9. https://doi.org/10.1200/JCO.2008.19.4969.
Sonoda Y, Yokosawa M, Saito R, Kanamori M, Yamashita Y, Kumabe T, Watanabe M, Tominaga T. O6-methylguanine DNA methyltransferase determined by promoter hypermethylation and immunohistochemical expression is correlated with progression-free survival in patients with glioblastoma. Int J Clin Oncol. 2010;15(4):352–8. https://doi.org/10.1007/s10147-010-0065-6.
Binabaj MM, Bahrami A, ShahidSales S, Joodi M, Joudi Mashhad M, Hassanian SM, Anvari K, Avan A. The prognostic value of MGMT promoter methylation in glioblastoma: a meta-analysis of clinical trials. J Cell Physiol. 2018;233(1):378–86. https://doi.org/10.1002/jcp.25896.
Chen Y, Hu F, Zhou Y, Chen W, Shao H, Zhang Y. MGMT promoter methylation and glioblastoma prognosis: a systematic review and meta-analysis. Arch Med Res. 2013;44(4):281–90. https://doi.org/10.1016/j.arcmed.2013.04.004.
Kanemoto M, Shirahata M, Nakauma A, Nakanishi K, Taniguchi K, Kukita Y, Arakawa Y, Miyamoto S, Kato K. Prognostic prediction of glioblastoma by quantitative assessment of the methylation status of the entire MGMT promoter region. BMC Cancer. 2014. https://doi.org/10.1186/1471-2407-14-641.
Vlassenbroeck I, Califice S, Diserens AC, Migliavacca E, Straub J, Di Stefano I, Moreau F, Hamou MF, Renard I, Delorenzi M, Flamion B, DiGuiseppi J, Bierau K, Hegi ME. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008;10(4):332–7. https://doi.org/10.2353/jmoldx.2008.070169.
Yang X, Gao L, Zhang S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief Bioinform. 2017;18(5):761–73. https://doi.org/10.1093/bib/bbw063.
Lin SY, Yeh KT, Chen WTL, Chen HC, Chen ST, Chiou HY, Chang JG. Promoter CpG methylation of tumor suppressor genes in colorectal cancer and its relationship to clinical features. Oncol Rep. 2004;11(2):341–8. https://doi.org/10.3892/or.11.2.341.
Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1(5):2353–64. https://doi.org/10.1038/nprot.2006.324.
Claus R, Wilop S, Hielscher T, Sonnet M, Dahl E, Galm O, Jost E, Plass C. A systematic comparison of quantitative high-resolution DNA methylation analysis and methylation-specific PCR. Epigenetics. 2012;7(7):772–80. https://doi.org/10.4161/epi.20299.
Dejeux E, Audard V, Cavard C, Gut IG, Terris B, Tost J. Rapid identification of promoter hypermethylation in hepatocellular carcinoma by pyrosequencing of etiologically homogeneous sample pools. J Mol Diagn. 2007;9(4):510–20. https://doi.org/10.2353/jmoldx.2007.060209.
Lee ES, Issa JP, Roberts DB, Williams MD, Weber RS, Kies MS, El-Naggar AK. Quantitative promoter hypermethylation analysis of cancer-related genes in salivary gland carcinomas: comparison with methylation-specific PCR technique and clinical significance. Clin Cancer Res. 2008;14(9):2664–72. https://doi.org/10.1158/1078-0432.CCR-07-1232.
Huntriss J, Woodfine K, Huddleston JE, Murrell A, Rutherford AJ, Elder K, Khan AA, Hemmings K, Picton H. Quantitative analysis of DNA methylation of imprinted genes in single human blastocysts by pyrosequencing. Fertil Steril. 2011;95(8):2564-2567.E8. https://doi.org/10.1016/j.fertnstert.2011.04.035.
Kishida Y, Natsume A, Toda H, Toi Y, Motomura K, Koyama H, Matsuda K, Nakayama O, Sato M, Suzuki M, Kondo Y, Wakabayashi T. Correlation between quantified promoter methylation and enzymatic activity of O6-methylguanine-DNA methyltransferase in glioblastomas. Tumor Biol. 2012;33(2):373–81. https://doi.org/10.1007/s13277-012-0319-1.
Johannessen LE, Brandal P, Myklebust TÅ, Heim S, Micci F, Panagopoulos I. MGMT gene promoter methylation status—assessment of two pyrosequencing kits and three methylation-specific PCR methods for their predictive capacity in glioblastomas. Cancer Genomics Proteomics. 2018;15(6):437–46. https://doi.org/10.21873/cgp.20102.
Kristensen LS, Michaelsen SR, Dyrbye H, Aslan D, Grunnet K, Christensen IJ, Poulsen HS, Grønbæk K, Broholm H. Assessment of quantitative and allelic MGMT methylation patterns as a prognostic marker in glioblastoma. J Neuropathol Exp Neurol. 2016;75(3):246–55. https://doi.org/10.1093/jnen/nlv024.
Reifenberger G, Malzkorn B, Acker T, Bettstetter M, Buslei R, von Deimling A, Dietmaier W, Dubbink HJ, Eigenbrod S, Garvalov BK, Gerstenmaier U, Giese A, Haase D, Hasselblatt M, Kirches E, Koch A, Marienfeld R, Mittelbronn M, Montesinos-Rongen M, Pagenstecher A, Riemenschneider MJ, Prinz M, Romeike B, Roos A, Spiegl-Kreinecker S, Schittenhelm J, Schlegel J, Thal DR, Tops BBJ, Weis J, Westphal G, Worm K, Felsberg J. Results of the international interlaboratory comparison of MGMT promoter methylation analysis involving twenty-three academic centers in Germany, Austria and The Netherlands. Neuro Oncol. 2014;16(suppl_3):iii49–50. https://doi.org/10.1093/neuonc/nou209.30.
Li LC, Dahiya R. Methprimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–31. https://doi.org/10.1093/bioinformatics/18.11.1427.
Brandes JC, Carraway H, Herman JG. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene. 2007;26(42):6229–37. https://doi.org/10.1038/sj.onc.1210433.
Chang MC, Chiang YC, Ho C-M, Chen YL, Chen CA, Cheng WF, Chou CY. New primers for methylation-specific polymerase chain reaction enhance specificity of detecting STAT1 methylation. Taiwan J Obstet Gynecol. 2012;51(1):43–9. https://doi.org/10.1016/j.tjog.2012.01.009.
Pandey RV, Pulverer W, Walter P, Kallmeyer R, Beikircher G, Pabinger S, Kriegner A, Weinhäusel A. MSRE-HTPrimer: a high-throughput and genome-wide primer design pipeline optimized for epigenetic research. Clin Epigenetics. 2016. https://doi.org/10.1186/s13148-016-0190-9.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. Methylight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8):e32–e0. https://doi.org/10.1093/nar/28.8.e32.
El-Maarri O, Herbiniaux U, Walter J, Oldenburg J. A rapid, quantitative, non-radioactive bisulfite-SNuPE-IP RP HPLC assay for methylation analysis at specific CpG sites. Nucleic Acids Res. 2002;30(6):e25. https://doi.org/10.1093/nar/30.6.e25.
Lee A, Youssef I, Osborn VW, Safdieh J, Becker DJ, Schreiber D. The utilization of MGMT promoter methylation testing in United States hospitals for glioblastoma and its impact on prognosis. J Clin Neurosci. 2018;2018(51):85–90. https://doi.org/10.1016/j.jocn.2018.02.009.
Yang H, Wei D, Yang K, Tang W, Luo Y, Zhang J. The prognosis of MGMT promoter methylation in glioblastoma patients of different race: a meta-analysis. Neurochem Res. 2014;39(12):2277–87. https://doi.org/10.1007/s11064-014-1435-7.
Ius T, Pignotti F, Della Pepa GM, Bagatto D, Isola M, Battistella C, Gaudino S, Pegolo E, Chiesa S, Arcicasa M, La Rocca G, Olivi A, Skrap M, Sabatino G. Glioblastoma: from volumetric analysis to molecular predictors. J Neurosurg Sci. 2020. https://doi.org/10.23736/S0390-5616.20.04850-X.
Alassiri AH, Alkhaibary A, Al-Sarheed S, Alsufani F, Alharbi M, Alkhani A, Aloraidi A. O6-methylguanine-DNA methyltransferase promoter methylation and isocitrate dehydrogenase mutation as prognostic factors in a cohort of Saudi patients with glioblastoma. Ann Saudi Med. 2019;39(6):410–6. https://doi.org/10.5144/0256-4947.2019.410.
von Rosenstiel C, Wiestler B, Haller B, Schmidt-Graf F, Gempt J, Bettstetter M, Rihani L, Wu W, Meyer B, Schlegel J, Liesche-Starnecker F. Correlation of the quantitative level of MGMT promoter methylation and overall survival in primary diagnosed glioblastomas using the quantitative MethyQESD method. J Clin Pathol. 2020;73(2):112–5. https://doi.org/10.1136/jclinpath-2019-206104.
Limam S, Missaoui N, Abdessayed N, Mestiri S, Selmi B, Mokni M, Yacoubi MT. Prognostic significance of MGMT methylation and expression of MGMT, P53, EGFR, MDM2 and PTEN in glioblastoma multiforme. Ann Biol Clin. 2019;77(3):307–17. https://doi.org/10.1684/abc.2019.1448.
Gutenberg A, Bock HC, Brück W, Doerner L, Mehdorn HM, Roggendorf W, Westphal M, Felsberg J, Reifenberger G, Giese A. MGMT promoter methylation status and prognosis of patients with primary or recurrent glioblastoma treated with carmustine wafers. Br J Neurosurg. 2013;27(6):772–8. https://doi.org/10.3109/02688697.2013.791664.
Jovanović N, Mitrović T, Cvetković VJ, Tošić S, Vitorović J, Stamenković S, Nikolov V, Kostić A, Vidović N, Krstić M, Jevtović-Stoimenov T, Pavlović D. The impact of MGMT promoter methylation and temozolomide treatment in serbian patients with primary glioblastoma. Medicina. 2019;55(2):34. https://doi.org/10.3390/medicina55020034.
Ma H, Zhao C, Zhao Z, Hu L, Ye F, Wang H, Fang Z, Wu Y, Chen X. Specific glioblastoma multiforme prognostic-subtype distinctions based on DNA methylation patterns. Cancer Gene Ther. 2020;27(9):702–14. https://doi.org/10.1038/s41417-019-0142-6.
Klopfenstein Q, Truntzer C, Vincent J, Ghiringhelli F. Cell lines and immune classification of glioblastoma define patient’s prognosis. Br J Cancer. 2019;120(8):806–14. https://doi.org/10.1038/s41416-019-0404-y.
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu X-Y, Fontebasso AM, Ryzhova M, Albrecht S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–37. https://doi.org/10.1016/j.ccr.2012.08.024.
Park AK, Kim P, Ballester LY, Esquenazi Y, Zhao Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019;21(1):59–70. https://doi.org/10.1093/neuonc/noy120.
Bernstock JD, Mooney JH, Ilyas A, Chagoya G, Estevez-Ordonez D, Ibrahim A, Nakano I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: clinical and translational implications. J Neurosurg. 2019;133(3):655–63. https://doi.org/10.3171/2019.5.JNS19364.
Thon N, Eigenbrod S, Kreth S, Lutz J, Tonn JC, Kretzschmar H, Peraud A, Kreth FW. IDH1 mutations in grade II astrocytomas are associated with unfavorable progression-free survival and prolonged postrecurrence survival. Cancer. 2012;118(2):452–60. https://doi.org/10.1002/cncr.26298.
Lutsik P, Feuerbach L, Arand J, Lengauer T, Walter J, Bock C. BiQ Analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res. 2011;39(Suppl_2):W551–6. https://doi.org/10.1093/nar/gkr312.
Therneau TM, Grambsch PM. Modeling survival data: extending the Cox model. Statistics for biology and health. New York: Springer; 2000. https://doi.org/10.1007/978-1-4757-3294-8.
Acknowledgements
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. We thank Sonja Hoffmann, Tatjana Pfander, Dalia Krauceviciene, Christina Lo Porto and Beate Schmitt for excellent technical assistance. We thank Dr. R. Weimann for sharing case material (Institute for Pathology, Winterberg 1, 66119 Saarbruecken).
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the German Epigenome Programme (DEEP) of the Federal Ministry of Education and Research in Germany (BMBF) (01KU1216F). MS is supported by the BMBF project de.NBI-epi (031L0101D) and the EU H2020 project SYSCID (733100).
Author information
Authors and Affiliations
Contributions
ST, WJW, and JBK contributed to conception and design; ST, WJW, JBK, and AL developed the methodology; AL, WJW, JBK, AW, DB, SU, CS, and JO contributed to acquisition of data (acquired and processed samples); ST, AL, WJW, JBK, and MS helped in analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis);: ST, WJW, JBK, AL, MS, WSS, and JW performed writing and review of the manuscript; SU, WSS, and JW supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The project was approved by the ethics committee from the Aerztekammer des Saarlandes (No.133/20). Consent to participate was collected for every patient in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Glioblastoma sample cohort with clinical monitoring data (blue), MSP (orange), PyroMark (yellow) and local deep sequencing (green) results; For MSP results, 1 equals the presence and 0 the absence of a methylated PCR product, respectively. Missing results are indicated with “-”. Discordant MSP results (Esteller vs. Felsberg primer pair; proposed primer pair vs. pyrosequencing or LDBS) are highlighted in light orange. Results obtained from the same tumor but two different DNA isolations are highlighted in grey. ø=average methylation.
Additional file 2: Table S2.
Average DNA methylation values, variances and p values for single CpGs and combinations of CpGs based on a logistic regression model; CpGs were analyzed methylation domain-wise, most significant neighboring CpGs, PyroMark and MSP-covered CpGs are highlighted in yellow, blue and red, respectively.
Additional file 3: Fig. S1.
MSP on human PBL and EpiTect Control methylated bisulfite-treated DNA with proposed primer pairs designed based on methylation domain modelling; electrophoretic separation of MSP reactions on a LONZA FlashGel system using 2.2% gels (Flash GelTM DNA cassettes, #57032, Biozym Scientific GmbH, Germany); Marker = Flash GelTM DNA marker (50bp–1.5kb #57033), PBL = peripheral blood leukocytes, bisulfite-converted DNA, EpiTect = EpiTect Control DNA (human), methylated and bisulfite-converted (Qiagen #59655); NTC = no template control.
Additional file 4: Fig. S2.
(A) DNA methylation heatmap of all CpGs in the MGMTup amplicon in malignant astrocytoma samples; CpGs are numbered relative to the TSS; grey lines represent missing data for the respective sample; CpGs present on the 450K/850K(EPIC) BeadChip arrays are indicated on top. CpGs grouped into five methylation domains (D1-D5) are highlighted in different colors with D1 and D2 represented by single CpGs (CpG -68 and CpG -67). Samples were ordered according to the sample clustering in MGMTe1 for clarity reasons. (B) Scatter plot with trend lines and confidence intervals based on local regression analysis; each dot represents the averaged DNA methylation of a sample per sequenced amplicon. Dots are colored based on the average methylation state in the exon 1 region: red=highly methylated (>30%), purple=intermediately methylated (between 9% and 30%), blue= lowly methylated (<9%).
Additional file 5: Table S3.
P values obtained for Cox regression survival analysis for each CpG in MGMTup, MGMTe1 and MGMTi1; 1st sheet: all samples; 2nd sheet: statistical outliers #18 and #23 were excluded from analysis.
Additional file 6: Fig. S3.
Kaplan–Meier survival plots including p values obtained from Cox regression model analysis and confidence intervals (blue/red); (A) average DNA methylation of CpG -48, CpG -61 and CpG -64, (B) TCGA 450K array-based DNA methylation of CpG -48 (cg01341123), (C) DNA methylation of CpG +18 in the presented cohort, (D) TCGA 450K array-based DNA methylation of CpG +18 (cg12981137).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tierling, S., Jürgens-Wemheuer, W.M., Leismann, A. et al. Bisulfite profiling of the MGMT promoter and comparison with routine testing in glioblastoma diagnostics. Clin Epigenet 14, 26 (2022). https://doi.org/10.1186/s13148-022-01244-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01244-4