
Abstract
Background
Nested nucleic acid amplification tests are often thought too sensitive or prone to generatingfalse positive results for routine use. The current study investigated the specificity and clinicalutility of a routine multiplex nested assay for mucosal herpetic infections.
Methods
Ninety patients, categorised into those clinically diagnosed to (a) have and (b) not haveherpetic infection, were enrolled. Swabs from oral and ano-genital sites were assayed by thenested assay and culture and the results assessed against clinical evaluation for diagnosingherpetic infections; cell content was also recorded.
Results
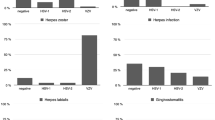
Twenty-six and 64 patients were thought to (a) have and (b) not have mucosal herpeticinfection. Taking the clinical evaluation as indicating the presence of herpetic infection, thenested polymerase chain reaction and culture had respective sensitivities of 19/26 (73%) and12/26 (46%) (Χ2 p = 0.02). There was no significant difference in specificities between nPCR62/64 (97%) and culture 63/64 (98%) (Χ2 p = 1.0). Cell content was important for viraldetection by nPCR (Χ2 p = 0.07) but not culture. Nesting was found necessary for sensitivity anddid not reduce specificity. Assay under-performance appeared related to sub-optimal cellcontent (20%) but may have reflected clinical over-diagnosis. The results suggest the need forvalidating specimen cell quality.
Conclusions
This study questions the value of routine laboratory confirmation of mucosal herpetic infection. The adoption of a more discriminatory usage of laboratory diagnostic facilities for genital herpetic infection, taking account of cell content, and restricting it to those cases where it actually affects patient management, may be warranted.
Similar content being viewed by others


Background
Herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) infections are among the commonest clinically encountered in medical practice and worldwide are the main causes of oral and genital ulceration [1–4]. HSV-2 is normally linked to genital ulceration but increasingly HSV-1 is also encountered and in Northern Ireland has become the predominant genital type recovered from women [5]. It is recognised that genital infections with either virus present with painful muco-cutaneous lesions in 95% of primary cases, and in 84% with non-primary episodes; constitutional upset accompanies 79% and 43% of primary and non-primary episodes respectively [6]. Although culture remains the gold standard for the direct diagnosis of HSV associated mucosal ulceration [7], increasingly HSV specific nucleic acid amplification test (NAT) assays are being recognised to improve test sensitivity [2, 8, 9]. Real-time assays are also becoming available [10–12] but are currently beyond the price range of many laboratories. We have previously reported a multiplex nested polymerase chain reaction (nPCR) assay for HSV detection, with significantly improved sensitivity when compared to standard virus culture [13]. However in routine practice there are two main concerns surrounding the use of a nested NAT format. First there is concern that the high sensitivity will result in the detection of clinically non-significant levels of infection, specifically asymptomatic shedding in the case of HSV. Secondly the assays are thought susceptible to generating false positive results [7, 14–16] and therefore will have an unacceptable level of specificity for routine practice. For these reasons we felt it necessary to validate the clinical performance of the multiplex nPCR assay we routinely use for HSV diagnosis. This study utilised the predictive value of clinical evaluation for diagnosing muco-cutaneous HSV infection by experienced medical personnel and was based in a genitourinary medicine clinic.
Methods
Patients
Over a 3 month period 90 patients who presented to the Department of Genitourinary Medicine, Royal Victoria Hospital, Belfast, were investigated for evidence of HSV infection. In addition to their routine assessment they were also selected for additional clinical categorisation by the attending consultants into those patients thought to (a) have, and (b) not have, clinical evidence in keeping with herpetic infection. Following a detailed history and examination patients with suspected herpetic infection were recorded as having : (i) primary HSV; (ii) first clinical episode of non-primary HSV; (iii) recurrent HSV. Those thought not to have herpetic infection involved patients with ulcerative or inflammatory lesions categorised on clinical examination as non-specific in nature.
Specimens
Swabs were taken from appropriate oral (2) and ano-genital sites (88) including penile, vulval, vaginal and anal specimens. The swabs were sent to the laboratory in 2 ml of viral transport medium consisting of phosphate buffered saline (PBS) pH 7.1, bovine serum albumin 7.5 μg/ml, penicillin G sodium 10 units/ml, streptomycin sulphate 10 μg/ml and amphotericin B 0.25 μg/ml. Each specimen was vortexed for 15 seconds on receipt in the laboratory to resuspend the cellular content, but otherwise received no further pre-treatment. A 50 μl volume was removed for virus culture and multiplex nPCR. A 1.5 ml volume of the remainder of the specimen was centrifuged in a screw-capped polypropylene tube (Sarstedt) at 15000 g for 10 minutes to assess the specimen cell content. Cell pellets of less than 2 mm in diameter were regarded as sub-optimal.
Virus culture
Virus culture was undertaken as reported previously [17]. Suspensions of 4 cell lines consisting of primary rhesus monkey kidney (CPHL, Porton Down, England), E6-vero (ATCC CRL1586), RD (ATCC CCL136) and HEp-2 (ATCC CCL23) were used. Four wells of 96 well U-bottomed tissue culture plates (Becton Dickinson, Oxford, England) were each inoculated with 10 μl of the specimen; all of the wells contained 90 μl of serum free Eagle's minimum essential medium (MEM) (Bio-Whittaker UK Ltd, Wokingham, England). Log dilutions from each inoculated well were made to 3 adjacent wells (in a column of 4) by the sequential transfer of 10 μl volumes. Each of the 4 cell suspensions (25 μl), in MEM with 10% fetal calf serum (Life Technologies, Paisley, Scotland, UK), was added to its corresponding column. The plates were incubated in a sealed box in a 5% CO2 atmosphere at 37°C for 7 days and read daily for the presence of cytopathic effect (CPE). Cells showing CPE were harvested and screened by the Syva MicroTrak HSV-1 and HSV-2 culture confirmation system (Behring, Milton Keynes, England).
Multiplex HSV-1 and HSV-2 nPCR
This was undertaken as previously described [13] using primers recognising the HSV-1 gpD gene and the HSV-2 gpG gene in a multiplex nPCR; HSV-1 and HSV-2 outer products were 221 and 184 base pairs (bp) while the inner products were 138 and 101 bp respectively. The assay has respective sensitivities of 0.01 and 0.1 TCID50 for HSV-1 and HSV-2. Briefly, hot-start was commenced in a Perkin Elmer GeneAmp 2400 thermal cycler with the cycling conditions as shown in Table 1. Specimens (10 μl) were added to 40 μl of mix and subjected to 35 first round cycles. One microlitre of product was transferred to 49 μl of second round mix for 25 second round cycles. A positive HSV control and a distilled water negative control were included in each run. First and second round products were visualised together on ethidium bromide-stained 2% agarose gels and photographed (Polaroid). Appropriately sized bands present on second round only, or present on both first and second rounds, were recorded as positive and strong positive respectively. All positive specimens were re-tested.
Statistical analysis
The clinical categorisation data for the patients was correlated with the results obtained by viral culture and nPCR. Difference in sensitivity and specificity between culture and nPCR and also the analysis of the impact of cell content on virus recovery in patients with clinical genital herpes infection versus those without evidence of HSV infection was determined by a two-tailed Chi-square (Χ2) test. Differences between herpes virus recovery by culture and nPCR in relation to the specimen cell content was compared using the Fisher exact test.
Results
Patients and specimens
Twenty-six of the 90 patients seen were thought on clinical grounds to have mucosal herpes while the remaining 64 were assessed as having non-specific lesions inconsistent with herpetic involvement. All patient specimens sent to the laboratory were received within 24 hours. Of the specimens received, 18/90 (20%) were found to be sub-optimal on the basis of a pellet size less than 2 mm in diameter after centrifugation.
Virus culture and nPCR
Taking the clinical diagnosis as the indicator of HSV infection, nPCR and culture had respective sensitivities of 19/26 (73%) and 12/26 (46%) (Χ2 p = 0.02) and specificities of 62/64 (97%) and 63/64 (98%) (Χ2 p = 1.0); all PCR positive specimens were repeatedly reactive. There was a similar percentage of specimens with sub-optimal cell content from patients with herpes infection diagnosed on clinical grounds compared to those without obvious evidence of HSV infection (4/26 (15%) versus 14/64 (22%) (Χ2 p = 0.48)). The cell content improved the identification of virus by nPCR in those patients with clinical herpetic infection (1/4 with poor cell content versus 17/22 with good cell content (Χ2 p = 0.07)) while this was not so for results obtained by viral culture (1/4 with poor cell content versus 11/22 with good cell content (Χ2 p = 0.59). The virus type by nPCR concurred with that isolated in all cases. There were 2 and 1 cases diagnosed by nPCR and culture respectively where the clinical diagnosis was not regarded as suggestive of HSV infection. HSV-2 was detected: (a) by nPCR and culture in a patient diagnosed with chronic candidiasis and (b) by nPCR alone in a second patient, diagnosed with an aphthous ulcer.
Discussion
The potential for amplicon contamination associated with NAT assay use is well recognised and makes mandatory the use of (a) separate, designated work stations and (b) a uni-directional work-flow from specimen preparation to amplified product analysis; this model of operation is standard in our laboratory . The increased sensitivity and additional amplification step of nested NAT assays is thought especially to be coupled with compromised specificity resulting from false positive results. Because of this they are commonly regarded as unsuitable for use in routine practice. This study was designed to address this belief. Its design relied on the predictive value of clinical judgement for diagnosing mucosal herpetic infection as the basis for estimating test specificity. In particular the setting of the study in a department of genitourinary medicine further strengthened the acumen of clinical diagnosis. False positive results, detection of asymptomatic shedding and clinical under-estimation of herpetic infection would be reflected by compromised assay specificity. The results confirmed high assay specificities with no significant difference between the nested NAT format and the current gold standard of virus culture (Χ2 p = 0.3). This provided convincing evidence of both (a) the appropriateness of the nested NAT assay's detection threshold for mucosal herpes infection and (b) its usefulness in a routine setting.
Two laboratory confirmed cases, clinically evaluated to lack evidence of herpetic infection, implied inappropriate detection in one, and/or a false positive result in the other; both cases involved HSV-2 detection. The first, treated as a chronic candidasis infection, was positive by both nPCR and culture (and also by immunofluorescence, though not part of this study). The second, treated clinically as a non-herpetic aphthous ulcer, was positive by nPCR alone. The 3 positive assays in the first case would indicate the infection was genuine, possibly obscured clinically by the presenting candidiasis . However it could also represent asymptomatic shedding of virus or atypical clinical presentation. In the second case the nPCR assay recorded a strong positive HSV-2 signal from the submitted specimen. Because the specimen was not extracted but reactive after direct addition into the first round reaction mix, we believe the likelihood of contamination at this step to be minimal. Also the detection was repeatable. In routine practice when clinical details and laboratory findings conflict we test a further specimen; a standard turn-around time of ≤ 24 hours for all NAT assays in our laboratory allows this approach.
While confirming the expected improvement in sensitivity ((19/26 (73%) and 12/26 (46%) (Χ2 p = 0.02)) of nPCR, the overall poor performance of both the nPCR and virus culture assays was unexpected. Since both have performed optimally in external quality assessment programs (NEQAS, run by the Public Health Laboratory Service for viral culture and the QCCA program run by the European Society for Clinical Virology for HSV PCR) we do not believe assay under-performance to be responsible. Additionally we have never encountered a specimen that yielded a virus on culture that was PCR negative, even without specimen extraction, indicating that PCR inhibitors are not involved or are very uncommon. A number of factors may have accounted for the observation. We believe the major impact was associated with specimens containing a sub-optimal cell content (20%). Our results indicated this had a bearing on the level of virus detected, especially by culture. Also it could be suggested that not pre-extracting the specimens may have compromised the nPCR assay. However from internal audit findings (unpublished) we have shown that pre-extraction does not increase the yield of HSV recovery from genital specimens with this assay. Finally, misdiagnosed herpetic infections may also have factored into the low yield. Against a recent report of syphilis increases [18] the potential for its misdiagnosis as atypical herpetic infection [19] is very real, underlying the importance of using tests of high sensitivity, which clearly do not include culture.
The results of this study indicated that a nested format of NAT assay was not only suitable for routine application but essential for achieving adequate sensitivity levels. The sensitivity levels of nested NAT formats therefore warrant their consideration by laboratories offering diagnostic facilities for herpetic infection. The other major advantage of this multiplex nested format is that it removes the necessity for pre-test extraction and automatically types the virus identified. Nesting with these primers has proven sufficient to overcome any inhibitors which would be expected in specimens of this nature and to date we have been unable through audit to provide evidence to support the need for specimen extraction, which for our other routine NAT assays is essential. However our results would suggest the need for validating specimen quality. For specimens sent in lysis buffer this is likely to require the quantitative ability associated with Real-time assays while for those in viral transport medium assessing cell content would suffice. While Real-time assays are becoming available they are expensive and are currently outside the cost range of many routine virology laboratories. In the meantime retention of culture for HSV diagnosis, or its replacement with standard PCR, will result in missed laboratory diagnosis and a compensatory over-reliance on uncorroborated clinical judgement.
More fundamentally these results bring into question the need for the laboratory confirmation of genital herpetic infection. Currently the laboratory confirmation and typing of herpetic involvement in genital herpes is recommended practice [20]. Our results indicate that laboratory results are adding little to clinical judgement in the majority of cases. They are at best typing the virus in approximately 70% of the cases investigated. While it is suggested that the virus type affects patient management through tailored counselling, it is questionable whether in the majority of cases the advice given will alter. By having a blanket approach to the use of laboratory methods for confirming herpetic infection, which constitutes a major component of most virus laboratories workload, scarce resources are being diverted from areas where they could be more useful, e.g. wider access to mutational analysis. We believe it is time to have a more discriminatory approach to the laboratory diagnosis of genital herpetic infection, restricting it to those cases where it actually affects patient management, and doing it well.
Conclusions
A nested amplification format for the laboratory diagnosis of mucosal herpetic infection was found to be necessary for adequate test sensitivity while not sacrificing test specificity. The unexpected poor sensitivity of both assays with comparison to clinical evaluation was probably related to the number of specimens with sub-optimal cell content (20%), but over-diagnosis of herpetic infection could have contributed to the findings. The results clearly suggest the need for validating specimen quality, either through cell count for specimens in viral transport medium or Real-time target quantification for those in lysis buffer. The results question the value of routine laboratory confirmation of mucosal herpetic infection. They suggest the need for a more discriminatory approach, taking account of the cell content, and restricting it to those cases where it actually affects patient management.
References
Beyrer C, Jitwatcharanan K, Natpratan C, Kaewvichit R, Nelson KE, Chen CY, et al: Molecular methods for the diagnosis of genital ulcer disease in a sexually transmitted disease clinic population in northern Thailand: predominance of herpes simplex virus infection. J Infect Dis. 1998, 178: 243-246.
do Nascimento MC, Sumita LM, de Souza VA, Pannuti CS: Detection and direct typing of herpes simplex virus in perianal ulcers of patients with AIDS by PCR. J Clin Microbiol. 1998, 36: 848-849.
Mertz KJ, Trees D, Levine WC, Lewis JS, Litchfield B, Pettus KS, et al: Etiology of genital ulcers and prevalence of human immunodeficiency virus coinfection in 10 US cities. The Genital Ulcer Disease Surveillance Group. J Infect Dis. 1998, 178: 1795-1798. 10.1086/314502.
Risbud A, Chan-Tack K, Gadkari D, Gangakhedkar RR, Shepherd ME, Bollinger R, et al: The etiology of genital ulcer disease by multiplex polymerase chain reaction and relationship to HIV infection among patients attending sexually transmitted disease clinics in Pune, India. Sex Transm Dis. 1999, 26: 55-62.
Christie SN, McCaughey C, McBride M, Coyle PV: Herpes simplex type 1 and genital herpes in Northern Ireland. Int J STD AIDS. 1997, 8: 68-69. 10.1258/0956462971918689.
Benedetti J, Corey L, Ashley R: Recurrence rates in genital herpes after symptomatic first-episode infection. Ann Intern Med. 1994, 121: 847-854.
Drake S, Taylor S, Brown D, Pillay D: Improving the care of patients with genital herpes. BMJ. 2000, 321: 619-623. 10.1136/bmj.321.7261.619.
Beards G, Graham C, Pillay D: Investigation of vesicular rashes for HSV and VZV by PCR. J Med Virol. 1998, 54: 155-157. 10.1002/(SICI)1096-9071(199803)54:3<155::AID-JMV1>3.0.CO;2-4.
Slomka MJ, Emery L, Munday PE, Moulsdale M, Brown DW: comparison of PCR with virus isolation and direct antigen detection for diagnosis and typing of genital herpes. J Med Virol. 1998, 55: 177-183. 10.1002/(SICI)1096-9071(199806)55:2<177::AID-JMV15>3.0.CO;2-F.
Espy MJ, Uhl JR, Mitchell PS, Thorvilson JN, Svien KA, Wold AD, et al: Diagnosis of herpes simplex virus infections in the clinical laboratory by LightCycler PCR. J Clin Microbiol. 2000, 38: 795-799.
Kessler HH, Muhlbauer G, Rinner B, Stelzl E, Berger A, Dorr HW, et al: Detection of Herpes simplex virus DNA by real-time PCR. J Clin Microbiol. 2000, 38: 2638-2642.
Schalasta G, Arents A, Schmid M, Braun RW, Enders G: Fast and type-specific analysis of herpes simplex virus types 1 and 2 by rapid PCR and fluorescence melting-curve-analysis. Infection. 2000, 28: 85-91. 10.1007/s150100050052.
Coyle PV, Desai A, Wyatt D, McCaughey C, O'Neill HJ: comparison of virus isolation, indirect immunofluorescence and nested multiplex polymerase chain reaction for the diagnosis of primary and recurrent herpes simplex type 1 and type 2 infections. J Virol Methods. 1999, 83: 75-82. 10.1016/S0166-0934(99)00108-1.
McDermott SS, McDermott PF, Skare J, Glantz M, Smith TW, Litofsky NS, et al: Positive CSF HSV PCR in patients with GBM: a note of caution. Neurology. 2000, 54: 746-749.
Hirsch HH, Bossart W: Two-centre study comparing DNA preparation and PCR amplification protocols for herpes simplex virus detection in cerebrospinal fluids of patients with suspected herpes simplex encephalitis. J Med Virol. 1999, 57: 31-35. 10.1002/(SICI)1096-9071(199901)57:1<31::AID-JMV5>3.0.CO;2-J.
Dumas AL, de Ancos E, Herbort CP: [Evaluation of the method of DNA amplification (PCR, polymerase chain reaction) for diagnosis of superficial ocular herpes],. Klin Monatsbl Augenheilkd. 1992, 200: 472-475.
O'Neill HJ, Russell JD, Wyatt DE, McCaughey C, Coyle PV: Isolation of viruses from clinical specimens in microtitre plates with cells inoculated in suspension. J Virol Methods. 1996, 62: 169-178. 10.1016/S0166-0934(96)02102-7.
Higgins SP, Sukthankar A, Mahto M, Jarvis RR, Lacey HB: Syphilis increases in Manchester, UK. Lancet. 2000, 355: 1466-
Graham D, Higgins SP: Patients with primary herpes should be screened for syphilis. BMJ. 2001, 322: 434-10.1136/bmj.322.7283.434.
National guideline for the management of genital herpes: Clinical Effectiveness Group (Association of Genitourinary Medicine and the Medical Society for the Study of Venereal Diseases). Sex Transm Infect. 1999, 75 Suppl 1: S24-S28.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/1/11/prepub
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Coyle, P.V., O'Neill, H.J., McCaughey, C. et al. Clinical utility of a nested nucleic acid amplification format in comparison to viral culture for the diagnosis of mucosal herpes simplex infection in a genitourinary medicine setting. BMC Infect Dis 1, 11 (2001). https://doi.org/10.1186/1471-2334-1-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-1-11