
Abstract
Background
Wolf-Hirschhorn syndrome is caused by distal deletion of the short arm of chromosome 4 (4p-). We report a case in which intrauterine growth restriction, hypospadias and foot deformity were detected by prenatal ultrasound examination at 29 weeks of gestation.
Case Presentation
A 31-year-old gravida 2 partus 1 woman was referred at 29 weeks' gestation with suspicion of intrauterine growth restriction. Sonographic examination revealed deformity of the right lower limb and undescended testes with an irregular distal penis. A cordocentesis was performed and chromosome analysis revealed a 46,XY,del(4)(p14) karyotype.
Conclusion
The prenatal detection of intrauterine growth restriction, hypospadias and foot deformity should lead doctors to suspect the presence of Wolf-Hirschhorn syndrome.
Similar content being viewed by others


Background
Wolf-Hirschhorn syndrome (WHS) is a well-known chromosomal disorder first described by Cooper and Hirschhorn in 1961 [1]. Since the first clinical description, more than 120 cases have been reported [2]. It is attributable to partial loss of material from the short arm of chromosome 4, with the majority of cases (87%) being de novo deletions of preferential paternal origin.
WHS is characterized by intrauterine growth restriction, mental retardation, characteristic facial dysmorphism, microcephaly, ear lobe anomalies and closure defects (cleft lip or palate, coloboma of the eye, and cardiac septal defects) [3].
Prenatal diagnosis of Wolf-Hirschhorn syndrome has been reported in fetuses karyotyped because of routine indications of chromosomal analysis or intrauterine growth restriction with or without associated anomalies [4]. We report a case in which congenital hypospadias and clubfoot was detected prenatally at 29 weeks' gestation in association with intrauterine growth restriction.
Case presentation
A 31-year-old gravida 2 partus 1 woman was referred at 29 weeks' gestation with suspicion of intrauterine growth restriction. The couple were healthy, nonconsanguineous, with unremarkable medical history. There was no family history of congenital defects. The woman denied cigarette smoking, use of alcohol, illicit drugs or medication, in addition to any intrauterine teratogenic or infectious exposure. Sonographic examination revealed a single live fetus. Fetal biparietal diameter, abdominal circumference and femur length measurements were compatible with 25 weeks' gestation. Amniotic fluid index was 15 cm. Doppler velocimetry of umbilical artery was normal. A 7.3 cm placental thickness was measured.
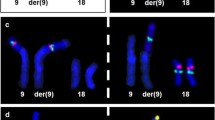
Examination of the fetal anatomy revealed a deformity of the right lower limb and undescended testes with an irregular distal penis (Figure 1,2). The parents were informed and a cordocentesis was performed. The karyotype analysis revealed a deletion of the short arm of chromosome 4, [46,XY,del(4)(p14)] (Figure 4).

Sonographic appearance of fetal genitalia. Note the irregular distal portion of fetal penis and undescended testes.

Ultrasound scan showing the deformity of foot.

Note the deletion of short arm of the chromosome 4
A FISH study to confirm the terminal deletion revealed a partial monosomy for 4p 15.2→4pter (Figure 5). After genetic counseling the family elected for termination of the pregnancy. Labor was induced and the woman delivered a stillborn male fetus weighing 900 g. The baby had prominent glabella, short philtrum, low-set ears, hypospadias, undescended testes and calcaneovalgus deformity of the right foot (Figure 3). Except for a large hydropic placenta, no additional anomaly was noted at autopsy. Chromosomal analysis of the woman revealed a normal karyotype (46,XX). Father refused a chromosomal analysis but he has 3 healthy children from his former spouse.

Photograph of the neonate with the Wolf-Hirschhorn syndrome. Note the hypospadias and calcaneovalgus deformity of the foot.

Fluorescent in situ hybridization. Chromosome 4 is green and the other chromosomes are blue. Neither of the chromosome 4 do not carry an extra piece of DNA.
Discussion
Wolf-Hirschhorn syndrome, caused by partial deletion of the short arm of chromosome 4, is characterised by severe growth restriction and mental defect, microcephaly, 'Greek helmet' facies, and midline fusion defects. The critical zone for development of this disorder is located distal to the Huntington disease-linked G8 (D4S10) marker [5]. Quarrel et al., [6] found that there was de novo deletion or rearrangement of 4p; in cases where the abnormality had arisen on the paternal chromosome. However, a paternal age effect was not observed. Goodship et al., [7] described a 2-year-old who presented with developmental delay and subtle dysmorphic features suggesting WHS: hypertelorism, prominent glabella, short philtrum, and carp-shaped mouth. Although high-resolution chromosomal analysis was normal in the child and in both parents, molecular analysis indicated that the child had not inherited a maternal allele for probes from 4p16. Fluorescence in situ hybridization (FISH) in the mother showed a submicroscopic translocation between chromosome 4 and 10. Chen et al [8] reported on the prenatal diagnosis of two sib female fetuses with a satellited short arm of chromosome 4 and a male fetus with a satellited long arm of chromosome X. The first two fetuses had a cryptic balanced translocation t(4;15)(p16;p11.1) inherited from a mother carrying a satellited 4p and having an affected child with the Wolf-Hirschhorn syndrome.
A family was reported with a balanced chromosomal translocation t(4;18)(p15.32;p11.21) in the father and an unbalanced translocation resulting in partial monosomy 4 and partial trisomy 18 in one living boy and a prenatally diagnosed male fetus. Both showed abnormalities consistent with WHS and had in addition aplasia of one umbilical artery [9].
WHS, a relatively common malformation pattern, has been exceptionally diagnosed in fetuses. Prenatal detection of abnormal male genitalia has been accomplished by ultrasonography. Hypospadias is one of the most common anomalies of the male genital tract. During the eighth to tenth week of embryologic development, incomplete fusion of the urethral folds results in an abnormal opening on the ventral surface of the penis. Hypospadias is classified as glandular, penile and scrotal type according to the location of the ectopic urethral meatus. It frequently occurs in isolation, although it can be associated with other anomalies in 8 per cent of cases. There is a recurrence risk of between 4 and 10 per cent. The prenatal diagnosis of hypospadias has been reported in fetuses with a family history [10]. Vinals et al. [11] documented the prenatal detection of congenital hypospadias in a case of Wolf-Hirschhorn syndrome.
Hypospadias is a common feature of several genetic syndromes: XXY and XXXXY and also associated with chromosomal abnormalities such as triploidy, trisomies 13 and 18. Furthermore it can be associated with anomalies of the urinary tract including exstrophy of the bladder, undescended testes, bifid glans, megalourethra and transposition.
Genetic syndromes such as Smith-Lemli-Opitz syndrome, Robinow syndrome, Fraser syndrome, Schprintzen syndrome, short-rib polydactyly syndrome and WHS may be associated with hypospadias [11].
Prenatal diagnosis of cases of monosomy 4p associated with severe IUGR has been reported. Cleft-lip and palate, diaphragmatic hernia, cystic hygroma and heart defect with right ventricular hypoplasia were detected by prenatal ultrasonography. At autopsy, midline fusion defects were observed in fetuses with WHS ranging from minor abnormalities such as scalp defect, hypertelorism, pulmonary isomerism, common mesentery, hypospadias and sacral dimple, to cleft palate, corpus callosum agenesis, ventricular septal defect and diaphragmatic hernia. Hypotrophic placentas with vascular lesions and a large placental chorioangioma have been reported previously [12]. In our case a large otherwise normal placenta was noted.
While a number of prenatally detected anomalies have been reported with WHS, to our knowledge, talipes calcaneovalgus has not been documented before [13].
Calcaneovalgus deformity is characterized by lateral deviation and eversion of the sole of the foot. Congenital club foot encompasses a wide spectrum of severity. It may arise from intrinsic factors, but it is frequently the consequence of restriction of fetal movements. It also commonly found in many syndromes and malformation clusters [14]. A variety of skeletal anomalies were found in patients with WHS, including split hand, clinodactyly, club feet, scoliosis and kyphosis, malformed toes, finger-like appearance of thumb, long slender fingers and delayed bone age [15].
Subtle abnormalities on ultrasound may suggest a chromosomal problem. Standard cytogenetics cannot always demonstrate a microdeletion. High-resolution banding and molecular analysis can help to confirm the diagnosis [16]. The prenatal detection of intrauterine growth restriction, hypospadias and clubfoot should raise the suspicion of WHS. Fetal skeletal and genital abnormalities detected by ultrasound should prompt the search for associated anomalies and karyotyping should be considered in fetuses with intrauterine growth restriction especially with a normal amniotic fluid volume.
References
Cooper H, Hirschhorn K: Apparent deletion of short arms of one chromosome (4 or 5) in a child with defects of midline fusion. Hum Chrom Newsl. 1961, 4: 14-16.
Verloes A, Schaaps JP, Herens C, Soyeur D, Hustin C, Dodinval P: Prenatal diagnosis of cystic hygroma and chorioangioma in the Wolf-Hirschhorn syndrome. Prenat Diagn. 1991, 11: 129-132.
Lurie IW, Lazjuk GL, Ussova I, Presman EB, Gurevich DB: The Wolf-Hirschhorn syndrome. I. Genetics. Clin Genet. 1980, 17: 375-384.
Snijders RJ, Sherrod C, Gosden CM, Nicolaides KH: Fetal growth retardation associated malformations and chromosomal abnormalities. Am J Obstet Gynecol. 1993, 168: 547-555.
Altherr MR, Bengtsson U, Elder FFB, Ledbetter DH, Wasmuth JJ, McDonald ME, et al: Molecular confirmation of Wolf-Hirschhorn syndrome with a subtle translocation of chromosome 4. Am J Hum Genet. 1991, 49: 1235-1242.
Quarrel OWJ, Snell RG, Curtis MA, Roberts SH, Harper PS, Shaw DJ: Paternal origin of the chromosomal deletion resulting in Wolf-Hirschhorn syndrome. J Med Genet. 1991, 28: 256-259.
Goodship J, Curtis A, Cross I, Brown J, Emslie J, Wolstenholme J, et al: A submicroscopic translocation, t(4;10), responsible for recurrent Wolf-Hirschhorn syndrome identified by allele loss and fluorescent in situ hybridisation. J Med Genet. 1992, 29: 451-454.
Chen CP, Devriendt K, Chern SR, Lee CC, Wang W, Lin SP: Prenatal diagnosis of inherited satellited non-acrocentric chromosomes. Prenat Diagn. 2000, 20: 384-9. 10.1002/(SICI)1097-0223(200005)20:5<384::AID-PD817>3.0.CO;2-2.
Kohlschmidt N, Zielinski J, Brude E, Schafer D, Olert J, Hallerman C, et al: Prenatal diagnosis of a fetus with a cryptic translocation 4p;18p and Wolf-Hirschhorn syndrome (WHS). Prenat Diagn. 2000, 20: 152-5. 10.1002/(SICI)1097-0223(200002)20:2<152::AID-PD738>3.0.CO;2-P.
Devesa R, Munoz A, Torrents M, Conmas C, Carrera JM: Prenatal diagnosis of isolated hypospadias. Prenat Diagn. 1998, 18: 779-788. 10.1002/(SICI)1097-0223(199808)18:8<779::AID-PD348>3.0.CO;2-3.
Vinals F, Sepulveda W, Selman E: Prenatal detection of congenital hypospadias in the Wolf-Hirschhorn (4p-) syndrome. Prenat Diagn. 1994, 14: 1166-1169.
Tachdjian G, Fondacci C, Tapia S, Huten Y, Blot P, Nessmann C: The Wolf-Hirschhorn syndrome in fetuses. Clin Genet. 1991, 42: 281-287.
Thompson P: Wolf-Hirschhorn syndrome. Review of the literature and three case studies. J Am Podiatr Med Assoc. 1998, 88: 192-197.
Romero R, Pilu G, Jeanty P, ed, et al: Prenatal Diagnosis of Congenital Anomalies, Appleton & Lange: Norwalk. 1988, 65-67.
Battaglia A, Carey JC, Cederholm P, Viskochil DH, Brothman AR, Galasso C: Natural history of Wolf-Hirschhorn: experience with 15 cases. Pediatrics. 1999, 103: 830-836.
De Keersmaecker B, Albert M, Hillion Y, Ville Y: Prenatal diagnosis of brain abnormalities in Wolf-Hirschhorn (4p-) syndrome. Prenat Diagn. 2002, 22: 366-70.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2393/3/1/prepub
Acknowledgments
Written consent was obtained from the patient or their relative for publication of the patient's details.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Aslan, H., Karaca, N., Basaran, S. et al. Prenatal diagnosis of Wolf-Hirschhorn syndrome (4p-) in association with congenital hypospadias and foot deformity. BMC Pregnancy Childbirth 3, 1 (2003). https://doi.org/10.1186/1471-2393-3-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2393-3-1