
Abstract
Background
Mutations in the transcription factor gene PAX6 have been shown to be the cause of the aniridia phenotype. The purpose of this study was to analyze patients with aniridia to uncover PAX6 gene mutations in south Indian population.
Methods
Total genomic DNA was isolated from peripheral blood of twenty-eight members of six clinically diagnosed aniridia families and 60 normal healthy controls. The coding exons of the human PAX6 gene were amplified by PCR and allele specific variations were detected by single strand conformation polymorphism (SSCP) followed by automated sequencing.
Results
The sequencing results revealed novel PAX6 mutations in three patients with sporadic aniridia: c.715ins5, [c.1201delA; c.1239A>G] and c.901delA. Two previously reported nonsense mutations were also found: c.482C>A, c.830G>A. A neutral polymorphism was detected (IVS9-12C>T) at the boundary of intron 9 and exon 10. The two nonsense mutations found in the coding region of human PAX6 gene are reported for the first time in the south Indian population.
Conclusion
The genetic analysis confirms that haploinsuffiency of the PAX6 gene causes the classic aniridia phenotype. Most of the point mutations detected in our study results in stop codons. Here we add three novel PAX6 gene mutations in south Indian population to the existing spectrum of mutations, which is not a well-studied ethnic group. Our study supports the hypothesis that a mutation in the PAX6 gene correlates with expression of aniridia.
Similar content being viewed by others


Background
Aniridia is a human congenital eye malformation with a population frequency of 1 in 60,000–100,000 [1]. Mutations in the transcription factor gene PAX6 cause blindness [2] through a spectrum of ocular manifestations among which aniridia and most probably foveal hypoplasia are the major signs [3–5].
The human PAX6 gene located on chromosome 11p13, was isolated by positional cloning as a candidate gene for aniridia [6]. Most of the point mutations in this gene will lead to premature truncation of protein [7]. Aniridia occurs due to decreased dosage of the PAX6 gene, which controls early events in the morphogenesis of the eye and brain [8, 9]. It exists in both sporadic and familial forms. One third of the aniridia cases are sporadic and two thirds are familial [3]. The sporadic cases can be part of WAGR syndrome (Wilms' tumor, aniridia, genitourinary anomalies, mental retardation), which is caused by hemizygous deletions of the PAX6 and WT1 genes. The PAX6 gene spans 22 kb in size and consists of 14 exons and 13 introns. The translation initiation codon is in exon 4 and the termination codon is in exon 13 [8]. The human PAX6 gene has paired box and homeobox motifs and encodes a highly conserved protein of 422 amino acids among metazoans. The function of the PAX6 protein is crucial for the development of human eye. The paired domain and the homeodomain are the two DNA binding domains, which are separated by a linker segment (LNK) and followed by a C-terminal region rich in proline, serine and threonine (PST). The presence of two DNA binding domains in PAX6 strongly suggests that the protein functions as a transcription factor to regulate the expression of other genes during embryogenesis [10, 11].
Mutations in the PAX6 gene have been reported in a growing number of aniridia patients and patients with other ocular malformations [12, 5]. The mutations are scattered throughout the gene. The vast majority of mutations reported so far are nonsense mutations, frameshift mutations or splicing errors that are predicted to cause premature truncation of the PAX6 protein, causing haploinsufficiency [12].
Even though the PAX6 gene has been studied in various ethnic populations, Indian aniridia mutations are not well studied. For this reason we decided to study PAX6 variations in an Indian population. Here we report five coding mutations of which three are novel mutations in sporadic cases of aniridia in south Indian population. Our study supports the hypothesis that the mutations in the human PAX6 gene appear to correlate with classic aniridia phenotype.
Methods
Clinical evaluation
This was done with informed consent and in accordance to the tenets of the Declaration of Helsinki. Twenty-eight members of six unrelated aniridia families and 60 healthy normal controls were recruited for this study. Examination included slit lamp, gonioscopy, IOP measurement and biomicroscopy.
Mutation screening and sequence analysis
Total genomic DNA was isolated by the salt precipitation method from peripheral blood [13]. The amplification of exons 4–13 to detect PAX6 gene mutations was carried out in a 20 μl reaction mixture containing 100 ng of genomic DNA, 100 pM of each primer, 1X PCR buffer (Promega USA) and 0.5 U of Taq DNA polymerase (Promega USA). The coding region of the human PAX6 gene was amplified with previously published primers [8]. PCRs were carried out in a MJ Research thermocycler for 35 cycles, consisting of 1 min at 95°C for denaturation, 1 min at 58°C for primer annealing and 10 min at 72°C for primer extension. Single strand conformation polymorphism analysis was carried out in 10% polyacrylamide with or without 2.5% glycerol as previously described [14]. The PCR products were sequenced to confirm the exact nucleotide change by ABI prism (Microsynth, Switzerland).
Allele specific clones
The superimposed mutant PCR products were amplified and cloned into the pGEM-T vector (Promega USA). Each transformed colony contains the mutant allele or the normal allele [15]. Exon-specific PCR-SSCP was performed from the transformed colonies. Two PCR reactions were performed in parallel. In the first reaction the 5' primer is complementary to the wild type sequence and in the second reaction the 5' primer is complementary to the mutant sequence. The elongation occurs only when primers and target sequence match completely, so only one allele of either mutant or wild type DNA is amplified [16, 17]. The plasmids were used as templates in amplification to generate PCR products for SSCP. The SSCP banding pattern was used to select allele-specific clone for sequencing (data not shown). The restriction digestion was performed for exon 6 with RsaI enzyme to differentiate between mutant and normal allele and visualized on a 2.5% agarose gel.
Results
We used the SSCP technique to search for mutations in exons 4–13 of the PAX6 gene (OMIM 106210) in probands with aniridia and normal controls. The clinical information for each sporadic aniridia patient is given in Table 1. Altered mobility shifts detected by SSCP were confirmed by repeating two or three times. All the mutants we describe here (except two that were analysed by direct sequencing) were cloned in to the pGEM-T vector followed by allele-specific PCR to pick up the correct clone using the plasmid DNA as template because the genomic PCR gave superimposed signals. Sequencing reactions were then carried out. Although this plasmid based SSCP analysis is time consuming, it reliably allows identification of each allele. The mutant sequences are deposited in Genbank [18]. (AY307164, AY289493, AY337853, AY337852, AY342394) and also in the PAX6 Allelic Variant database [19]. The summary of our findings is given in Table 2.
Novel mutations in the PAX6gene
In proband 28–1 the plasmid-based clones were sequenced, revealing an insertion with duplication of five bases at position 715 in codon 118 (c.715ins5), which resulted in a novel frameshift mutation in PAX6 gene. The phenotypic expression of aniridia is shown in Fig. 1A. The pedigree and the SSCP pattern and the unusual band shift detected in exon 6 is given in Fig. 2A. The insertion of 5 bases duplicates the cDNA sequence (Genbank M93650) from bases 711–715 of PAX6 gene (Fig. 3A). De novo change was observed at position 715 in the proband, which is a site for most common reported mutations [20–22]. The PCR product of 300 bp was digested with RsaI enzyme, which resulted in a 220 bp and 80 bp products in normal whereas the mutant gives 220 bp and 85 bp (data not shown).

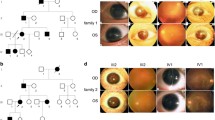
Phenotypic expression of aniridia with PAX6 gene mutation. The aniridic probands showed typical features of sclerocornea with nystagmus in proband 28–1 (A); Foveal hypoplasia in proband 27–1 (B); Ptosis, microcornea with dislocated cataractous lens in proband 10–1 (C); Ectopia lentis in proband 16–1 (D).

The pedigree and SSCP analysis of aniridia probands. Pedigree shows the affected (arrow) and unaffected individuals to the corresponding SSCP gel; unaffected family members (1,2,3), filled square (male) and filled circle (female) are affected members. N- unrelated healthy controls. Arrow indicates the mobility shift in SSCP gel.

Electropherogram showing the mutant and normal controls of PAX6 gene. Chromatogram showing the sequence variations in PAX6 gene is indicated by arrow. Plasmid based sequencing revealed c.715ins5 in 28–1 (A), c.1201delA in 27–1 (B), c.901delA in 10–1 (C) and IVS9-12C>T in 18–1 (F). Direct sequencing revealed c.482C>A in 21–1 (D), and c.830G>A in 16–1 (E), N-Normal control, Box- duplication of sequence.
The phenotypic expression of proband 27–1 is shown in Fig. 1B. The patient showed an abnormal band shift when compared with the unaffected family members in SSCP analysis (Fig. 2B). The plasmid-based sequencing results showed a deletion of single base at position 1201; codon 280 (c.1201delA) in PST region is shown in Fig. 3B. The deletion of adenosine residue at exon 10 predicts the premature truncation at exon 12 alters the conservation of amino acids from serine at codon 280. The proband had another unique change, A>G at position 1239 (c.1239A>G), 38 bases downstream of the deletion on the same allele in exon 10 (data not shown). The two changes were identified by allele specific PCR with repeated sequencing of different clones, since the direct sequencing of genomic PCR gave superimposed signals. Two changes on one allele have been previously reported [23].
The morphology of aniridia due to deletion in the LNK region is shown in Fig. 1C. In proband 10–1, an obvious bandshift on the SSCP gel was seen (Fig. 2C), which on sequencing (plasmid-based) revealed the mutation c.901delA in the coding region of exon 8 at codon 180 (Fig. 3C). The single adenosine residue deletion results in a frameshift mutation, generating a stop codon within the LNK region. The consequences of this mutation would be to produce a truncated protein, lacking the homeobox domain.
The PCR-SSCP followed by direct sequencing revealed a nucleotide substitution in proband 21–1 (Fig. 2D). The heterozygous transversion (C>A) was observed at position 482 (c.482C>A), which disrupts the cysteine (TGC) to stop (TGA) at codon 40 (C40X) in exon 5 in the paired domain (Fig. 3D). As a consequent, the nonsense mutation C40X produced a truncated protein, lacking the LNK, homeobox and PST regions.
Another nonsense mutation (W156X), which was detected in proband 16–1, resulted in expression of aniridia phenotype (Fig. 1D). The SSCP gel analysis (Fig. 2E), and the chromatogram revealed G>A transition at position 830 (c.830G>A) which changes the tryptophan (TGG) to stop (TGA) at codon 156 in LNK region (Fig. 3E). We report these nonsense mutations for the first time in a south Indian population, although they have been reported before in French population [24, 25].
PAX6gene polymorphism in aniridia
SSCP analysis of exon 10 in the family of proband 18–1 is shown in Fig. 2F. The sequencing analysis showed an intronic change at position -12 of intron 9 (IVS9-12C>T) is shown in Fig. 3F. This polymorphism has been previously reported in Indian [26] and Caucasian [27] populations. Even though the mutation falls in the splice acceptor site of exon 9 it seems to be a neutral polymorphism. The unaffected parents have normal sequences in all the exons of PAX6 gene.
Discussion
Mutations of the PAX6 gene are known to cause many cases of inherited and sporadic aniridia. The PAX6 gene is clearly vital for the normal development of the eye [28]. The phenotype presumably results from heterozygous insufficiency such that if only one copy of the gene is active insufficient protein is produced to support normal development of the eye. The protein products of the mutant genes could theoretically have DNA binding activity and compete with the wild type protein. However, the fact that aniridia caused by intragenic mutation is phenotypically indistinguishable from aniridia caused by deletion of entire PAX6 gene makes it likely that the mutant alleles are in fact null alleles [4, 25].
The pathological mutations will be found throughout the gene including the promoter and the other regulatory regions. In these patients all the mutations if translated, would result in truncated protein products. The translated proteins would be unable to carry out normal PAX6 function because functional domains are deleted. The two DNA binding domains are important for the normal activity of the protein [7, 10]. Proband 27–1 with c.1201delA also has another change, c.1239A>G downstream of the deletion, however its impact on transcriptional product is unknown. The downstream mutation may be less important because it is likely that the other mutation (c.1201delA) is the one responsible for aniridia. Though the two nonsense mutations C40X and W156X were previously reported in other population, we report for the first time in aniridic patients in south Indian population and thus it could be a recurrent mutation.
The change in the splice acceptor region of intron 9 (IVS9-12C>T) is a neutral polymorphism that has been observed before and the C>T substitution in the splice acceptor consensus sequence is not predicted to have any effect on function [29]. In addition, other studies with this change in patients with aniridia have a clear pathological mutation elsewhere in the gene [19]. It is likely that the patient 18–1 might have a pathological mutation elsewhere in the PAX6 gene, perhaps in one of the cis regulatory sequences.
Analysis of PAX6 mutants showed that the pathogenicity effects appear to be loss-of-function mutations, which leads to aniridia (Fig. 1) and other variable phenotypes [25]. The mutations showed varying phenotypic severity of the disease. The proband with c.715ins5 mutation showed the presence of sclerocornea with nystagmus in both eyes. A similar phenotype (Foveal hypoplasia) was observed in probands with c.1201delA and c.482C>A. Proband 10–1 with c.901delA revealed ptosis, microcornea with dislocated cataractous lens. Marfan syndrome and ectopia lentis was observed in both eyes of the proband 16–1 with c.830G>A. Even though Marfan syndrome is rarely associated with aniridia [3] we observed the syndrome in the proband. The peripheral corneal pannus with bullous keratopathy and corneal ectasia was observed in IVS9-12C>T. It is worthwhile to note that all the mutations observed in this study are associated with common phenotypes.
Our data provide evidence that some of the PAX6 gene mutations identified in aniridia patients lead to disruption of PAX6 gene expression due to the premature termination and most of which are assumed to cause loss of activity of one allele [30]. The Nonsense mutations would be predicted to result in truncated proteins due to a mechanism called 'nonsense-mediated decay' which degrades mRNAs containing premature stop codons (nonsense mutations). Therefore, PAX6 mRNAs that contain nonsense mutations would be degraded and would not be translated [31]. Identifying new mutations contributes valuable information for carrier detection for specific effect mutation and genetic counseling.
Conclusions
In summary we add three novel frameshift mutations, two nonsense mutations and a polymorphism to the existing spectrum of PAX6 mutations in south India in patients with the characteristic phenotype of aniridia. The two nonsense mutations are identified for the first time in south Indian population, which is not a well-studied ethnic group. Our genetic analysis provides further evidence that haploinsuffiency of the PAX6 gene causes the classic aniridia.
References
Shaw MW, Falls HF, Neel JV: Congenital aniridia. Am J Hum Genet. 1960, 12: 389-415.
Hanson I, van Heyningen V: Pax6: More than meets the eye. Trends Genet. 1995, 11: 268-272. 10.1016/S0168-9525(00)89073-3.
Nelson LB, Spaeth GL, Nowinski TS, Margo CE, Jackson L: Aniridia: a review. Surv Ophthalmol. 1984, 28: 621-642.
Crolla JA, van Heyningen V: Frequent chromosome aberrations revealed by molecular cytogenetic studies in probands with aniridia. Am J Hum Genet. 2003, 71: 1138-1149. 10.1086/344396.
Schroeder HW, Orth U, Meyer-Konig E, Gal A: Hereditary foveal hypoplasia – clinical differentiation. Klin Monatsbl Augenheilkd. 2003, 220: 559-562. 10.1055/s-2003-41874.
Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M, Royer-Pokora B, Collins F, Swaroop A, Strong LC, Saunders GF: Positional cloning and characterization of a paired-box and homeobox-containing gene from the aniridia region. Cell. 1991, 67: 1059-1074. 10.1016/0092-8674(91)90284-6.
Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL: PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994, 7: 463-471.
Glaser T, Walton DS, Maas RL: Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992, 2: 232-239.
Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, Moore AT, van Heyningen V: PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet. 2001, 28: 214-216. 10.1038/90042.
Epstein JA, Glaser T, Cai J, Jepeal L, Walton DS, Maas RL: Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 1994, 8: 2022-2034.
Chi N, Epstein JA: Getting your Pax straight: Pax proteins in development and disease. Trends Genet. 2002, 18: 41-47. 10.1016/S0168-9525(01)02594-X.
Prosser J, van Heyningen V: PAX6 mutations reviewed. Hum Mutat. 1998, 11: 93-108. 10.1002/(SICI)1098-1004(1998)11:2<93::AID-HUMU1>3.3.CO;2-J.
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res. 1988, 16: 1215-
Neethirajan G, Hanson IM, Krishnadas SR, Vijayalakshmi P, Anupkumar K, Sundaresan P: A novel PAX6 gene mutation in an Indian aniridia patient. Mol Vis. 2003, 9: 205-209.
Nollau P, Wagener C: Methods for detection of point mutations: performance and quality assessment. IFCC Scientific Division, Committee on Molecular Biology Techniques. Clin Chem. 1997, 43: 1114-1128.
Gibbs RA, Nguyen PN, Caskey CT: Detection of single DNA base differences by competitive oligonucleotide priming. Nucelic Acids Res. 1989, 17: 2437-2448.
Wu DY, Ugozzoli L, Pal BK, Wallace RB: Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc Natl Acad Sci USA. 1989, 86: 2757-2760.
Entrez Nucleotide. [http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide]
Human PAX6 Allelic Variant Database. [http://pax6.hgu.mrc.ac.uk/Tables/tableall.htm]
Gronskov K, Rosenberg T, Sand A, Brondum-Nielsen K: Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet. 1999, 7: 274-286. 10.1038/sj.ejhg.5200308.
Sonoda S, Isashiki Y, Tabata Y, Kimura K, Kakiuchi T, Ohba N: A novel PAX6 gene mutation (P118R) in a family with congenital nystagmus associated with a variant form of aniridia. Graefes Arch Clin Exp Ophthalmol. 2000, 238: 552-558. 10.1007/s004170000124.
Malandrini A, Mari F, Palmeri S, Gambelli S, Berti G, Bruttini M, Bardelli AM, Williamson K, van Heyningen V, Renieri A: PAX6 mutation in a family with aniridia, congenital ptosis, and mental retardation. Clin Genet. 2001, 60: 151-154. 10.1034/j.1399-0004.2001.600210.x.
Azuma N, Hotta Y, Tanaka H, Yamada M: Missense mutations in the PAX6 gene in aniridia. Invest Ophthalmol Vis Sci. 1998, 39: 2524-2528.
Brown A, McKie M, van Heyningen V, Prosser J: The human PAX6 mutation database. Nucl Acids Res. 1998, 26: 259-264. 10.1093/nar/26.1.259.
Vincent MC, Pujo AL, Olivier D, Calvas P: Screening for PAX6 mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet. 2002, 11: 163-169. 10.1038/sj.ejhg.5200940.
Dharmaraj N, Reddy A, Kiran V, Mandal A, Panicker S, Chakrabarti S: PAX6 gene mutations and genotype-phenotype correlations in sporadic cases of aniridia from India. Ophthalmic Genet. 2003, 24: 161-165. 10.1076/opge.24.3.161.15607.
Axton R, Hanson I, Danes S, Sellar G, van Heyningen V, Prosser J: The incidence of PAX6 mutation in patients with simple aniridia: an evaluation of mutation detection in 12 cases. J Med Genet. 1997, 34: 279-286.
Singh S, Mishra R, Arango NA, Deng JM, Behringer RR, Saunders GF: Iris hypoplasia in mice that lack the alternatively spliced Pax6(5a) isoform. Proc Natl Acad Sci USA. 2002, 99: 6812-6815. 10.1073/pnas.102691299.
Krawczak M, Reiss J, Cooper DN: The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992, 90: 41-54.
Hanson IM, Seawright A, Hardman K, Hodgson S, Zaletayev D, Fekete G, van Heyningen V: PAX6 mutations in aniridia. Hum Mol Genet. 1993, 2: 915-920.
Byers PH: Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest. 2002, 109: 3-6. 10.1172/JCI200214841. Review
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/5/9/prepub
Acknowledgements
The authors are very much thankful to the participants in this study. This study was supported by a grant from the Indian Council of Medical Research (ICMR), New Delhi, India. Authors thank Dr. Isabel Hanson, University of Edinburgh, Molecular Medicine Centre for much helpful discussion, critical reading of the manuscript. Dr. Edwin Stone, Dept of Molecular Ophthalmology, University of IOWA, USA for the kindly gifting the primers. We thank Dr. VR. Muthukkaruppan, Director of Research and Dr. Reena Chandrashekhar, Sr. Scientist, Aravind Medical Research Foundation for their valuable suggestions. We also thank V.R. Muthulakshmi, T.P. Vasanthi, for technical assistance. Mrs. Jeyalakshmi for typing & Mr. R. Jeyakrishnan for photographic work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors' contributions
GN carried out the molecular genetic analysis. SRK, PV, and SS contributed for the clinical diagnosis of patients. PS conceived the study carried out the molecular analysis with GN and drafted the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Neethirajan, G., Krishnadas, S.R., Vijayalakshmi, P. et al. PAX6 gene variations associated with aniridia in south India. BMC Med Genet 5, 9 (2004). https://doi.org/10.1186/1471-2350-5-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-5-9