
Abstract
Background
The first high quality draft of the grape genome sequence has just been published. This is a critical step in accessing all the genes of this species and increases the chances of exploiting the natural genetic diversity through association genetics. However, our basic knowledge of the extent of allelic variation within the species is still not sufficient. Towards this goal, we constructed nested genetic core collections (G-cores) to capture the simple sequence repeat (SSR) diversity of the grape cultivated compartment (Vitis vinifera L. subsp. sativa) from the world's largest germplasm collection (Domaine de Vassal, INRA Hérault, France), containing 2262 unique genotypes.
Results
Sub-samples of 12, 24, 48 and 92 varieties of V. vinifera L. were selected based on their genotypes for 20 SSR markers using the M-strategy. They represent respectively 58%, 73%, 83% and 100% of total SSR diversity. The capture of allelic diversity was analyzed by sequencing three genes scattered throughout the genome on 233 individuals: 41 single nucleotide polymorphisms (SNPs) were identified using the G-92 core (one SNP for every 49 nucleotides) while only 25 were observed using a larger sample of 141 individuals selected on the basis of 50 morphological traits, thus demonstrating the reliability of the approach.
Conclusion
The G-12 and G-24 core-collections displayed respectively 78% and 88% of the SNPs respectively, and are therefore of great interest for SNP discovery studies. Furthermore, the nested genetic core collections satisfactorily reflected the geographic and the genetic diversity of grape, which are also of great interest for the study of gene evolution in this species.
Similar content being viewed by others

Background
The study of natural allelic diversity has proved fruitful in understanding the genetic basis of complex traits [1–6]. However, exploiting it successfully through association genetics requires basic knowledge of the extent of allelic variation within a species. One of the most interesting ways to achieve this goal consists of developing high-density diversity maps like the those developed in human and chicken, which allow the identification of causal polymorphisms for important traits [7–10]. The recent publication of the first high quality draft of the grapevine genome sequence opens the way to building such a diversity map [11]. Like in animals or in other perennial plant species where genetic approaches based on the study of segregating populations are hampered by a long biological cycle, association genetics is of particular interest in grapevine.
The development of diversity map relies on the discovery of sequence polymorphisms in the genome in a small set of genotypes that are as representative as possible of available genetic diversity. Such a concept was first proposed by Frankel and Brown under the name of core collection [12]. Core collections can be built using different types of markers. For example, molecular markers were used for rice, wheat and potato, while for yam a core collection was built using the origin of cultivars, eating quality, tuber shape, tuber flesh colour, and morphotype [13–16]. Different strategies have been proposed to assist the construction of core collections including the M-Method developed by Schoen and Brown and implemented in the software MSTRAT [17–20]. This strategy has been successfully used for the construction of core collections in Arabidopsis thaliana and Medicago truncatula and was also proposed as a preliminary step in association genetics [21–24].
Large collections of genetic resources are available for grapevine especially in Europe [25]. The largest one is held by INRA in France at the domain of Vassal: this collection contains 7000 accessions of Vitis genus of worldwide origin [26]. The genotyping of the whole collection using 20 well-scattered SSR markers is complete Laucou et al. (in prep). The cultivated compartment (V. vinifera L. subsp. sativa) is represented by 3900 accessions corresponding to 2262 unique genotypes (Laucou et al, in prep), from 38 different countries. It represents about a half of the known grapevine cultivars [27]. The Vassal collection was highly diverse for V. vinifera L. subsp. sativa, exhibiting a total of 326 alleles for the 20 SSRs markers with an average of 16.3 SSR alleles per locus (Laucou et al, in prep). Moreover a large proportion of these alleles (17%) were present at very low frequency (freq < 0.05%).
A first core collection (M-core) in grape was recently developed based on 50 morphological traits on 1759 accessions from the Vassal collection [28]. It was used for a preliminary study of the extent of linkage disequilibrium (LD) in V. vinifera L. as well as for association studies [28, 29]. However, the size of the M-core (141 individuals) limits its use for the analysis of wide sequence diversity.
Here we present the use of the data set obtained by Laucou et al. (in prep) to develop four nested genetic core collections (G-cores) suitable for the search for allelic diversity. The ability of retaining the SSR genetic diversity using different sample sizes was studied and compared to the SSR diversity present in the M-core and in the Vassal collection. Finally, the allelic diversity captured at the sequence level in the different sub-cores was analysed by sequencing three gene fragments. This work provides the foundation required for the development of a detailed map of haplotypic diversity in grapevine.
Results
Construction of nested core collections representing the available germplasm diversity of cultivated V. vinifera L
We first determined the optimal size of a core collection by retaining the 271 alleles showing a frequency above 0.05%. Forty-eight cultivars were necessary to capture 100% of the 271 alleles (Figure 1A). Within this core collection of 48 cultivars (G-48), we then determined the two most diverse samples of 12 (G-12) and 24 (G-24) cultivars (Table 1). In order to assess the robustness of these nested core collections, we calculated the percentage of identical varieties among the G-12, G-24 and G-48 core collections obtained in the second run using the same process, which corresponded to 83.3% (10) of the varieties selected in the two G-12 to 83.3% (20) of the varieties selected in the two G-24 and to 60.24% (29) of the varieties selected in the two G-48. Among these two sets of samples, the G-48 core collection presenting the highest Nei's index was chosen as the reference core collection (Table 2).

Redundancy curves obtained using MSTRAT software. Redundancy curves with standard deviation obtained using MSTRAT software (five independent samplings). Determination of the optimal size allowed the capture of all alleles of the original sample. A. For the 271 alleles of the restricted Vassal collection using the M-method (blue dot) and random sampling method (pink dot). B. For the 326 alleles of the Vassal collection using the G-48 core as core using the M-method (blue dot) and random sampling method (pink dot).
The G-48 core was used as a core to build the final core collection retaining the 326 alleles found in the cultivated compartment of Vitis vinifera L. represented in the Vassal collection by Laucou et al. (in prep). The optimal size of this final core collection was 92 individuals (Fig. 1B). The cultivars added at this step contained only rare alleles (freq < 0.05%, present on less than 3 copies), which corresponded to less choice for the selection of varieties. Indeed, only two alternative samples were proposed by MSTRAT, with only one individual differing between the two samples: Rich baba rose faux versus Kizil. Again, we selected the G-92 presenting the highest value for the Nei's index as the reference core collection for the cultivated compartment of V. vinifera L; the resulting final core collection is listed in Table 2.
In order to estimate the gain of SSR allelic diversity, we compared the number of alleles captured in samples obtained by the M-method and by random sampling. In each case, when using the M-method, we observed a gain (Table 3), the greatest of which being obtained for the selection of the G-48.
Analysis of the diversity retained in the nested core collections using different descriptors
The reference nested core collections for the cultivated compartment of Vitis vinifera were described for several characteristics and compared to the Vassal collection and to the M-core collection (141 individuals) defined by Barnaud et al. [28].
SSR diversity
The nested core collections represented 58% to 100% of the total SSR diversity of the Vassal collection and 70% to 100% of the restricted SSR diversity of the Vassal collection (only considering alleles with frequencies higher than 0.05%) (Table 1). All the SSR alleles with a frequency of more than 5% within the Vassal collection are present in the G-12 core and all those with a frequency of more than 3.5% within the Vassal collection are present in the G-24. The values of the unbiased Nei's diversity index and the level of unbiased observed heterozygosity for the G-12 core, G-24 core and G-48 core collection were quite similar and slightly higher than those calculated for the G-92 core collection. These values were slightly higher than those of the Vassal collection and of the M-core (Table 1). We also compared allele frequencies of the SSR markers in the three G-cores and in the M-core with the frequencies observed in the Vassal collection: the best correlation was obtained between the Vassal collection and the M-core (r2 = 0.98) and the G-92 (r2 = 0.92) core collections (Table 1).
Geographic origin and final uses
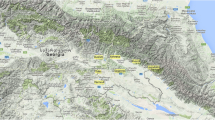
The definition of the true geographical origin of grapevine cultivars is sometimes difficult due to many migration events with humans [30]. Based on current knowledge, the cultivars held in the Vassal collection originated from 38 countries, with about half of them from Western Europe (France, Iberian Peninsula and Italy). The cultivars selected in the nested core collection originated from 27 different countries (Table 2). However 10 varieties of the G-92 sample could not be assigned to a precise geographical origin. Among them, 9 varieties were recent crosses between varieties from different countries (indicated by * in the Table 2) and one have an unknown origin (Moscatel de Oeiras faux). Moscatel de Oeiras faux microsatellite data seemed to indicate a Western Europe origin when compared to the whole collection. The origins of the 82 remaining varieties were well distributed (Figure 2): 21 (25%) came from the Caspian region (Dagestan, Georgia, Armenia and Azerbaijan) and the Middle East (Iran) which corresponds to the center of domestication, and 35 (42%) came from Western Europe and North Africa (Iberian Peninsula, Morocco, Algeria, Tunisia, Italy and France) (Table 4). Interestingly, five varieties (6% of the G-92) originated from Central Asia and Asia despite their very limited representation in the whole collection (less than 2%).

Probable geographic origin of the varieties contained in the nested genetic core collections. Each triangle corresponds to one variety, red triangles correspond to the first sub-sample of the nested genetic core collection (G-12), yellow triangles to the second sub-sample (G-24), black triangles to the third sub-sample (G-48) and green triangles to the fourth sub-sample (G-92). Ten varieties belonging to the Core G-92 did not have a precise geographical origin and are not shown on this map.
No differences were observed between the M-core (22 countries) and the Vassal collection (Table 4) whereas all the G-cores differed from the Vassal collection. Indeed the number of cultivars from Western Europe and the center of domestication were more balanced in the G-92 core with a very good representation of the whole set of geographic origins. The same trend was observed in the different sub-cores (Table 4).
We also compared the G-92 core collection, the M-core and the Vassal collection with respect to the final use of the cultivars: wine making (wine cultivars), fruit consumption (table cultivars) or both (wine/table cultivars). The M-core and the different G-cores all resembled the Vassal collection (Table 4).
Evaluation of the capture of unlinked diversity in the nested core collections
Next, we assessed the ability of the nested G-core samples to capture diversity unlinked to the SSR markers used to build the nested core collection. Barnaud et al. estimated using 38 SSR markers mapped on five different linkage groups (LG) with a maximum distance of 30 cM that LD in grape extends only within LG and is around 16.8 cM maximum [28]. We analysed the polymorphism of three gene fragments mapped further than 16.8 cM from the SSR markers in the same linkage group. DFR mapped in LG 18, 25.3 cM from the SSR marker VVIn16; L-DOX mapped in LG 8, 26 cM from the SSR marker VMC1b11 and BURP mapped in LG 3, 26 cM from the SSR marker VVMD28.
Forty-one nucleotide polymorphisms (40 substitutions and 1 in/del) were observed in the G-92, ranging from 12 to 15 depending on the gene fragment (Table 5). The total polymorphism is thus one SNP for 49 nucleotides. The number of SNPs per base also varied between the three gene fragments: one SNP for every 58 nucleotides for DFR, one SNP for every 42 nucleotides for L-DOX and one SNP for every 50 nucleotides for BURP. The difference of genetic diversity between coding and non coding region of the sequences was estimated only for the DFR sequence which has a quite similar length of the two types of regions. For this gene the polymorphism was different between coding and non-coding regions with a ratio of 3.2 (one SNP for every 127 nucleotides for coding region versus one SNP for every 39 nucleotides for non-coding region). Considering all genes together, the number of SNPs detected increased from 32 to 36 between the G-12 and the G-24 cores and from 36 to 40 between the G-24 and the G-48 cores. Only one more SNP was discovered in the G-92 core than in the G-48 core for the L-DOX gene fragment (this SNP is present in two varieties: Œil de Dragon and Badagui). The higher number of SNPs in the G-24 than in the G-48 cores was due to two genotypes: Yapincack with three additional SNPs in the DFR gene fragment and Kisilowy with one additional SNP in the L-DOX gene fragment.
Estimation of the ability to capture unlinked diversity of the G-24 core and G-12 core was performed by comparing their SNP diversity with SNP diversity in five random samples of 24 individuals in the G-48 core and 12 individuals in the G-24 core. The number of SNPs in the different random samples varied from 35 to 37 SNPs for the five random samples of 24 individuals and from 30 to 34 SNP for the five random samples of 12 individuals. In order to compare SNP distribution, we also calculated the unbiased Nei's index, which varied from 0.24 to 0.25 for the five random samples of 24 individuals and from 0.30 to 0.32 for the five random samples of 12 individuals. The unbiased Nei's index of the G-24 and G-12 cores was respectively 0.28 and 0.33.
Estimating the unlinked diversity within the whole Vassal collection (2262 cultivars) would have been very fastidious. Consequently we compared the capture of unlinked diversity in the nested core collections and in the M-core developed only on morphological traits. The total number of SNPs in the M-core (25 SNPs; Table 5) was smaller than in any of the nested G-core samples, even the G-12 core (32 SNPs; Table 5). Moreover, none of the SNPs observed in the M-core was new compared to those found in the nested core collections.
Discussion
In the present work, we developed a set of nested core collections from the cultivated compartment of the Vassal collection, using the M-method and SSR diversity data obtained on 2262 unique genotypes. However, in this way we did not take into account the somatic variants present within V. vinifera L. cultivated germplasm. The usefulness of core collections is due to their ability to capture the diversity of the whole species. Even the smallest nested core collections were more efficient in capturing allelic diversity than the M-core with its 141 accessions. The Vassal collection, which formed the basis of this work, includes around 3900 cultivars which correspond to 2262 unique SSR genotypes from 38 countries, including from the main domestication area. This represents more than half the varieties found world wide [27]. A core collection developed from Vassal collection is thus of major interest for the scientific community, and thanks to the vegetative propagation ability of grape, could be easily multiplied and distributed.
Construction of the core collections
The first result of our work is the fact that only a small number of cultivars (92 individuals, 4% of the Vassal collection) are needed to represent the whole diversity and an even smaller number of cultivars are needed to capture all the most frequent alleles (48 individuals, 2.1%). The comparison with other models is not easy, as they have different biological characteristics, the original collection did not reach the same global diversity of the species, and the analyses are seldom performed in the same way. Nevertheless, the core collections developed for A. thaliana (18%) or M. truncatula (31%) using the same method required higher percentages of individuals selected to represent all the genetic diversity [21, 22]. In our study we only considered the cultivated compartment which tends to be less diverse than wild compartments [31]. But the high level of heterozygosity of the grapevine is probably also one of the factors that allow a lower number of individuals than homozygous species like the two plant species mentioned above. Finally, the small number of individuals needed to represent the genetic diversity of the cultivated grapevine also pinpointed the high redundancy of the Vassal collection where many kingroups were highlighted and the interest in using such core collections to optimize the study of the phenotypic and genetic diversity in grapevine [32, 33].
Nested core collections are of great interest for identifying the sequence diversity that exists in the cultivated compartment of the V. vinifera species
The total genetic diversity revealed in the sequences of three gene fragments (2010 bp) in the G-92 core was quite high with 41 SNPs, i.e. one SNP for every 49 nucleotides. This is substantially higher than the level of genetic diversity observed in the M-core on the same gene set. Moreover, it is higher that the level of genetic diversity observed on an other set of 25 gene fragments totalling 12 kilobases sequenced on seven cultivated individuals (one SNP for every 118 nucleotides) by Salmaso et al. and on a set of 230 gene fragments, what represents the analysis of over 1 Mb of grape DNA sequence 11 grape genotypes (one SNP for every 64 nucleotides) by Lijavetzky et al. [34, 35]. This comparison thus emphasises the interest of such a core collection for the discovery of genetic diversity.
Among cultivated species, polymorphism in grape is relatively high compared to Zea mays (one SNP every 100 nucleotides), Pinus pinaster (one SNP for every 102 nucleotides) and Hordeum vulgare (one SNP for every 78 nucleotides), while it is relatively low compared to wild species such as A. thaliana (one SNP for every 32 nucleotides) [21, 36–38].
G-48 core is highly diverse and non-redundant
The G-92 core was built taking into account extremely rare alleles. Considering the rapid evolution of SSR markers, we assumed that the alleles present in two cultivars or less in the collection did not adequately represent gene diversity and they were thus removed when we built the G-48 core [39–41]. Indeed, only one additional SNP was revealed in the G-92 sample (present in two cultivars and not in the M-core) compared to the G-48, thus validating our assumption. On one hand, the gain in the unlinked diversity was high in the G-48, probably due to the decrease in redundancy compared to the Vassal collection (revealed by the number of kingroups). On the other hand, when compared to a random sampling, the gain was much higher using the M-method. The final G-48 core is highly non-redundant and highly diverse. Moreover the G-48 core optimized the unlinked diversity in the three different regions sequenced compared to the M-core, whose individuals coming from Vassal collection were not selected based on their genotypes, by consequent they could be consider as a less optimized sampling within the Vassal collection.
The G-12 and G-24 cores already include respectively 78% and 88% of the SNPs markers present in the G-92 core (80% and 90% of the G-48 core). They also include 58% and 73% of all the SSRs markers identified within the Vassal collection, representing a gain of 6% to 8% compared to random sampling from the G-48 or G-24 core. From a technical point of view, the size of the G-12 and G-24 cores is better suited for high throughput genomic studies and consequently highly suitable for ambitious projects of SNP discovery.
Geographic origin and final uses of the varieties within the G-core
Interestingly the nested core collections constructed in the present work reflect the distribution of grapes in Europe and around the Mediterranean Sea but with over-representation of the cultivars originating from the Caspian region and Middle East, and under-representation of the cultivars from Western Europe (Iberian peninsula, France and Italy) compared to the Vassal collection. We compared the SSR allele frequencies of the nested core collections and of the Vassal collection and found low correlations. This result further emphasizes the decrease in redundancy in the core collections compared with the Vassal collection, but also reflected the relative high number of cultivars originating from Western Europe in the Vassal collection, whereas the main domestication center is the Middle East [30]. These two regions may thus represent important sources of genetic diversity for the V. vinifera L. species. They represent the cradle of viticulture and the first migration of cultivars by Greeks and Etruscans, and a second domestication center in Western Mediterranean region [42]. Finally, despite their low representation in the Vassal collection, the presence of cultivars from Asia and Central Asia in the nested core collections could also indicate an underexploited center of diversification worthy of prospection and analysis.
The proportion of table varieties, wine varieties and table/wine varieties was very well conserved in the nested core collections compared to the M-core and to the Vassal collection. The distinction between these three categories of cultivars is based on morphological traits such as berry size, bunch size and compacity but also on other traits such as the sugar/acid balance at maturity [43]. Previous studies have shown that there is strong genetic differentiation between these three groups of varieties that may be due either to divergent selection based on the same gene pool or to the use of specific gene pools for the development of the three types of varieties [44, 27].
As a consequence, if the samples are well suited for analysis of allelic diversity, other uses can also be proposed for the cores, for example, the nested core collection could help understand the evolution of grape. Both G-12 and G-24 cores contained more frequent alleles representing ancient alleles while G-48 and G-92 may constitute subsequent diversification of cultivars in recent periods.
Conclusion
In the present work, we developed a set of robust nested core collections of V. vinifera L. (cultivated compartment) that will facilitate the discovery of allelic diversity by the scientific community. Moreover, this is an important basic tool for the development of projects of association mapping in grapevine. In conclusion, even if these nested core collections are statistically too small to study correlations between phenotype and nucleotide diversity, their use for preliminary tests of hypothesis will speed up the selection of suitable candidates (for example by discarding unsuitable candidates) and for SNP discovery. Due to the perennial nature of grape and the ease of vegetative propagation, these nested core collections could easily be disseminated worldwide for analyses (by simple request at request-vassal@supagro.inra.fr).
Methods
Plant material and DNA extraction
For each genotype of the four nested core collections, an accession of the Vassal collection (Domain de Vassal, Herault, France) was selected (Table 1) and a batch of young leaves was collected and lyophilized for long-term conservation. Lyophilized leaves were ground twice for 1 min at 20 Hz using a Qiagen-Retsch MM300 crusher. DNA was extracted using the Qiagen DNeasy Plant mini kit (Qiagen) following the manufacturer's instructions with minor modifications: addition of 1% w/v of PVP-40 to the AP1 solution, addition of 180 μl AP2 instead of 130 μl and an additional step of 10 minutes centrifugation at 6000 rpm after incubation on ice, which enabled the majority of the cellular remains and aggregates formed after the addition of AP2 to be pelleted.
Methods for the construction of the core collection
The dataset obtained by Laucou et al. (in prep) on the 2262 unique genotypes from the Vassal collection was used. The M-method proposed by Schoen and Brown and implemented in the MSTRAT software by Gouesnard at al. was used to generate the nested genetic core collections that maximize the number of observed alleles in the SSR data set [19, 20]. The efficiency of the sampling strategy was assessed by comparing the total number of alleles captured using MSTRAT in samples of increasing size with the number of alleles captured in randomly chosen collections of the same size (five independent samplings). After having determined the optimal size of the nested core collections, 200 core collections were generated independently for each sample size. Putative core collections exhibiting the same allelic richness (determined by the total number of alleles represented) were ranked using Nei's index as the second criterion [45].
PCR primer design
The gene sequences that were analysed were derived from three genes located on three separate chromosomes (Table 6). Two were involved in the anthocyanin metabolic pathway: the dihydroflavonol 4-reductase (DFR, gi 499017) present in one copy in the genome of V. vinifera L. and the leucoanthocyanidin dioxygenase (L-DOX gi 22010674) present at least in three copies in the genome of V. vinifera L. based on the NCBI database. The third gene codes for a BURP domain protein presenting a differential expression in a natural mutant of berry development compared to the wild type (VvBURP1; gi 22014825) [46–48]. Specific PCR primers (Table 2) were designed for the amplification of fragments of these three genes using Primer3 software and tested for amplification on the genomic DNA of the 12 individuals of the core G-12 [49].
PCR amplification, sequencing, sequence analysis and SNP detection
The 25 μl PCR reaction mixtures contained 20 ng of genomic DNA, 50 mM KCl, 10 mM TRIS-HCl (pH 8.3), 0.4 mM of each primer, 125 μM of each dNTP, 1.5 mM MgCl2 and 2.5 U of Taq polymerase (Qiagen). PCR amplifications were performed in a MJ Research PTC 100 Thermal Cycler programmed as follows: 5 min denaturation at 94°C, 35 cycles of 94°C for 30 s, 52°C for 45 s, and 72°C for 1 min, followed by an extension step at 72°C for 8 min. The PCR products were purified using the Agencourt AMPure method (Beckman Coulter) and directly sequenced in the two ways using the Big Dye Sequencing kit according to the manufacturer's specifications (Applied Biosystems Inc.). The sequence products were purified using the Agencourt CleanSEQ method (Beckman Coulter) and loaded onto an ABI PRISM® 3130 XL (Applera) capillary sequencer. The DNA sequences were analysed using the Staden Package [50]. Heterozygous SNPs were identified as double pics on the chromatograms and coded according to international codes (nucleotide codes of the International Union of Biochemistry). Insertion/Deletions were easily identified by overlapping sequences. Sequencing both strands enable to deal with such events. Only SNPs present on both forward and reverse sequences were validated.
Statistical analysis
Different indices were used in this study. The selection of reference core collections among those constructed using MSTRAT and exhibiting the same allelic richness (determined by the total number of alleles represented) was performed using Nei's index (Nei, 1987) as the second criterion. Nei's index is given for one locus by:INeij = 1- ∑ pij2where pijrepresents the i allele frequency of the jlocus. The Nei diversity index for all the loci is the sum of indices for each locus given by INei = ∑j INeij2. The more the allelic frequencies are equilibrated within a sample, the higher the value of Nei's index
As the samples compared were of different size (M-core, nested core collections and the whole collection) the comparison was performed using the unbiased observed heterozygosity and the unbiased Nei's index [45]. The unbiased Nei's index for the locus jis given by: HNeij = (2n/2n-1) (1- ∑ pij2) where nrepresents the number of individuals and where pijrepresents the iallele frequency of the jlocus, the unbiased Nei diversity index for all the loci studied is given by HNei = (1/C) ∑ jHNeij2where Cis the number of loci studied. The more the allelic frequencies are equilibrated within a sample, the higher the value of the unbiased Nei's index. The observed heterozygosity for the jlocus is given by Hobsj = 1- ∑ xij2where xij represents the homozygote frequency for iallele of the jlocus. The unbiased observed heterozygosity for the jlocus is: Hunobsj = (2n/2n-1) (1- ∑ xij2) >where n represents the number of individuals and the unbiased observed heterozygosity for all loci studied is H unobs = (1/C) ∑ jHunobsj2where Cis the number of loci studied.
We compared the SSR frequencies found in the M-core and the nested G core-collections with those of the Vassal collection using the R2 correlation coefficient. R2 is given by R2 = (Cov ij /σiσj) 2 where Covij is the covariance between the two samples compared and σi and σjare the variance of samples i and j respectively.
References
Roses AD: A model for susceptibility polymorphisms for complex diseases: apolipoprotein E and Alzheimer disease. Neurogenetics. 1997, 1: 3-11.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G: Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001, 411 (6837): 599-603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH: A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001, 411 (6837): 603-6.
Maloof JN, Borevitz JO, Dabi T, Lutes J, Nehring RB, Redfern JL, Trainer GT, Wilson JM, Asami T, Berry CC, Weigel D, Chory J: Natural variation in light sensitivity of Arabidopsis. Nat Genet. 2001, 29 (4): 441-6.
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES: Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001, 28: 286-289.
Szalma SJ, Buckler ES, Snook ME, McMullen MD: Association analysis of candidate genes for maysin and chlorogenic acid accumulation in maize silks. Theor Appl Genet. 2005, 110: 1324-1333.
The international HapMap Project. Nature. 2003, 426: 789-796.
International Chicken Polymorphism Map Consortium: A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature. 2004, 432: 717-22.
Glinsky GV: Integration of HapMap-based SNP pattern analysis and gene expression profiling reveals common SNP profiles for cancer therapy outcome predictor genes. Cell Cycle. 2006, 5 (22): 2613-25.
Pal P, Xi H, Sun G, Kaushal R, Meeks JJ, Thaxton CS, Guha S, Jin CH, Suarez BK, Catalona WJ, Deka R: Tagging SNPs in the kallikrein genes 3 and 2 on 19q13 and their associations with prostate cancer in men of European origin. Hum Genet. 2007, 122 ((3–4)): 251-9.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pè ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quétier F, Wincker P, French-Italian Public Consortium for Grapevine Genome Characterization: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449 (7161): 463-7.
Frankel OH, Brown AHD: Plant genetic resources today: a critical appraisal. Crop Genetic Resources: Conservation and Evaluation. Edited by: Holden JHW, Williams JT. 1984, London Georges Allen & Unwin Ltd, 249-257.
Zhang H, Sun J, Wang M, Liao D, Zeng Y, Shen S, Yu P, Mu P, Wang X, Li Z: Genetic structure and phylogeography of rice landraces in Yunnan, China, revealed by SSR. Genome. 2007, 50 (1): 72-83.
Hao CY, Zhang XY, Wang LF, Dong YS, Shang XW, Jia JZ: Genetic diversity and core collection evaluations in common wheat germplasm from the Northwestern Spring Wheat Region in China. Molecular Breeding. 2006, 17: 69-77.
Ghislain M, Andrade D, Rodríguez F, Hijmans RJ, Spooner DM: Genetic analysis of the cultivated potato Solanum tuberosum L. Phureja Group using RAPDs and nuclear SSRs. Theor Appl Genet. 2006, 113 (8): 1515-27.
Lebot V, Malapa R, Molisale T, Marchand JL: Physico-chemical characterisation of yam (Dioscorea alata L.) tubers from Vanuatu. Genet Resour Crop Evol. 2005, 53: 1199-1208.
Wang JC, Hu J, Liu NN, Xu HM, Zhang S: Investigation of Combining Plant Genotypic Values and Molecular Marker Information for Constructing Core Subsets. J Int Plant Biol. 2006, 48 (11): 1371-1378.
Jansen J, van Hintum T: Genetic distance sampling: a novel sampling method for obtaining core collections using genetic distances with an application to cultivated lettuce. Theor Appl Genet. 2007, 114: 421-428.
Schoen DJ, Brown AHD: Conservation of allelic richness in wild crop relatives is aided by assessment of genetic markers. Proc Natl Acad Sci. 1993, 90: 10623-10627.
Gouesnard B, Bataillon TM, Decoux G, Rozale C, Schoen DJ, David JL: MSTRAT: an algorithm for building germplasm core collections by maximizing allelic or phenotypic richness. J Hered. 2001, 92: 93-4.
McKhann HI, Camilleri C, Berard A, Bataillon T, David JL, Reboud X, Le Corre V, Caloustian C, Gut IG, Brunel D: Nested core collections maximizing genetic diversity in Arabidopsis thaliana. Plant J. 2004, 38 (1): 193-202.
Ellwood SR, D'Souza NK, Kamphuis LG, Burgess TI, Nair RM, Oliver RP: SSR analysis of the Medicago truncatula SARDI core collection reveals substantial diversity and unusual genotype dispersal throughout the Mediterranean basin. Theor Appl Genet. 2006, 112 (5): 977-83.
Ronfort J, Bataillon T, Santoni S, Delalande M, David JL, Prosperi JM: Microsatellite diversity and broad scale geographic structure in a model legume: building a set of nested core collection for studying naturally occurring variation in Medicago truncatula. BMC Plant Biol. 2006, 13: 6-28.
Whitt SR, Buckler ES: Using natural allelic diversity to evaluate gene function. Methods in Molecular Biology, Plant Functional Genomics: Methods and Protocols. Edited by: Grotewald E. 2003, Totowa, NJ: Humana Press Inc, 236: 123-139.
Bioversity International. [http://www.bioversityinternational.org]
Domaine de Vassal. [http://www.montpellier.inra.fr/vassal]
This P, Lacombe T, Thomas MR: Historical origins and genetic diversity of wine grapes. Trends Genet. 2006, 22 (9): 511-9.
Barnaud A, Lacombe T, Doligez A: Linkage disequilibrium in cultivated grapevine, Vitis vinifera L. Theor Appl Genet. 2006, 112 (4): 708-716.
This P, Lacombe T, Cadle-Davidson M, Owens CL: Wine grape (Vitis vinifera L.) color associates with allelic variation in the domestication gene VvmybA1. Theor Appl Genet. 2007, 114 (4): 723-730.
Mc Govern PE: Ancient Wine: the search for the origins of viniculture. 2003, Princeton: Princeton University Press
Bartsch D, Lehnen M, Clegg J, Pohl-Orf M, Schuphan II, Ellstrand NC: Impact of gene flow from cultivated beet on genetic diversity of wild sea beet populations. Mol Eco. 1999, 8 (10): 1733-41.
Bowers J, Boursiquot JM, This P, Chu K, Johansson H, Meredith C: Historical Genetics: The Parentage of Chardonnay, Gamay, and Other Wine Grapes of Northeastern France. Science. 1999, 285 (5433): 1562-1565.
Staraz DM, Bandinelli R, Boselli M, This P, Boursiquot JM, Laucou V, Lacombe T, Varès D: Genetic Structuring and Parentage Analysis for Evolutionary Studies in Grapevine: Kin Group and Origin of the Cultivar Sangiovese Revealed. J Am Soc Hortic Sci. 2007, 132: 514-524.
Salmaso M, Faes G, Segala C, Stefanini M, Salakhutdinov I, Zyprian E, Toepfer R, Stella Grando M, Velasco R: Genome diversity and gene haplotypes in the grapevine (Vitis vinifera L.) as revealed by single nucleotide polymorphisms. Molecular Breeding. 2004, 14: 385-395.
Lijavetzky D, Cabezas JA, Ibáñez A, Rodríguez V, Martínez-Zapater JM: High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genomics. 2007, 8: 424-434.
Ching A, Caldwell KS, Jung M, Dolan M, Smith OS, Tingey S, Morgante M, Rafalski AJ: SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 2002, 7: 3-19.
Dantec LL, Chagné D, Pot D, Cantin O, Garnier-Géré P, Bedon F, Frigerio JM, Chaumeil P, Léger P, Garcia V, Laigret F, De Daruvar A, Plomion C: Automated SNP detection in expressed sequence tags: statistical considerations and application to maritime pine sequences. Plant Mol Biol. 2004, 54 (3): 461-70.
Russell J, Booth A, Fuller J, Harrower B, Hedley P, Machray G, Powell W: A comparison of sequence-based polymorphism and haplotype content in transcribed and anonymous regions of the barley genome. Genome. 2004, 47 (2): 389-98.
Thuillet AC, Bru D, David J, Roumet P, Santoni S, Sourdille P, Bataillon T: Direct estimation of mutation rate for 10 microsatellite loci in durum wheat, Triticum turgidum (L.) Thell. ssp durum desf. Mol Biol Evol. 2002, 19 (1): 122-5.
Donnelly P: The coalescent and microsatellite variability. Microsatellites – evolution and applications. Edited by: Goldstein DB, Schlötterer C. 1999, Oxford: Oxford University Press, 116-128.
Valdes AM, Slatkin M, Freiner NB: Allele frequencies at microsatellite loci: the stepwise mutation model revisited. Genetics. 1993, 133: 737-749.
Arroyo-García R, Ruiz-García L, Bolling L, Ocete R, López MA, Arnold C, Ergul A, Söylemezoğlu G, Uzun HI, Cabello F, Ibáñez J, Aradhya MK, Atanassov A, Atanassov I, Balint S, Cenis JL, Costantini L, Goris-Lavets S, Grando MS, Klein BY, McGovern PE, Merdinoglu D, Pejic I, Pelsy F, Primikirios N, Risovannaya V, Roubelakis-Angelakis KA, Snoussi H, Sotiri P, Tamhankar S, This P, Troshin L, Malpica JM, Lefort F, Martinez-Zapater JM: Multiple origins of cultivated grapevine (Vitis vinifera L. ssp. sativa) based on chloroplast DNA polymorphisms. Mol Eco. 2006, 15 (12): 3707-14.
Boursiquot JM, Dessup M, Rennes C: Distribution of the Main Phenological, Agronomical and Technological Characters of Vitis-Vinifera L. Vitis. 1995, 34 (1): 31-35.
Aradhya MK, Dangl GS, Prins BH, Boursiquot JM, Walker MA, Meredith CP, Simon CJ: Genetic structure and differentiation in cultivated grape, Vitis vinifera L. Genet Res. 2003, 81 (3): 179-92.
Nei M: Molecular Evolutionary Genetics. 1987, New York: Columbia University Press
Boss PK, Davies C, Robinson SP: Expression of anthocyanin biosynthesis pathway in red and white grapes. Plant Mol Biol. 1996, 32: 565-569.
Gollop R, Even S, Colova-Tsolova V, Perl A: Expression of the grape dihydroflavonol reductase gene and analysis of its promoter region. Journ Exp Bot. 2002, 373 (53): 1397-1409.
Fernandez L, Torregrosa L, Terrier N, Sreekantan L, Grimplet J, Davies C, Thomas MR, Romieu C, Ageorges A: Identification of genes associated with flesh morphogenesis during grapevine fruit development. Plant Mol Biol. 2007, 63 (3): 307-23.
Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-86.
Bonfield JK, Smith KF, Staden RA: new DNA sequence assembly program. Nucleic Acids Research. 1995, 23: 4992-4999.
Acknowledgements
This project (Trilateral project TRI017 CoreGrapeGen) was funded by Genoplante, the French Ministry of Research, the INRA Genetic and Breeding Department, and the region of Languedoc-Roussillon. We thank D. Vares and the technical staff of the Vassal domain for plant management and also A. Doligez and T. Bataillon for critical reading of the manuscript. We acknowledge Daphne Goodfellow for improving the English.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LLC carried out the sequence, participated in the sequence alignment, performed the statistical analysis and drafted the manuscript. AF-L carried out the sequence and participated in the sequence alignment. VL carried out the SSR analysis. SV carried out the sequence. TL participated in the design of the study and performed the ampelographic analysis. A-FA participated in the design and coordination of the study and helped to draft the manuscript. J-MB participated in the design of the study and performed the ampelographic analysis. PT conceived of the study, participated in the design and coordination of the study and helped to draft the manuscript. All authors read and approved the final manuscript
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Le Cunff, L., Fournier-Level, A., Laucou, V. et al. Construction of nested genetic core collections to optimize the exploitation of natural diversity in Vitis vinifera L. subsp. sativa. BMC Plant Biol 8, 31 (2008). https://doi.org/10.1186/1471-2229-8-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-8-31