
Abstract
Background
Soybean lipoxygenases (Lxs) play important roles in plant resistance and in conferring the distinct bean flavor. Lxs comprise a multi-gene family that includes GmLx1, GmLx2 and GmLx3, and many of these genes have been characterized. We were interested in investigating the relationship between the soybean lipoxygenase isozymes from an evolutionary perspective, since soybean has undergone two rounds of polyploidy. Here we report the tetrad genome structure of soybean Lx regions produced by ancient and recent polyploidy. Also, comparative genomics with Medicago truncatula was performed to estimate Lxs in the common ancestor of soybean and Medicago.
Results
Two Lx regions in Medicago truncatula showing synteny with soybean were analyzed. Differential evolutionary rates between soybean and Medicago were observed and the median Ks values of Mt-Mt, Gm-Mt, and Gm-Gm paralogs were determined to be 0.75, 0.62, and 0.46, respectively. Thus the comparison of Gm-Mt paralogs (Ks = 0.62) and Gm-Mt orthologs (Ks = 0.45) supports the ancient duplication of Lx regions in the common ancestor prior to the Medicago-Glycine split. After speciation, no Lx regions generated by another polyploidy were identified in Medicago. Instead tandem duplication of Lx genes was observed. On the other hand, a lineage-specific duplication occurred in soybean resulting in two pairs of Lx regions. Each pair of soybean regions was co-orthologous to one Lx region in Medicago. A total of 34 Lx genes (15 MtLxs and 19 GmLxs) were divided into two groups by phylogenetic analysis. Our study shows that the Lx gene family evolved from two distinct Lx genes in the most recent common ancestor.
Conclusion
This study analyzed two pairs of Lx regions generated by two rounds of polyploidy in soybean. Each pair of soybean homeologous regions is co-orthologous to one region of Medicago, demonstrating the quartet structure of the soybean genome. Differential evolutionary rates between soybean and Medicago were observed; thus optimized rates of Ks per year should be applied for accurate estimation of coalescence times to each case of comparison: soybean-soybean, soybean-Medicago, or Medicago-Medicago. In conclusion, the soybean Lx gene family expanded by ancient polyploidy prior to taxon divergence, followed by a soybean- specific duplication and tandem duplications, respectively.
Similar content being viewed by others


Background
Lipoxygenases (LOXs) have been intensively studied for the past century and have been reported in yeast, algae, fungi, animals, and plants [1]. In higher plants, LOXs are almost ubiquitous and involved in various physiological processes. Importantly, the oxidized products by these enzymes are involved in the traumatic acid and jasmonic acid (JA) pathways, which confer various biotic and abiotic resistance traits to plants [2–4]. Soybean lipoxygenases have received significant attention, since the oxidized compounds cause soy products to have an unpleasant flavor [1, 5, 6]. Many lipoxygenase isozymes have been isolated and are well characterized, but these studies were mainly focused on LOX 1, 2 and 3, the lipoxygenases preferentially expressed in seeds. Moreover, an analysis of soybean mutant lines lacking the isozymes has shown that the Lx1 and Lx2 loci are tightly linked, while the Lx3 locus is independent of the other two loci [7, 8]. Additionally, several vegetative Lx genes, such as Lx4, Lx5, Lx6, Lx7, and Lx8, have been detected and characterized [9, 10].
A number of Lx loci have been identified in the fully sequenced Arabidopsis thaliana and almost completed Medicago genomes. This suggests that there are more Lx genes not yet reported in the soybean genome, and that the number of soybean Lxs would outnumber those in Arabidopsis and Medicago. The expansion of a gene family is related to gene duplication events, such as whole genome duplication, tandem duplication, and transposition [11]. An investigation of the Lx gene family expansion is also of interest in the context of soybean genome evolution, since soybean is known to have undergone two rounds of polyploidy events by analyses of ESTs [12, 13]. The duplicated soybean genome has been investigated by RFLP mapping and more than two regions have been detected by RFLP probes [14]. Furthermore, analyses of homeologous BAC clones, anchored by FAD2 and HCBT genes, revealed highly conserved regions produced by a recent duplication 14.5 million years ago (MYA) [15, 16]. The presence of duplicated soybean chromosomal regions was substantiated by analyzing seventeen homeologous BACs [17]. These studies also showed the genome rearrangement of homeologous regions after whole genome duplication.
Genome duplication events are common in most crop plants [18]. Even model plants, such as Arabidopsis and Medicago, have undergone at least one round of genome doubling [12, 19]. A comparative genomic approach using Medicago has provided insights into complex legume genomes, which cannot be satisfactorily studied provided by the model plant A. thaliana [20]. The divergence of soybean and Medicago from a common ancestor which experienced genome duplication was estimated at 50 MYA [14]. Medicago and soybean, two closely related legume plants, have had two bursts of gene duplication, but it is not clear whether they had a shared polyploidy event before taxon divergence. To clarify this issue, a phylogenetic analysis of gene families was performed, and a hypothesis of shared polyploidy prevailed over the alternative hypothesis of taxon divergence prior to duplication [21]. Later, the early duplication of soybean before the Medicago-soybean split was supported by the whole genome duplication which predated speciation between Medicago and Lotus japonicus [22].
With regard to genome conservation in legumes, many studies have shown broad-scale conservation of legume genomes and gene order [23, 24]. Not only among soybean, Medicago and Arabidopsis [24] but microsynteny was also observed among three genomes [25]. Mudge et al. (2005) [26] identified very high synteny between 3 Mb of soybean DNA sequences and 2 Medicago chromosomes. A recent study also demonstrated a network of synteny within conserved regions among Arabidopsis, Medicago and soybean [27].
In this study the evolutionary expansion of the soybean Lx gene family was demonstrated to occur by two rounds of polyploidy and the evolutionary relationships of nineteen Lx genes in four homeologous chromosomal regions were explored. Moreover, the differential rates of evolution in orthologous and paralogous regions of the Lx gene regions between soybean and Medicago reflect the history of the paleopolyploid soybean genome.
Results
Assembly and Mapping of Soybean BACs and Scaffolds
PCR-based screens of the gmw1 BAC library identified a total of six BAC clones: gmw1-45b2 and gmw1-91g6 for both Lx1 and Lx2; gmw1-6b18, gmw1-9c4, gmw1-22a20, and gmw1-22f19 for Lx3. In other words, Lx1 and Lx2 are located on the same BACs, but Lx3 is on different BACs, which is in accordance with many previous reports [8, 28]. The six BAC clones were sequenced using 454 sequencing technology and the average read length was 250 bp. The number of contigs varied from 1 to 25 and the largest assembled contig was 36 kb (see Additional file 1). Two BAC clones, gmw1-22a20 and gmw1-22f19, were fully sequenced. The remaining gaps were closed by hybridization assemblies, adding ABI-Sanger sequences amplified across the gaps [29]. Here, we mainly used gmw1-9c4 containing Lx3 and gmw1-91g6 containing Lx1 and Lx2 for further analysis (Figure 1). More than ten scaffolds containing lipoxygenase genes were identified from the 7× whole genome sequencing (WGS) assembly from early 2008 http://www.phytozome.com/soybean and selected scaffolds showing synteny with BAC clones were analyzed for further study. Sequences of gmw1-9c4 and gmw1-91g6 were embedded in Scaffold 88 and Scaffold 134, respectively. Also, the sequences of Scaffold 146 and Scaffold 215 were highly identical to each other and showed colinearity with those BAC clones (Figures 1 and 2). Scaffold 88, Scaffold 134, Scaffold 146, and Scaffold 215 were named GmA, GmA', GmB, and GmB', respectively. A total of 13 soybean Lx genes were searched on NCBI and nine of them were included in GmA, GmA', GmB, and GmB'. Two of the soybean Lx genes did not have proper scaffolds with high scores, and the scaffold containing GmLOX9 did not show any synteny except for the GmLOX9 gene itself (see Additional file 2).

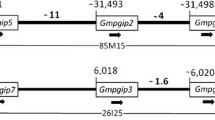
Comparative map of six Lx regions from soybean and Medicago. In silico genetic mapping with SSR markers placed the soybean Lx regions into four different linkage groups (LGs). Two BAC clones, gmw1-9c4 and gmw1-91g6, were selected containing Lx1, Lx2 and Lx3, and these genes were embedded in Scaffolds 88 and 134. Two blocks showing synteny with selected BACs were detected in Medicago and contig 148 on Medicago chromosome 2 was more similar to the BACs. Blast search with the sequence of contig 214 on Medicago chromosome 8 resulted in two more soybean Lx scaffolds. Soybean QTLs for each Lx region are denoted as brown characters and some of them are conserved among the four regions.

Dot plot alignments of six Lx regions between soybean and Medicago. MtA shows a high level of similarity with GmA and GmA'. Both GmB and GmB' have an inversion block, and their sequences end with repetitive lipoxygenases like MtB. Common sequences among the six regions are highlighted with blue dotted lines.
Genetic mapping was achieved by identification of lipoxygenase genes and simple sequence repeat (SSR) markers placed on the composite map http://www.soybase.org. Previously, GmLx1 and GmLx2 on Scaffold 134 were mapped to linkage group (LG) F and GmLx3 on Scaffold 88 was mapped to LG E [7, 28, 30]. By in silico mapping based on sequence, the four scaffolds were placed on four different LGs: Scaffold 88 was anchored by Satt575, Satt213, Sat_112, and Satt411 on LG E; Scaffold 134 contained Sat_090, Satt656, and Sat_417 on LG F; Scaffold 146 had Sat_115, Sat_199, Sat_129, Sat_233, and Satt089 on LG A2; Scaffold 215 was mapped to LG M by Sat_389, Satt404 and Sat_391.
Numerous QTLs have been related to these four Lx regions in soybean and some of them have been associated with more than one region: corn earworm resistance (CEW) and yield QTLs on part of LG E and LG F [31–34]; sucrose content QTLs on LG A2 and LG M [35], oil QTLs on LG E and LG A2 [36, 37]. These mutually conserved QTLs indicate that specific genes associated with CEW, yield, sucrose, and oil have been retained across homeologous genomic regions after genome duplication (Figure 1). Additionally, the carbon isotope discrimination (CID) on LG F and soybean cyst nematode resistance (SCN) on LG A2 have been reported [32, 38].
Comparison of Lx Regions in G. max and M. truncatula
Two Lx regions colinear to these two soybean BACs were detected on Medicago chromosomes 2 and 8 in Medicago pseudomolecule 2.0 http://www.medicago.org/genome and named MtA and MtB, respectively (Figure 2). MtA consists of five BAC clones: AC148918, AC137554, AC146308, AC136955 and AC155896. MtB is comprised of four BAC clones: AC149580, AC140032, AC149638 and AC174341. A dot-plot analysis of the six Lx regions between soybean and Medicago revealed that all showed synteny with some genome rearrangement by insertion, deletion, and tandem duplication. MtA shared most of the genes with the two soybean BACs; however, Mt8 contig 214 showed synteny with only short regions of the both ends of the soybean BACs, with tandem duplicated Lxs being observed instead. Also, a search in the Medicago database http://www.tigr.org/tdb/e2k1/mta1/ identified 32 Lx gene loci. Only 15 Lxs in these two regions were further analyzed because the remaining loci did not show any synteny with soybean Lx regions.
Detailed gene structure and comparisons of the six Lx regions are shown by blue dotted lines (Figure 2) and BLASTZ (Figure 3). The Ks values between homologous genes were calculated (see Additional file 3). Full annotation of the genes is available in Additional file 4. A total of 15 pairs of combinations between the six regions were compared based on their Ks values (Table 1). By comparing the median Ks values of common genes among the six regions, differential evolutionary rates between Medicago and soybean were observed. The median Ks value between MtA and MtB was 0.75, which was close to the Medicago older peak estimated by other analyses [12, 13, 22]. The median Ks value between Gm-Gm paralogs was similar to previous reports [12, 13]. However, the median Ks value between Gm-Mt paralogs was smaller than Mt-Mt paralogs, but larger than Gm-Gm paralogs (Tables 1 and 2). The median Ks value of Gm-Mt orthologs was almost the same as that of Gm-Gm paralogs. The median Ks value of GmA-GmA' and GmB-GmB' were 0.11 and 0.10, respectively, suggesting they were produced by a recent polyploidy in soybean like the event defining the FAD2 gene family and HCBT gene regions [15, 16].

Diagrammatic representation of gene conservation between the six Lx regions by BLASTZ. The sequence highlighted with blue dotted lines in Figure 2 was analyzed in detail with gene prediction. The length and orientation of predicted genes are represented as arrows, and homologous sequences are connected with grey boxes. Each lipoxygenase is depicted as a black box and white arrow, and soybean Lx genes registered in GenBank are denoted with their gene names. A total of 34 Lx loci, 15 from Medicago and 19 from soybean, were detected.
The gene density of the six Lx regions was similar: one gene per 7.06 kb in MtA; one gene per 8.11 kb in MtB; one gene per 7.27 kb in GmA; one gene per 7.55 kb in GmA'; one gene per 7.59 kb in GmB; one gene per 7.62 kb in GmB'. The density of these regions in Medicago was not significantly different from that of the homologous regions in soybean, consistent with previous reports of one gene per 6 kb or 5.8–6.7 kb [16, 26, 39]. The average GC content was approximately the same among those regions: 32.68% in MtA; 32.52% in MtB; 32.14% in GmA; 32.05% in GmA'; 31.96% in GmB; 31.17% in GmB'. Among the six Lx regions in this study, GmA and GmA' were more similar to MtA, whereas GmB and GmB' were closer to MtB (Figs. 2, 3).
Phylogenetic Analysis of Lx Genes in Soybean and Medicago
A total of 34 Lxs were detected from the six homologous Lx regions: 2 in MtA; 13 in MtB; 3 in GmA; 4 in GmA'; 7 in GmB; 5 in GmB' (Figure 3). For convenience, each Lx gene was named according to its species, chromosomal region, and physical order. Thus, their designated names are different from their GeneIDs in GenBank. Because the Lx gene structures were very similar, their evolutionary relationships were uncovered by calculating their Ks values. The Ks values among ten Lxs (from MtB_Lx2 to MtB_Lx11) ranged from 0.3440 to 0.6393, indicating extensive tandem duplication of Lx genes after whole genome duplication in Medicago. Phylogenetic analysis using parsimony of 34 Lx genes in the six regions classified these 34 Lx genes into two clades denoted as black and white squares (Figure 4). The grouping of Lx genes showed that GmB Lxs were more similar to MtB Lxs than to GmA or GmA' Lxs. In other words, the divergence time between GmA and GmB was earlier than the time of speciation between the two species. After taxon divergence, GmA and GmB regions were duplicated resulting in GmA, GmA', GmB, and GmB'. In Medicago, the tandem duplication of Lx genes was observed instead of another polyploidy.

Phylogenetic analysis of 34 Lx proteins. A parsimony tree was generated using bootstrap analysis with 1,000 replicates and branch swapping. Bootstrap values larger than 50 are denoted on each branch. The tree was rooted using Arabidopsis LOX1 (AtLOX1) and rice LOX1(OsLOX1). Soybean and Medicago Lxs in homologous regions represented as black and white squares fall into two major clades. For convenience, each Lx gene is named according to its species, chromosomal region and physical order instead of its GeneID. The Lxs in GmA diverged earlier from Lxs in GmB and MtB and later Lxs in GmB and MtB are separated, suggesting duplication before taxon divergence.
Discussion
Ancient polyploidy in the Lxregions of common ancestor
Previously, it had not been clear whether soybean and Medicago shared a polyploidy event because the old peak of paralog Ks in Medicago did not overlap with that of the soybean [12, 13]. To explain the gap between the soybean and Medicago paralog Ks peaks, Blanc and Wolfe hypothesized that the soybean lineage split from one of the allopolyploid genomes of Medicago [12]. Later, an analysis of gene families provided a framework of shared polyploidy prior to taxon divergence [21]. A total of 56% of gene families also supported a shared soybean-Medicago duplication before the split, whereas the remaining gene families supported alternative hypotheses, including taxon divergence prior to the ancient polyploidy [40]. In addition, a lower synonymous substitution rate in soybean was suggested to explain the difference between Medicago and soybean Ks value peaks [13, 40].
Our data corroborates the hypothesis that the two peaks of median Ks values in soybean and Medicago actually represent the same event but show differential synonymous substitution rates. While orthologs refer to homologous genes that have been generated by speciation, paralogs are homologous genes generated via duplication [41]. The median Ks values of Mt-Mt paralogs, Gm-Mt paralogs, and Gm-Gm paralogs revealed the differential evolutionary rates between the two species (Table 1). The median Ks value of Mt-Mt paralogs (Ks = 0.75) is greater than that of Gm-Gm paralogs (Ks = 0.46), while the Gm-Mt value is intermediate. Thus, to decide the chronological order of duplication and taxon divergence without bias produced by differential evolutionary rates, it is absolutely crucial to compare the values within the same category. A comparison of Gm-Mt paralogs (Ks = 0.62) and Gm-Mt orthologs (Ks = 0.45) indicates that ancient duplication occurred prior to speciation (Table 2). In conclusion, the Mt-Mt, Gm-Mt and Gm-Gm paralogs actually represent the same duplication event, although their absolute values look different.
Recently, a large-scale duplication between Medicago and L. japonicus was proven to have occurred before speciation [22]. The Ks distribution of ancient duplication between Medicago and Lotus was not significantly different, even though Medicago had a narrower peak and Lotus showed a broader peak. The median Ks value of older polyploidy in Medicago and Lotus had been estimated to be 0.7 to 0.9 [12, 13, 22]. In our study, the Ks value of older polyploidy in soybean was much smaller, consistent with previous studies (Table 2) [12, 13, 21]. Thus, optimized rates of Ks per year should be applied for balanced estimation of coalescence times to each case of comparison: soybean-soybean, soybean-Medicago or Medicago-Medicago.
Most crop legumes belong to the Hologalegina and phaseoloid-millettioid clades [42]. The earlier duplication between Medicago and Lotus is the duplication event in the common ancestor of the Hologalegina clade, which includes Medicago, Lotus, and Pisum. Soybean belongs to the phaseoloid-millettioid clade, which contains Glycine, Phaseolus, and Vigna. Taken together, our data support an ancient duplication event in the common ancestor of the Hologalegina and phaseoloid-millettioid clades.
Evolutionary change of soybean and Medicagoafter speciation
It has been suggested that the younger peak in Medicago did not correspond to another polyploidy but a series of tandem duplications because the peak was too broad [21, 40]. Also, there was no clear Ks peak suggesting large scale duplication after the Medicago-Lotus split [22]. In this study, only two Medicago Lx regions produced by ancient polyploidy were detected, and no chromosomal region generated by recent duplication was identified. Instead, ten occurrences of extensive single gene duplication were observed in one Medicago Lx region. The colinearity between MtA and MtB was not high except for repetitive Lxs and a few flanking genes (Figure 2). It is thought that these duplicated regions were differentiated by a diploidization process.
A total of four soybean chromosomal regions were anchored by three to seven Lx genes. Among the four Lx regions, the level of similarity and sequence conservation was high between regions produced by the recent duplication (Figures. 2, 3). These two pairs of Lx regions were generated by two rounds of polyploidy in soybean. With respect to the conservation level of sequence and structure, both inter- and intra-pairs showed synteny (e.g. GmA-GmB, GmA-GmB', GmA'-GmB, and GmA'-GmB'). The level of diploidization in soybean Lx regions generated by ancient polyploidy was lower than that of Medicago. Moreover, the conserved QTLs among the four regions- sucrose, oil, yield, and corn earworm resistance- support their duplicated origin (Figure 1).
Until now, sequence-based analyses of the soybean genome have been focused on regions produced by recent polyploidy [15–17, 43]. The comparative genomics approach used in this paper furthered our understanding of the soybean genome and allowed us to speculate on chromosomal regions produced by both recent and ancient duplication events. Furthermore, each pair of Lx regions in soybean was close to an Lx region in Medicago. Co-orthologs refer to genes generated by a lineage-specific duplication [41]. Thus, GmA/GmA' and GmB/GmB' are the co-orthologous chromosomal regions to MtA and MtB, respectively (Figure 1). In this case, it is difficult to conclude whether Medicago and soybean, are allo- or autopolyploids. But it is clear that soybean and Medicago share both of the genome, rejecting the hypothesis of Medicago allopolyploid history after the Medicago-soybean split.
Expansion and functional divergence of the Lxgene family
Phylogenetic analysis divided 34 Lx genes identified in six Lx regions from soybean and Medicago into two clades (Figure 4). We expect two distinct Lx genes in the most recent common legume ancestor. This parsimony tree showed that the Lx genes in the GmB region were closer to the Lx genes in MtB than those of GmA, suggesting duplication prior to taxon divergence. After the split, tandem duplication of Lx genes occurred in MtB, whereas the soybean Lx genes duplicated to GmA Lx/GmA' Lx and GmB Lx/GmB' Lx. This evolution of the Lx gene family in the homologous regions is diagrammatically represented in Figure 5.

Expansion of the Lx gene family in soybean and Medicago homologous regions in relation to the evolutionary events in the six regions. No direct evidence of a recent polyploidy event in Medicago was detected; instead, tandem duplication of Lxs was observed in MtB. In soybean, two pairs of Lx regions generated by two rounds of polyploidy were identified and analyzed. Each pair of soybean regions is co-orthologous to the region in Medicago, suggesting co-orthologous regions produced by lineage-specific duplication in soybean.
Duplicated genes have been reported to undergo non-functionalization, neo-functionalization, or sub-functionalization [44]. Among the 19 Lx genes in the four soybean regions, nine were previously characterized and confirmed functional (see Additional file 2). In addition, the duplicated lipoxygenase genes had different activities at different pH values and different substrate specificities, suggesting differential functional specificities among lipoxygenase isoforms [9, 45]. Moreover, the patterns of cellular and subcellular localization in pod walls were distinct among the isoforms, indicating independent functions [46]. Specialized isoforms are expected to improve the plant's flexibility to various environmental conditions.
Retention of multiple copies of Lxs in soybean, Medicago, and their common ancestor are reasonable from an evolutionary perspective because lipoxygenases confer various biotic and abiotic resistance traits to plants. Plant lipoxygenases have been reported to conferred resistance to stresses such as herbivores and wounding [47, 48]. Also, clusters of genes related to resistance and disease response have been reported in soybean [15, 49]. In grape (Vitis vinifera), the gene family encoding the grapevine phytoalexin is comprised of 43 genes, 20 of which were previously shown to be expressed [50]. Numerous Lx genes will increase protein or mRNA dosage, leading to resistance in plants. The beneficial effects of increased dosage of genes involved in defense or resistance has been reported in various studies: resistance to glyphosate in plants, protection against heavy metals in hamsters, and decreased susceptibility to HIV infection in humans [51–53]. This mechanism of gene family expansion and functional divergence of duplicated genes may also be relevant to understanding the evolution of other gene families.
A systematic approach is required for crop improvement and modification because most crops have more than one gene copy in their genomes. It is absolutely essential to investigate the number of loci of a particular gene of interest in the breeding of polyploid crops. Further understanding and insights into the paleopolyploid crop genome will lead to more efficient crop improvement and molecular breeding.
Conclusion
In this study, multiple Lx genes anchored in four soybean regions and two Medicago regions were analyzed at the sequence level. Differential evolutionary rates between soybean and Medicago were revealed among the six regions, with Medicago showing a greater synomymous substitution rate than soybean. This fact suggests that an optimized coalescence estimation is needed for each comparison: Gm-Gm, Gm-Mt or Mt-Mt. The four soybean Lx regions are comprised of two pairs of recently duplicated regions, and each pair is co-orthologous to one region in Medicago. These results support an ancient polyploidy in the common ancestor of soybean and Medicago, which preceded separation of the Hologalegina and phaseoloid-millettioid clades. Based on the tetrad soybean genome structure, four copies of duplicated genes or four homeologous regions in soybean are theoretically expected. Phylogenetic analysis showed that the Lx gene family basically expanded by whole genome duplication. Moreover, Lx genes underwent extensive tandem gene duplication.
Methods
Lipoxygenase BAC selection and mining of soybean super contigs
Three specific PCR primers were designed to select BAC clones that contained the target genes, Lx1, Lx2 and Lx3, based on GenBank acc. numbers J02795, J03211, and U50081, respectively. The primer sequences were: Lx1 forward, 5'-TTA ATG CTT TCT TGG GCC CTA-3' and Lx1 reverse, 5'-CGC TCT CCC GTT CCA TTT CC-3'; Lx2 forward, 5'-GCT ATA AAT CAC GTT TCG TTA C-3' and Lx2 reverse, 5'-TAT GCC CTC CTC CTC TGT TC-3'; Lx3 forward, 5'-GTAGTGTTGGTGGGTTGCAAAGATG-3' and Lx3 reverse, 5'-GCA AAC AAA GTG GAT GCT TCC ATG-3'.
A pilot experiment was performed with G. max cultivar Williams 82, prior to BAC selection to optimize PCR conditions using a PTC-225-DNA gradient cycler from MJ research (Watertown, MA., USA). Williams 82 was used as a positive control during the selection procedure. The amplification reaction was 11 μl in volume and contained 100 ng of Williams 82 genomic DNA, 15 pmol of each forward and reverse primer, 10.0 mM of dNTP mix, 1 μl of 10× buffer, 6.9 μl of dd-H2O, and 0.2 unit of Taq DNA polymerase (Vivagen, Sungnam, Korea). The PCR conditions were 94°C for 2 min, 35 cycles of 94°C for 30 sec, annealing temperature for 30 sec, 72°C for 30 sec, and a final extension of 2 min at 72°C.
The Williams 82 G. max BAC clone library [54] was PCR-screened using the same conditions as described above for the genomic DNA of Williams 82. The final PCR screen was conducted with 0.2 μl of the candidate BACs as a template from a working copy of the library.
Soybean super contigs (scaffolds) were identified by BLAST search with lipoxygenase genes against the soybean genome sequence produced by the Soybean Genome Project, DOE Joint Genome Institute http://www.phytozome.com/soybean.
In silicomapping of BACs and super contigs
Genetic markers for Lx1, Lx2 and Lx3 were defined on the consensus soybean genetic map (December, 2006; http://soybase.org) and the sequences of the accessions from which the SNP-containing sequence tagged site was developed were compared with BAC clone sequences using BLAST2 http://www.ncbi.nlm.nih.gov/blast. Simple sequence repeat (SSR) markers in BAC clones and scaffolds were identified by BLAST search against genome survey sequence (GSS) records restricted to soybean SSR-containing clones http://www.ncbi.nlm.nih.gov/blast. Thus, the genetic map positions of the selected BAC clones and scaffolds were determined by the loci of lipoxygenase and SSR markers.
BAC sequencing and assembly
Six BACs, gmw1-45b2 (EU028318), gmw1-91g6 (EU028319), gmw1-6b18 (EU028314), gmw1-9c4 (EU028315), gmw1-22a20 (EU028316) and gmw1-22f19 (EU028317), were sequenced using Genome Sequencer (GS)-FLX. Sequence data were assembled using Phred, Phrap, and Consed to diminish the number of contigs. The remaining gaps were closed by hybrid assembly [29], adding ABI-Sanger sequences from the end of the contigs.
Sequence analysis and annotation
Repetitive sequences were screened using RepeatMasker http://www.repeatmasker.org/.
Gene prediction of soybean and Medicago sequences was performed using FgeneSH on an Arabidopsis matrix, because the results were better suited for BLASTZ results than that of the Medicago matrix http://www.softberry.com. Each predicted gene was annotated by BLASTP searches against UniProt. Syntenic regions in M. truncatula were detected using the BLASTN program with nucleotide collection restricted to M. truncatula. These syntenic regions were compared with Pipmaker [55], BLASTZ program, and visualized using SynBrowse http://www.synbrowse.org and GBrowse http://www.gmod.org/ggb/gbrowse.shtml.
Nucleotide substitution rates, dating of duplication events, and phylogenetic analysis
The Ks values between putative homologues were calculated using the PAML package [56]. Sequences of lipoxygenases in the six Lx regions of soybean and Medicago were compiled and aligned using ClustalX and sequence overhang at the 5'- and 3'-end of alignments were removed. A parsimony tree was generated using bootstrap analysis with 1,000 replicates and branch swapping in PAUP* 4.0 [57] and rooted with Arabidopsis and rice as out-groups.
References
Forster C, North H, Afzal N, Domoney C, Hornostaj A, Robinson DS, Casey R: Molecular analysis of a null mutant for pea (Pisum sativum L.) seed lipoxygenase-2. Plant Mol Biol. 1999, 39 (6): 1209-1220. 10.1023/A:1006173313548.
Gardner HW: 9-Hydroxy-traumatin, a new metabolite of the lipoxygenase pathway. Lipids. 1998, 33 (8): 745-749. 10.1007/s11745-998-0265-z.
Kolomiets MV, Chen H, Gladon RJ, Braun EJ, Hannapel DJ: A leaf lipoxygenase of potato induced specifically by pathogen infection. Plant Physiol. 2000, 124 (3): 1121-1130. 10.1104/pp.124.3.1121.
Veronesi C, Rickauer M, Fournier J, Pouenat ML, Esquerre-Tugaye MT: Lipoxygenase gene expression in the tobacco-Phytophthora parasitica nicotianae interaction. Plant Physiol. 1996, 112 (3): 997-1004. 10.1104/pp.112.3.997.
Bell E, Mullet JE: Characterization of an Arabidopsis lipoxygenase gene responsive to methyl jasmonate and wounding. Plant Physiol. 1993, 103 (4): 1133-1137. 10.1104/pp.103.4.1133.
Davies CS, Nielsen SS, Nielsen NC: Flavor improvement of soybean preparations by genetic removal of lipoxygenase-2. Journal of the American Oil Chemists Society. 1987, 64 (10): 1428-1433. 10.1007/BF02636994.
Kim MY, Ha BK, Jun TH, Hwang EY, Van K, Kuk YI, Lee SH: Single nucleotide polymorphism discovery and linkage mapping of lipoxygenase-2 gene (Lx2) in soybean. Euphytica. 2004, 135 (2): 169-177. 10.1023/B:EUPH.0000014909.40136.20.
Kitamura K, Kumagai T, Kikuchi A: Inheritance of lipoxygenase-2 and genetic-relationships among genes for lipoxygenase-1, lipoxygenase-2 and lipoxygenase-3 isozymes in soybean seeds. Japanese Journal of Breeding. 1985, 35 (4): 413-420.
Kato T, Ohta H, Tanaka K, Shibata D: Appearance of new lipoxygenases in soybean cotyledons after germination and evidence for expression of a major new lipoxygenase gene. Plant Physiol. 1992, 98 (1): 324-330. 10.1104/pp.98.1.324.
Saravitz DM, Siedow JN: The differential expression of wound-inducible lipoxygenase genes in soybean leaves. Plant Physiol. 1996, 110 (1): 287-299. 10.1104/pp.110.1.287.
Kong H, Landherr LL, Frohlich MW, Leebens-Mack J, Ma H, dePamphilis CW: Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50 (5): 873-885. 10.1111/j.1365-313X.2007.03097.x.
Blanc G, Wolfe KH: Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004, 16 (7): 1667-1678. 10.1105/tpc.021345.
Schlueter JA, Dixon P, Granger C, Grant D, Clark L, Doyle JJ, Shoemaker RC: Mining EST databases to resolve evolutionary events in major crop species. Genome/National Research Council Canada = Genome/Conseil national de recherches Canada. 2004, 47 (5): 868-876.
Shoemaker RC, Polzin K, Labate J, Specht J, Brummer EC, Olson T, Young N, Concibido V, Wilcox J, Tamulonis JP, Kochert G, Boerma HR: Genome duplication in soybean (Glycine subgenus soja). Genetics. 1996, 144 (1): 329-338.
Schlueter JA, Scheffler BE, Schlueter SD, Shoemaker RC: Sequence conservation of homeologous bacterial artificial chromosomes and transcription of homeologous genes in soybean (Glycine max L. Merr.). Genetics. 2006, 174 (2): 1017-1028. 10.1534/genetics.105.055020.
Schlueter JA, Vasylenko-Sanders IF, Deshpande S, Yi J, Siegfried M, Roe BA, Schlueter SD, Scheffler BE, Shoemaker RC: The FAD2 gene family of soybean: insights into the structural and functional divergence of a paleopolyploid genome. Crop Science. 2007, 47 (Supplement_1): 14-26.
Schlueter JA, Lin JY, Schlueter SD, Vasylenko-Sanders IF, Deshpande S, Yi J, O'Bleness M, Roe BA, Nelson RT, Scheffler BE, Jackson SA, Shoemaker RC: Gene duplication and paleopolyploidy in soybean and the implications for whole genome sequencing. BMC genomics. 2007, 8: 330-10.1186/1471-2164-8-330.
Udall JA, Wendel JF: Polyploidy and crop improvement. Crop Science. 2006, 46-
Wendel JF: Genome evolution in polyploids. Plant Mol Biol. 2000, 42 (1): 225-249. 10.1023/A:1006392424384.
Thoquet P, Gherardi M, Journet EP, Kereszt A, Ane JM, Prosperi JM, Huguet T: The molecular genetic linkage map of the model legume Medicago truncatula: an essential tool for comparative legume genomics and the isolation of agronomically important genes. BMC Plant Biol. 2002, 2: 1-10.1186/1471-2229-2-1.
Pfeil BE, Schlueter JA, Shoemaker RC, Doyle JJ: Placing paleopolyploidy in relation to taxon divergence: a phylogenetic analysis in legumes using 39 gene families. Syst Biol. 2005, 54 (3): 441-454. 10.1080/10635150590945359.
Cannon SB, Sterck L, Rombauts S, Sato S, Cheung F, Gouzy J, Wang X, Mudge J, Vasdewani J, Schiex T, Spannagl M, Monaghan E, Nicholson C, Humphray SJ, Schoof H, Mayer KF, Rogers J, Quetier F, Oldroyd GE, Debelle F, Cook DR, Retzel EF, Roe BA, Town CD, Tabata S, Peer Van de Y, Young ND: Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci USA. 2006, 103 (40): 14959-14964. 10.1073/pnas.0603228103.
Zhang XC, Wu X, Findley S, Wan J, Libault M, Nguyen HT, Cannon SB, Stacey G: Molecular evolution of lysin motif-type receptor-like kinases in plants. Plant Physiol. 2007, 144 (2): 623-636. 10.1104/pp.107.097097.
Yan HH, Mudge J, Kim DJ, Larsen D, Shoemaker RC, Cook DR, Young ND: Estimates of conserved microsynteny among the genomes of Glycine max, Medicago truncatula and Arabidopsis thaliana. Theor Appl Genet. 2003, 106 (7): 1256-1265.
Cannon SB, McCombie WR, Sato S, Tabata S, Denny R, Palmer L, Katari M, Young ND, Stacey G: Evolution and microsynteny of the apyrase gene family in three legume genomes. Mol Genet Genomics. 2003, 270 (4): 347-361. 10.1007/s00438-003-0928-x.
Mudge J, Cannon SB, Kalo P, Oldroyd GE, Roe BA, Town CD, Young ND: Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biol. 2005, 5: 15-10.1186/1471-2229-5-15.
Schlueter JA, Scheffler BE, Jackson S, Shoemaker RC: Fractionation of Synteny in a Genomic Region Containing Tandemly Duplicated Genes across Glycine max, Medicago truncatula, and Arabidopsis thaliana. The Journal of heredity. 2008
Reinprecht Y, Poysa VW, Yu K, Rajcan I, Ablett GR, Pauls KP: Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome/National Research Council Canada = Genome/Conseil national de recherches Canada. 2006, 49 (12): 1510-1527.
Wicker T, Schlagenhauf E, Graner A, Close TJ, Keller B, Stein N: 454 sequencing put to the test using the complex genome of barley. BMC genomics. 2006, 7: 275-10.1186/1471-2164-7-275.
Cregan PB, Kollipara KP, Xu SJ, Singh RJ, Fogarty SE, Hymowitz T: Primary trisomics and SSR markers as tools to associate chromosomes with linkage groups in soybean. Crop Science. 2001, 41 (4): 1262-1267.
Rector BG, All JN, Parrott WA, Boerma HR: Quantitative trait loci for antibiosis resistance to corn earworm in soybean. Crop Science. 2000, 40 (1): 233-238.
Specht JE, Chase K, Macrander M, Graef GL, Chung J, Markwell JP, Germann M, Orf JH, Lark KG: Soybean response to water: A QTL analysis of drought tolerance. Crop Science. 2001, 41 (2): 493-509.
Terry LI, Chase K, Jarvik T, Orf J, Mansur L, Lark KG: Soybean quantitative trait loci for resistance to insects. Crop Science. 2000, 40 (2): 375-382.
Wang D, Graef GL, Procopiuk AM, Diers BW: Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor Appl Genet. 2004, 108 (3): 458-467. 10.1007/s00122-003-1449-z.
Maughan PJ, Maroof MAS, Buss GR: Identification of quantitative trait loci controlling sucrose content in soybean (Glycine max). Molecular Breeding. 2000, 6 (1): 105-111. 10.1023/A:1009628614988.
Diers BW, Keim P, Fehr WR, Shoemaker RC: RFLP analysis of soybean seed protein and oil content. Theor Appl Genet. 1992, 83 (5): 608-612. 10.1007/BF00226905.
Mansur LM, Lark KG, Kross H, Oliveira A: Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor Appl Genet. 1993, 86 (8): 907-913.
Heer JA, Knap HT, Mahalingam R, Shipe ER, Arelli PR, Matthews BF: Molecular markers for resistance to Heterodera glycines in advanced soybean germplasm. Molecular Breeding. 1998, 4 (4): 359-367. 10.1023/A:1009673422067.
Triwitayakorn K, Njiti VN, Iqbal MJ, Yaegashi S, Town C, Lightfoot DA: Genomic analysis of a region encompassing QRfs1 and QRfs2: genes that underlie soybean resistance to sudden death syndrome. Genome/National Research Council Canada = Genome/Conseil national de recherches Canada. 2005, 48 (1): 125-138.
Shoemaker RC, Schlueter J, Doyle JJ: Paleopolyploidy and gene duplication in soybean and other legumes. Curr Opin Plant Biol. 2006, 9 (2): 104-109. 10.1016/j.pbi.2006.01.007.
Koonin EV: Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005, 39: 309-338. 10.1146/annurev.genet.39.073003.114725.
Lavin M, Herendeen PS, Wojciechowski MF: Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol. 2005, 54 (4): 575-594. 10.1080/10635150590947131.
Van K, Kim DH, Cai CM, Kim MY, Shin JH, Graham MA, Shoemaker RC, Choi BS, Yang TJ, Lee SH: Sequence Level Analysis of Recently Duplicated Regions in Soybean [Glycine max (L.) Merr.] Genome. DNA Res. 2008, 15 (2): 93-102. 10.1093/dnares/dsn001.
Lynch M, Conery JS: The evolutionary fate and consequences of duplicate genes. Science. 2000, 290 (5494): 1151-1155. 10.1126/science.290.5494.1151.
Fuller MA, Weichert H, Fischer AM, Feussner I, Grimes HD: Activity of soybean lipoxygenase isoforms against esterified fatty acids indicates functional specificity. Arch Biochem Biophys. 2001, 388 (1): 146-154. 10.1006/abbi.2000.2269.
Dubbs WE, Grimes HD: Specific lipoxygenase isoforms accumulate in distinct regions of soybean pod walls and mark a unique cell layer. Plant Physiol. 2000, 123 (4): 1269-1280. 10.1104/pp.123.4.1269.
Wang R, Shen W, Liu L, Jiang L, Liu Y, Su N, Wan J: A novel lipoxygenase gene from developing rice seeds confers dual position specificity and responds to wounding and insect attack. Plant Mol Biol. 2008, 66 (4): 401-414. 10.1007/s11103-007-9278-0.
Wu J, Wang L, Baldwin IT: Methyl jasmonate-elicited herbivore resistance: does MeJA function as a signal without being hydrolyzed to JA?. Planta. 2008, 227 (5): 1161-1168. 10.1007/s00425-008-0690-8.
Graham MA, Marek LF, Shoemaker RC: Organization, expression and evolution of a disease resistance gene cluster in soybean. Genetics. 2002, 162 (4): 1961-1977.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quetier F, Wincker P, French-Italian Public Consortium for Grapevine Genome Characterization: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449 (7161): 463-467. 10.1038/nature06148.
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK: The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005, 307 (5714): 1434-1440. 10.1126/science.1101160.
Palmiter RD: Protection against zinc toxicity by metallothionein and zinc transporter 1. Proc Natl Acad Sci USA. 2004, 101 (14): 4918-4923. 10.1073/pnas.0401022101.
Widholm JM, Chinnala AR, Ryu JH, Song HS, Eggett T, Brotherton JE: Glyphosate selection of gene amplification in suspension cultures of 3 plant species. Physiol Plant. 2001, 112 (4): 540-545. 10.1034/j.1399-3054.2001.1120411.x.
Marek LF, Shoemaker RC: BAC contig development by fingerprint analysis in soybean. Genome/National Research Council Canada = Genome/Conseil national de recherches Canada. 1997, 40: 420-427.
Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W: PipMaker-a web server for aligning two genomic DNA sequences. Genome Res. 2000, 10 (4): 577-586. 10.1101/gr.10.4.577.
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997, 13 (5): 555-556.
Swofford DL: PAUP*. Phylogenetic analysis using parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts 2003.
Acknowledgements
This research was supported by a grant (grant no. 305005-4) for BAC clone seletion funded by the Agricultural R&D Promotion Center, the Ministry of Food, Agriculture, Forestry and Fisheries, the Republic of Korea, in part by a grant (code no. CG3121) for genetic mapping from the Crop Functional Genomics Center of the 21st Century Frontier Research Program funded by the Ministry of Education, Science and Technology (MEST), the Republic of Korea and a grant from the BioGreen 21 Project (code no. 20080401034010) for DNA sequencing, Rural Development Administration, the Republic of Korea. K. Van and K.D. Kim are the recipients of a fellowship from the BK21 program granted by MEST, the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JHS designed this study, selected BACs, produced the phylogenetic tree and analyzed the sequences. KV estimated Ks values and helped to design and draft the manuscript. DHK identified and sequenced BAC selections and annotated BACs. KDK mined syntenic regions in Medicago truncatula, performed comparative genomics as well as Ks value estimation and mapped BACs and scaffolds in silico. BSC sequenced BACs and assembled their sequences. YEJ helped to design primers and select BAC clones. MYK helped to draft the manuscript. SHL helped to design this study as well as draft the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12870_2008_341_MOESM1_ESM.doc
Additional File 1: Assembly statistics of six BAC clones from GS-FLX. This data provided show the assembly statistics of six BAC clones from GS-FLX and the remaining gaps were closed by hybridization assemblies, adding ABI-Sanger sequences amplified across the gaps. (DOC 23 KB)
12870_2008_341_MOESM2_ESM.doc
Additional File 2: List of GenBank GeneIDs corresponding to soybean Lx genes with their phylogenetic relationships. A total of 13 soybean Lx genes were searched on NCBI and nine of them were included in GmA, GmA', GmB, and GmB' (DOC 82 KB)
12870_2008_341_MOESM3_ESM.doc
Additional File 3: Pairwise comparisons of Ks values between homologous genes. These Ks values of common genes among the six homologous regions show differential evolutionary rates between Medicago and soybean. (DOC 144 KB)
12870_2008_341_MOESM4_ESM.doc
Additional File 4: Descriptions of predicted genes based on UniRef results within the six Lx regions. This table provides descriptions of predicted genes and Lx genes are highlighted with green color. (DOC 112 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shin, J.H., Van, K., Kim, D.H. et al. The lipoxygenase gene family: a genomic fossil of shared polyploidy between Glycine max and Medicago truncatula. BMC Plant Biol 8, 133 (2008). https://doi.org/10.1186/1471-2229-8-133
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-8-133