
Abstract
Background
The discovery of genetic networks and cis-acting DNA motifs underlying their regulation is a major objective of transcriptome studies. The recent release of the maize genome (Zea mays L.) has facilitated in silico searches for regulatory motifs. Several algorithms exist to predict cis-acting elements, but none have been adapted for maize.
Results
A benchmark data set was used to evaluate the accuracy of three motif discovery programs: BioProspector, Weeder and MEME. Analysis showed that each motif discovery tool had limited accuracy and appeared to retrieve a distinct set of motifs. Therefore, using the benchmark, statistical filters were optimized to reduce the false discovery ratio, and then remaining motifs from all programs were combined to improve motif prediction. These principles were integrated into a user-friendly pipeline for motif discovery in maize called Promzea, available at http://www.promzea.org and on the Discovery Environment of the iPlant Collaborative website. Promzea was subsequently expanded to include rice and Arabidopsis. Within Promzea, a user enters cDNA sequences or gene IDs; corresponding upstream sequences are retrieved from the maize genome. Predicted motifs are filtered, combined and ranked. Promzea searches the chosen plant genome for genes containing each candidate motif, providing the user with the gene list and corresponding gene annotations. Promzea was validated in silico using a benchmark data set: the Promzea pipeline showed a 22% increase in nucleotide sensitivity compared to the best standalone program tool, Weeder, with equivalent nucleotide specificity. Promzea was also validated by its ability to retrieve the experimentally defined binding sites of transcription factors that regulate the maize anthocyanin and phlobaphene biosynthetic pathways. Promzea predicted additional promoter motifs, and genome-wide motif searches by Promzea identified 127 non-anthocyanin/phlobaphene genes that each contained all five predicted promoter motifs in their promoters, perhaps uncovering a broader co-regulated gene network. Promzea was also tested against tissue-specific microarray data from maize.
Conclusions
An online tool customized for promoter motif discovery in plants has been generated called Promzea. Promzea was validated in silico by its ability to retrieve benchmark motifs and experimentally defined motifs and was tested using tissue-specific microarray data. Promzea predicted broader networks of gene regulation associated with the historic anthocyanin and phlobaphene biosynthetic pathways. Promzea is a new bioinformatics tool for understanding transcriptional gene regulation in maize and has been expanded to include rice and Arabidopsis.
Similar content being viewed by others

Background
A key objective of global gene expression studies is the identification of transcription factors and their DNA binding sites responsible for co-expression of genes. DNA binding sites can be predicted in silico by searching regulatory regions of co-expressed genes for overrepresented motifs [1, 2]. Recently, the genome sequence of maize (Zea mays L.) was released [3], facilitating searches for cis-acting motifs in one of the world’s most important crops. Useful motif discovery tools already exist for maize including Grassius [4] and PlantPAN [5], but they retrieve only known, experimentally defined motifs from databases such as PLACE [6] or PlantTFDB [7]. There remains a need for software that predicts de novo motifs from co-expressed genes in maize including from microarray data.
In general, two major types of algorithms exist to search co-regulated genes for de novo motifs. The first approach, consensus searching, consists of searching sets of genes for similar sequences. This consensus method limits motif searches to 12 bases in length (because of the calculation time necessary to search longer motifs) and allows for a few substitutions [8]. Weeder [8] is a widely used program that applies consensus-based sampling. The second type of search algorithm is probabilistic and uses a position weight matrix (PWM) to define a motif [9]. In the PWM, the probability of occurrence of each of the four possible nucleotides is calculated for every position within a predicted motif. Motif PWMs are first identified by scanning regulatory sequences for similar motifs. Predicted motifs are reported if the probability of the motif occurrence is statistically non-random compared to the background. Widely used software programs that apply a probabilistic algorithm are BioProspector [10] and MEME (Multiple Expectation-maximization for Motif Elicitation) [11]. These programs employ different statistical approaches. BioProspector uses Gibbs sampling [12] which randomly picks subsequences of a defined length and iteratively searches within input promoters until a high probability match is found, defined as having PWM values that are significantly different from the input background sequences. By contrast, MEME divides sequences into sub-segments, and all sub-segments are systematically processed as a possible motif. The probability that each sub-segment occurs non-randomly within input promoters is calculated based on its PWM values (Expectation, E) which is then refined based on the probability of occurrence of each nucleotide at each position within the sub-segment (Maximization, M). The sub-segment with the highest probability after EM is chosen and modified by iterating the EM algorithm until a candidate motif cannot be improved [11].
The various motif discovery programs have significant limitations. For example, one limit of Gibbs sampling and hence BioProspector [10], is that different motifs are often obtained at each run. In contrast, MEME predictions are consistent [11]. The main problem with all the current motif discovery programs is their low accuracy. The best motif discovery program thus far was shown to be only 17.4% accurate, in E.coli, with many known motifs being missed [13]. In order to overcome the problem of low prediction accuracy, motif discovery programs have been combined to increase their effectiveness, creating what has been termed an ensemble algorithm [13]. One of the first ensemble algorithms was the BEST program [14] which combined the advantages of three motif discovery programs. Other ensemble tools also exist to define de novo motifs in Arabidopsis and rice, for example MotifVoter [15] that clusters the best motifs from 10 motif discovery tools. However, most ensemble algorithms are conservative because they report only motifs that are retrieved by more than one of the motif discovery programs [15]. To help researchers evaluate motif discovery programs objectively, benchmark data sets have been created, in which known motifs are embedded into diverse sequences [16]. Each motif discovery program can then be compared based on the rate of true and false predictions.
Ideally, a motif discovery program for maize should be validated by its ability to retrieve transcription factor binding sites that have been experimentally validated. Some of the best studied transcription factor targets in maize are those of C1 and P, transcription factors which upregulate the biosynthetic enzymes responsible for production of the red-purple pigments, anthocyanin and phlobaphene, respectively [17–20]. C1 and P are homologous proteins belonging to the R2R3 Myb family of regulators [21], and they have been shown to interact with identical cis-acting motifs in the A1 promoter [18, 22].
In this study, first, a benchmark data set was used to compare and evaluate the accuracy of the three most used motif discovery programs, Weeder, BioProspector and MEME. Improvements were then created to reduce the limitations of each program. These improvements were incorporated into a comprehensive motif discovery pipeline customized for maize called Promzea. Promzea was then validated by asking whether it could retrieve known binding sites of maize C1 and P transcription factors [18–20, 22].
Promzea accurately identified these binding sites, in particular those for P, using only a small number of input genes from these pathways. Interestingly, in a genome-wide scan, Promzea retrieved these binding sites in additional genes, including upstream genes that may help to regulate these pathways. Promzea was also tested against the Maize Development Atlas, a tissue-specific microarray dataset resource for maize [23].
Implementation
Overview of Promzea
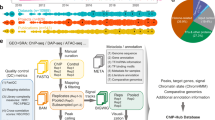
An online pipeline called Promzea was developed to discover de novo cis-acting elements in maize (Figure 1) using a user-friendly interface created in Perl. Promzea is publicly available at http://www.promzea.org. The tool was subsequently expanded to include rice and Arabidopsis. For rationale and complete methodological details, see Additional file 1. Here only an overview of Promzea is provided, along with key parameters below. Briefly, using the online interface, the user first submits either a list of co-expressed cDNA FASTA sequence files, a microarray probe-set ID (in the case of maize), gene ID list or a BED file [24], for example with chromosome coordinates corresponding to peaks from ChIP-seq experiments [25]. In the case of a cDNA file, the sequences are BLAST searched against the chosen plant genome. A list of corresponding promoters to the user input is retrieved from a maize promoter database (Additional file 1). A command line version of the program is also available in the Discovery Environment of the iPlant Collaborative [26]; in this version, users can use as input a BED file allowing them to search for motifs within peaks discovered by ChIP-seq or ChIP-chip experiments [25]. The promoter data set is then searched for shared motifs using three motif discovery programs: MEME, BioProspector and Weeder (Table 1). These motif discovery programs were chosen based on using algorithms that allowed for fast and accurate and/or complimentary searching. The justification for combining multiple motif discovery programs is described in Additional file 1. The motif results are filtered, combined from all three programs, ranked and then displayed for the user along with a ranking score (MNCP, see below; Additional file 1). Finally, Promzea searches the chosen plant genome for genes containing each candidate motif, providing the user with the complete gene list and corresponding gene annotations, along with other forms of validation for the user to analyze (see Generating Promzea, below).

Flow chart of the Promzea motif discovery pipeline. Abbreviations: HG, hypergeometric distribution; MNCP, Mean Normalized Conditional Probability score.
Parameters of motif discovery programs used in Promzea
MEME was set to search for ten motifs with a maximum length of 10 nucleotides on both DNA strands. BioProspector was set to search for 10-nucleotide long motifs and retain only the first ten motifs found. Weeder was set to search for motifs ranging in length from 6–10 nucleotides (medium option). In addition, FIMO [27], PSCAN [28] and Clover [29] were used to retrieve motifs from the maize genome.
Defining filters for each standalone program within Promzea using benchmark data sets
As noted above, within Promzea, a custom filter was designed for each of the three motif discovery programs employed; the purpose was to reduce the false discovery ratio (nFDR) while preserving the true positives as measured using the nucleotide Correlation Coefficient (nCC score). Both nFDR and nCC are defined in Additional file 1. The filter parameters were optimized using the Sandve et al. (2007) benchmark data set [16] based on limiting the probability (pB or pH, respectively for Binomial or hypergeometric test p-values - see Additional file 1) that a motif prediction could occur randomly; the best filters were chosen based on their impact on the nFDR and nCC scores. For BioProspector, pB thresholds at 0.3, 0.5 and 0.7 significantly reduced the average nFDR score (from 0.92 with unfiltered motif discovery data to 0.82, 0.86 and 0.86, respectively, Friedman’s test p-value <0.01; Figure 2A). Though the average nCC scores between the filtered data were not significantly different from one another, the filter pB = 0.7 was chosen for BioProspector as it caused the least absolute reduction in the nCC score average compared to the unfiltered data (from 0.097 to 0.084; Figure 2A). For MEME, a significance level of 0.05 was chosen as it achieved the best balance between a significant reduction in the nFDR average (from 0.96 to 0.85, Friedman’s test p-value < 0.05) and a significant increase in the nCC average (from 0.065 to 0.073, p-value < 0.01; Figure 2B). For Weeder, a significance level of 0.3 was selected as it similarly achieved the best balance between a significant reduction in the average nFDR score (from 0.97 to 0.95, p-value < 0.001) and the largest absolute increase in the average nCC score (from 0.054 to 0.071, p-value < 0.001; Figure 2C).

Optimization of motif filtering for each standalone motif discovery program. The performance of each motif discovery program, applied to the Sandve et al. (2007) benchmark data set, was measured using the nucleotide Correlation Coefficient score mean (nCC, grey bar) and the nucleotide False Discovery Ratio mean (nFDR, black line). Shown is the performance of each original program (unfiltered) and after motif filtering at three probability cut-offs (p) for: (A) BioProspector, using the binomial distribution; (B) MEME using the hypergeometric distribution; and (C) Weeder using the binomial distribution. FDR and nCC error bars indicate the mean confidence intervals.
Defining the ranking of post-filtered motifs
In order to rank the predicted remaining motifs after filtering and then combining the results of all three motif discovery programs, Promzea incorporates a published metric, the Mean Normalized Conditional Probability or MNCP [30] (for details, see Additional file 1). Briefly MNCP is based on the biological principle that if a promoter/first intron contains multiple occurrences of a given motif, then the chance that motif is non-random is higher. Specifically, the MNCP score allows one to determine if the mean occurrence of any given motif in the data set (where the motif has been defined) is higher than its mean occurrence in a random set of promoters/first introns (e.g. whole genome). A motif with a higher MNCP score has a lower probability of being false.
Generating the Promzea software pipeline
The above filtering and ranking principles were integrated into the Promzea software pipeline (Figure 1; Additional file 1: Supplementary materials and methods). To match the user input cDNA to the maize genome, full-length cDNAs were retrieved from the maize, rice and Arabidopsis genomes using their GFF files and respective genome data [3, 31, 32]. For each predicted gene, the corresponding promoters were compiled into a list: the flat file containing ≤1 kb of upstream sequences consisted of 39,656 predicted promoters in the case of maize, 27,416 promoters for Arabidopsis and 58,058 promoters for rice (in Additional file 2: Table S1). At least 70% of the maize genome and 35% of the rice genome are composed of transposable elements [3, 31] which could generate false-positives. In order to overcome this problem, repeat-masked sequences were used to create the promoter flat files. Another problem in motif prediction is the presence of distal cis-acting elements possibly located up to 50 kb from the transcription starting site [33, 34]. However, a maximum length of 1 kb was chosen because motif discovery algorithms struggle with larger search spaces which dilute the signal strength, and it is difficult to anticipate the exact position of a distal cis-acting element. Taking these limitations into account, for motif discovery in Promzea, we applied the same parameters for motif discovery and filtering as used in the Sandve et al. (2007) benchmark validation (Additional file 1: Supplementary materials and methods). In Promzea, the final filtered set of motifs is represented for the user as consensus sequence logos using Weblogo Software [35]. The predicted motifs are ranked using their MNCP scores (see above, and Additional file 1). As false positives were observed in the predictions using the benchmark data set, Promzea gives the user quality control visualizations to validate each predicted motif. One such validation is whether the motif is located at a similar position(s) within promoters of different genes. The frequency of motif occurrence at each position, as defined by each motif discovery program, is shown as a graphic using the Chart: Clicker Perl module [36]. Another validation is whether Promzea retrieves promoters of genes consistent with a common genetic pathway, by searching the maize genome for promoters containing each candidate motif. For this form of validation using gene annotations, all the genes having a defined Gene Ontology annotation were compiled into flat files using data from the Gene Ontology project of each genome.
Results
In silicovalidation of filtering then combining motif discovery programs using benchmark data sets
To generate a motif discovery tool, the effectiveness of existing motif discovery tools was first analyzed using benchmark data sets containing known motifs from Sandve et al. (2007). When BioProspector (alone, unfiltered) was applied to the three types of benchmark data sets from Sandve et al. (2007), the average number of true positive motifs (nTPs) predicted was 1191 while the number of false positives (nFPs) was 10,785 (Figure 3A-C, Table 2). Unfiltered MEME predicted an average of 1145 nTPs correctly, but also 29,982 nFPs. By contrast, unfiltered Weeder predicted two-fold more nTPs (2083 on average) but a very high average number of nFPs (99,561; Table 2). However, each of the three standalone motif discovery programs appeared to identify different sets of motifs (see Additional file 3). It was thus hypothesized that combining the programs (an ensemble-type algorithm) would increase the total number of true positives. In fact, combining the programs increased the number of nTPs to 3185, a >50% increase compared to the best standalone program, Weeder, under the software parameters chosen (Figure 3A-C, Table 2). However, combining the programs also increased the number of nFPs compared to each standalone program. Filtering each motif discovery program separately (from Figure 2, earlier) before combining the results reduced the average nFPs by 25.7% compared to the combined unfiltered data yet only reduced nTPs by 8.7% (Figure 3A-C, Table 2). The nCC score after combining all three filtered programs was not significantly different compared to each standalone program, likely because nTPs and nFPs both increased (Additional file 4).

Effectiveness of combining different motif discovery programs. (A-C) The performance of each motif discovery program, applied to the Sandve et al. (2007) benchmark data set, was measured using the total number of true positive nucleotides (nTP, grey bars) and the total number of false positive nucleotides (nFP, black lines). Shown are scores for the three types of data sets that comprise the Sandve dataset: (A) synthetic (Algorithm Markov), (B) semi-synthetic (Algorithm Real), and (C) real promoters (Model Real). Shown are the scores of each standalone unfiltered program, as well as the scores after combining the outputs of the three programs without filtering (combined) or with filtering (combined filt). (D) The performance of each standalone program or the combined programs was compared using the average nucleotide sensitivity (nSn). Shown are the mean nSn scores for the synthetic data (AM: Algorithm Markov), semi-synthetic data (AR: Algorithm Real) and real data (MR: Model Real). The asterisks (***) indicate that the average nSn score of the combined filtered programs is statistically higher than the average nSn score using Weeder alone at p < 0.01. Each error bar represents the 95% mean confidence interval. (E) The partition of final true positives found by the three motif discovery tools after filtering is shown. Shared results are motif nucleotides retrieved by at least two of the standalone programs. Filtering and combining the standalone programs are the basis of Promzea.
Compared to each standalone program, combining all three filtered programs also significantly improved the ratio of software-predicted true positives versus the actual number of real motif nucleotides (sensitivity, nSn; Dunn’s Multiple Comparisons Test, p < 0.01). The nSn increased by 22% compared to the most sensitive standalone program, Weeder, under the conditions used (Figure 3D; in Additional file 2: Table S2).
The effectiveness of our strategy was further demonstrated by examining the origin of the final predicted nTPs after all three filtered results had been combined. Of the final number of nTPs retrieved from the benchmark data set, 41% were found to have been discovered by Weeder alone, 16% from MEME alone and 10% from BioProspector alone (Figure 3E). Only 33% of nTPs had been found by two or three of the standalone programs. This result confirms that widely used motif discovery programs retrieve distinct sets of motifs and that combining the predictions increases the chance of discovering new regulatory motifs.
Concerning motif ranking using the MNCP score, the analysis using the benchmark Model Real data set showed that as the MNCP score of a predicted motif increased, the chance that it was composed of nucleotide false positives decreased (in Additional file 2: Table S3).
Validation of Promzea by comparing motif predictions to experimentally defined motifs in the maize anthocyanin and phlobaphene biosynthetic pathways
The effectiveness of Promzea was tested based on its ability to detect experimentally defined binding sites for the maize transcription factors, C1 and P, which upregulate enzymes responsible for the biosynthesis of anthocyanin and phlobaphene, respectively (Figure 4) [17–20]. Eight gene promoters containing the C1 and P binding sites were selected (Figure 4, red labels). The corresponding cDNAs (including all close homologs, 12 in total; see Additional file 5 for a list of sequences), were used as input into Promzea following the parameters described (Additional file 1: supplementary materials and methods). Promzea retrieved 29 genes that matched these cDNAs after BLAST searching (in Additional file 2: Table S4); from the corresponding promoters, five motifs were identified along with their MNCP scores (Figure 5).

The maize anthocyanin and phlobaphene biosynthesis pathways regulated by transcription factors C1 and P. Genes encoding biosynthetic enzymes regulated by C1 are shown in red text; those also regulated by P are underlined. C1 and P are homologous proteins [21], and they have been shown to interact with identical binding sites in the A1 promoter [18, 22].

Motifs predicted by Promzea for genes encoding the maize anthocyanin biosynthesis pathway. Promzea searched for motifs in sequences upstream (−200 bp to +1) of the genes indicated in Figure 4 as well as their closest DNA sequence paralogs (see Methods). Shown are the sequence logos, the motif discovery program that identified each motif and the corresponding MNCP score. BioP, BioProspector.
Of the five motifs predicted by Promzea with MNCP scores >1, two matched the experimentally defined P binding sites (Motif1 and Motif5, Figure 6). The partially related C1 motif was found in Motif4 as described below. Based on STAMP [37], Promzea Motif1 and Motif5 were found to be highly similar to the two versions of the experimentally defined binding site of the P-protein (e-value = 2.00e-10 and 2.91e-10; Figure 6) [18, 20, 38]. Interestingly, Motif1 and Motif5 were overrepresented in the −60 to −40 and −80 to −60 promoter regions respectively (Figure 6), consistent with the experimentally defined −65 to −55 binding site of P in the A1 promoter [18]. Motif1 was also overrepresented in the −120 to −100 promoter region (Figure 6), which was consistent with the other experimentally binding sites of P in the A1 promoter at −123 to −88 [18, 20]. Promzea-predicted Motif1 or Motif5 were also retrieved in four out of the five input promoters shown experimentally to contain a P binding site in their promoters (Figure 4, underlined red labels); copies of the P binding site were also predicted in the first 200 bp of the promoter of PAL1, encoding phenylalanine ammonia lyase (Figure 6).

Motifs predicted by Promzea compared to experimentally defined motifs in the literature. Shown are the motif binding sites for transcription factor P (and C1, see text) in the phlobaphene and anthocyanin biosynthetic pathways. The preferential position of each motif predicted by Promzea is indicated in the fourth column from the right. The e-value for STAMP is indicated by the False Discovery Ratio (FDR). The superscript number in the extreme right column represents the number of motif copies present in the promoter of the indicated gene (−200 bp to +1).
Promzea-predicted Motif2 was statistically close (e-value = 4.50e-07) to the MRE binding site identified in an Arabidopsis chalcone synthase promoter [19, 39] (Figure 6). In Arabidopsis, the MRE motif mediates light responsiveness [39]. Motif2 was retrieved by Promzea in the maize chalcone synthase (C2) promoter but also in six out of seven other input gene promoters, validating this Promzea prediction (Figure 6).
Promzea-predicted Motif4 was similar to motif ACIIPVPAL2 (e-value = 6.50e-08; Figure 6) discovered in beans [40]. The ACIIPVPAL2-like element was found in the promoter of PAL2 (Phenylalanine Ammonia Lyase 2), an ortholog of the maize PAL genes necessary for the biosynthesis of phenylpropanoid secondary metabolites including anthocyanins. PAL1 is the rate-limiting step in anthocyanin biosynthesis. Promzea retrieved the ACIIPVPAL2-like motif in the promoters of PAL1 and four additional anthocyanin genes (C2, A1, A2 and Bz1), again validating Promzea predictions. Interestingly, the CA-rich region at the beginning of Motif4 was related to the C1 consensus binding site (CAACCACCAGTCAA GAC) that was previously defined experimentally [20].
The ability of Promzea to retrieve promoter motifs associated with the anthocyanin pathway that were defined experimentally not only in maize, but in also in other plant species, validates Promzea as an accurate tool for motif discovery.
A novel candidate motif in the anthocyanin pathway and expansion of the regulatory network to the branched amino acid metabolic pathway
Promzea also retrieved Motif3 as a candidate motif in the anthocyanin biosynthetic pathway, a motif not previously defined experimentally (Figure 6). Promzea Motif3 was retrieved from the promoter of A1 and additional paralogs of genes in the anthocyanin pathway (in Additional file 2: Table S4). Motif 3 was over-represented in the −40 to −20 promoter regions of these promoters (Figures 6 and 7). In a subsequent search of the maize genome, Motif 3 was retrieved in a total of 762 promoters (in Additional file 2: Table S5); the over-represented GO annotations of the corresponding genes, based on the hypergeometric test, identified these genes as being related to zinc ion binding (p =2.71e-04) and branched chain family amino acid metabolic processes (p = 4.63e-03) (Figure 7; Additional file 6). The latter annotation was also enriched in the four other predicted motifs (Additional file 6). As anthocyanin and phlobaphene are derived from phenylalanine, a branched amino acid, this finding appears to validate novel Motif3 as well as the Promzea pipeline, and predicts that anthocyanin biosynthesis may be transcriptionally coordinated with branched chain amino acid biosynthesis.

Example of the Promzea output for anthocyanin pathway Motif3. For each predicted motif, the following outputs are displayed: (A) the sequence logo (upper) and the plain consensus sequence (lower); (B) the frequency of occurrence of the motif at each upstream position range from the user input data set; (C) summary of annotations of genes containing the motif from the genome-wide retrieval (when applicable). A user can click on the Gene List link and Over-Represented Annotation link to retrieve lists of genes containing the motif and detailed gene annotations, respectively.
Promzea retrieved additional genes that contain the same candidate motifs as the anthocyanin input promoters
As noted above for Motif3, each motif predicted by Promzea from the anthocyanin pathway was used to search the genome to retrieve genes containing that motif (Additional file 6; in Additional file 2: Table S5, anthocyanin pathway genes removed). Interestingly, the five motifs were associated with the same GO annotations: branched chain family amino acid metabolic process, heat shock protein binding, myosin complex or motor activity (Additional file 6). In total, Promzea retrieved between 131 genes (Motif1) and 762 genes (Motif3) with promoters enriched for any one of these motifs (in Additional file 2: Table S5).
Interestingly, Promzea retrieved 127 genes with promoters that contained all five motifs in the −200 bp regions of their promoters (Table 3; Additional file 6; in Additional file 2: Table S6). This list included genes encoding: PAL1, the rate-limiting step in phenylpropanoid biosynthesis which includes anthocyanins; branched amino acid enzymes (as already noted anthocyanin is derived from the branched amino acid phenylalanine); ABC-type transporters (which have been implicated in anthocyanin transport across vacuolar membranes); and regulatory proteins including transcription factors and kinases. Intriguingly, all five anthocyanin promoter motifs were also predicted in the promoters of genes similar to those involved in coordinating sugar, light, cold-temperature and low phosphate dependent activation of anthocyanin biosynthesis, namely: genes similar to gibberellin receptor GID1L2 and gibberellin 20 oxidase; genes similar to those encoding the light-regulatory pathway proteins COP1 and PIF3 (Phytochrome Interacting Factor 3) and numerous sugar transfer/modification enzymes (Table 3; in Additional file 2: Table S6).
These data demonstrate that the genome-wide motif retrieval function of Promzea may allow researchers to predict new genes that may be part of a broader co-regulated network.
Testing of Promzea using the maize development atlas
To further test the Promzea pipeline using data similar to a typical user, microarray data was used from the Maize Development Atlas, a microarray data set of tissue-specific gene expression [23]. Select motifs associated with each tissue are presented (Figure 8) as well as all predicted motifs (Additional file 7).

Promzea predictions of promoter motifs associated with tissue-specific gene expression from the maize development atlas [23]. Tissue-specific microarray data was used as input into Promzea, and selected motif predictions are shown and compared to previously identified promoter motifs. Please see Additional file 7 for all input sequence data and results.
As one case study, a list of 48 embryo-specific transcripts was used as input into Promzea (Additional file 7) from which 13 associated promoter motifs were predicted (Additional file 7). Using Clover, Promzea then retrieved genes associated with promoters in the genome that contained these motifs along with their associated GO annotation terms: genes enriched with any one of nine of the 13 motifs were annotated as having nutrient reservoir activity (Figure 8; Additional file 7), consistent with the embryo being part of the seed. Predicted embryo Motif2 and Motif6 were highly similar to the ABADESI2 cis-acting element (p = 5.06e-08 and p = 1.10e-11 respectively, Figure 8), known to be involved in ABA dependent desiccation during seed maturation [41].
As another case study, a total of 134 tassel-specific transcripts were investigated using Promzea, from which 11 motifs were predicted (Additional file 7). Genes enriched with any one of 9 out of the 11 motifs in their promoters were annotated as being involved in sexual reproduction (GO:0019953) consistent with the function of the tassel (Figure 8; Additional file 7).
From another reproductive tissue, the silk, 12 tissue-specific transcripts were entered into Promzea (Additional file 7). Promzea predicted 10 promoter motifs enriched in the promoters of the associated genes, of which six motifs were enriched in promoters retrieved from genome-wide searches, associated with genes involved in sucrose metabolism; other motifs were enriched in genes associated with defence responses to fungi (Figure 8), which is consistent with this tissue (e.g. against Fusarium which can enter through silks).
Interestingly, motifs similar to the Nonamer motif or NONAMERATH4 motif (AGATCGACG) were most frequently predicted by Promzea in silks (four out of 10 motifs), roots (3 out of 10 motifs) and leaves (one out of six motifs) (Figure 8; Additional file 7 - STAMP outputs). This motif was discovered in the promoter of the Arabidopsis gene encoding Histone 4 [42]. A mutation in Histone 4 was shown to be deleterious to cell specificity of gene expression [42].
These results appear to confirm that Promzea retrieves meaningful motifs associated with co-expressed, tissue-specific genes in data sets that would be typical of users.
Discussion
Promzea provides the plant community with a customized interface to detect de novo cis-acting motifs that are over-represented in the promoters or introns of co-expressed maize genes. By filtering and combining the results of multiple standalone motif discovery programs, Promzea predicts more true motifs than current individual programs without increasing the false discovery ratio (Figure 3). For each run output, Promzea provides a ranking of the predicted motifs based on their MNCP scores (Figure 5). An MNCP score of ≤1 means that the motif is more frequently present in a random set of maize sequences than the user data set of co-expressed genes. MNCP scores can help eliminate motifs that have a general function in the plant and that are not necessary specific to a condition (e.g. tissue specificity). False positives caused by transposons and retro-elements, which are abundant in the maize and rice genomes [43], were reduced by the use of repeat masked promoter data in addition to the use of MNCP scores. False positives are a problem in any motif discovery program; furthermore, cis-acting motifs regulate genes at different biological levels that may or may not be of interest (e.g. developmental cue versus an environmental stimulus). Given these caveats, Promzea generates additional outputs to help a user decide which motif(s) to pursue, placing the emphasis back on the user. Promzea searches the maize genome for genes that contain each predicted motif; the corresponding gene annotations are summarized so that a user can decide whether the predicted motif is relevant to the input gene cluster (e.g. belongs to the biological pathway of interest; Figure 7C; in Additional file 2: Table S5). As gene annotations can be limiting, Promzea also generates the complete list of genes that contain each predicted motif (in Additional file 2: Table S5); a user can then search the list using relevant keywords to determine whether a predicted motif retrieves expected genes. Promzea thus narrows the number of candidate cis- acting motifs for subsequent experimental validation. Promzea should be especially useful to molecular biologists for the prediction of specific promoters for transgene research and targeted maize improvement; few such promoters currently exist for the maize community.
Users can maximize the utility of Promzea. First, prior to using Promzea, it is critical for the user to define robust clusters of co-expressed genes since motif discovery can be diluted by the presence of extra genes that are not part of the real gene network of interest [44, 45]. Second, it is important for the user to know that Promzea employs algorithms that are stochastic in nature, including BioProspector and the selection of random background sequences required for the filtering process. As a result, each Promzea run can generate slightly different outputs. Users are recommended to run Promzea multiple times to verify the uniformity of their results. Finally, Promzea does not compare predicted motifs to motifs previously defined by the research community; for this, the user is encouraged to use STAMP to match a motif to online databases [37], or Matalign [38] for comparisons to motifs found in the literature (Figures 6 and 8). Matalign may also be used to compare the different motifs predicted by Promzea to determine if there are likely duplicates.
In this study, the Promzea pipeline was validated, first, by its ability to retrieve experimentally defined binding sites for transcription factors that regulate the maize anthocyanin and phlobaphene biosynthetic pathways (Figure 4) [18–22, 46–48]. Our case study revealed that Promzea could potentially identify motifs not only from co-expression data, but also from a virtual data set, which might be expected to have a common cis-acting motif, such as in promoters of genes belonging to a specific biochemical pathway (Figure 4). Our case study also demonstrated that Promzea could not only retrieve valid cis-acting motifs, but could make novel predictions about the corresponding biological network, as 127 genes in the maize genome had promoters containing all five predicted motifs in the first 200 bp of their promoters (Table 3; in Additional file 2: Table S6). Promzea has thus predicted a broader putative co-regulated gene network than has been identified experimentally, a finding that will need further investigation.
Promzea was also tested using tissue-specific microarray data from the Maize Development Atlas [23] since this type of data is similar to that of a typical Promzea user (Figure 8). GO annotations of genes enriched for promoter motifs predicted by Promzea appeared to be logical for the specific tissue (Figure 8; Additional file 7): for instance, the GO term ‘sexual reproduction’ was over-represented in 9 out of 11 motifs predicted for tassel-specific transcripts, while the GO term ‘nutrient reserve’ was over-represented in 11 out of 13 embryo predicted motifs. Motifs in some tissues were associated with GO annotations that were not expected, or else there were multiple GO annotations, perhaps suggesting the importance of biological sampling: for example, separating cell types may be critical for software to predict meaningful cis-acting elements.
As a final lesson, it is noteworthy that mutants in maize transcription factors C1 and P were isolated and characterized 100 years ago [49]. The genes encoding these transcription factors began to be isolated 70–80 years later [48, 50]. The binding sites for C1 and P were defined biochemically one decade later [18, 20, 22]. Our study shows that the bioinformatics prediction of cis-acting motifs may help to uncover genetic relationships even in well-studied biological pathways, in this case additional genes that are putatively co-regulated with genes encoding anthocyanin and phlobaphene biosynthetic enzymes.
Conclusions
There was a need for a software program to help maize researchers identify de novo cis-acting motifs underlying co-expressed suites of genes. Here, we analyzed the accuracy of the most widely used motif discovery programs and showed that they had limited accuracy and retrieved distinct sets of motifs. We applied statistical filters to reduce the false discovery ratios of these programs and then combined the search results to improve motif prediction, and validated this approach using benchmark data. These principles were integrated into an online software program for motif discovery that was customized for maize called Promzea. Promzea was subsequently expanded to include rice and Arabidopsis. Promzea was able to retrieve experimentally defined binding sites of maize transcription factors known to regulate the anthocyanin and phlobaphene biosynthetic pathways. Interestingly, the genome-wide motif discovery function of Promzea predicted a broader network of co-regulated genes. Promzea was also tested using tissue specific microarray data from maize as input. Promzea should be a useful tool for de novo predictions of cis-acting motifs from transcriptome data. Promzea is publicly available at http://www.Promzea.org and on the Discovery Environment of the iPlant Collaborative website.
Availability and requirements
Promzea is accessible at http://www/promzea.org and was tested on Firefox web browsers.
Project Name: Promzea
Project Home Page: http://www.promzea.org
Operating system(s): Platform independent
Other requirements: None
Programming language: Perl
License: Freely available for use
Any restrictions to use by non-academics: Promzea uses programs that require a licence for non-academics users; refer to the individual program licences.
Abbreviations
- HG:
-
Hypergeometric distribution
- MEME:
-
Multiple Expectation-maximization for Motif Elicitation
- MNCP:
-
Mean Normalized Conditional Probability
- nCC:
-
Score, nucleotide correlation coefficient
- nFDR:
-
Nucleotide false discovery ratio
- nFP:
-
Nucleotide false positive
- nTP:
-
Nucleotide true positive
- PWM:
-
Position weight matrix
References
Vandepoele K, Quimbaya M, Casneuf T, De Veylder L, Van de Peer Y: Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol. 2009, 150 (2): 535-546. 10.1104/pp.109.136028.
MacLean D, Jerome C, Brown A, Gray J: Co-regulation of nuclear genes encoding plastid ribosomal proteins by light and plastid signals during seedling development in tobacco and Arabidopsis. Plant Mol Biol. 2008, 66 (5): 475-490. 10.1007/s11103-007-9279-z.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA: The B73 maize genome: complexity, diversity, and dynamics. Science. 2009, 326 (5956): 1112-1115. 10.1126/science.1178534.
Yilmaz A, Nishiyama MY, Fuentes BG, Souza GM, Janies D, Gray J, Grotewold E: GRASSIUS: a platform for comparative regulatory genomics across the grasses. Plant Physiol. 2009, 149 (1): 171-180. 10.1104/pp.108.128579.
Chang W-C, Lee T-Y, Huang H-D, Huang H-Y, Pan R-L: PlantPAN: Plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genomics. 2008, 9 (1): 561. 10.1186/1471-2164-9-561.
Higo K, Ugawa Y, Iwamoto M, Korenaga T: Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27 (1): 297-300. 10.1093/nar/27.1.297.
Zhang H, Jin J, Tang L, Zhao Y, Gu X, Gao G, Luo J: PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39 (suppl 1): D1114-D1117.
Pavesi G, Zambelli F, Pesole G, Weeder H: An algorithm for finding conserved regulatory motifs and regions in homologous sequences. BMC Bioinformatics. 2007, 8 (1): 46. 10.1186/1471-2105-8-46.
Stormo GD: Consensus patterns in DNA. Methods Enzymol. 1990, 183: 211-221.
Liu X, Brutlag D, Liu J: BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pacific Symposium on Biocomputing 2001. Edited by: Altman RB, Dunker AK, Hunter L, Klein TE. Hackensack, New Jersey, USA: World Scientific Press; 2001: 127-138.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. Menlo Park, California: AAAI Press; 1994: 28-36.
Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC: Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993, 262 (5131): 208-214. 10.1126/science.8211139.
Hu J, Yang Y, Kihara D: EMD: an ensemble algorithm for discovering regulatory motifs in DNA sequences. BMC Bioinformatics. 2006, 7 (1): 342. 10.1186/1471-2105-7-342.
Che D, Jensen S, Cai L, Liu JS: BEST: Binding-site estimation suite of tools. Bioinformatics. 2005, 21 (12): 2909-2911. 10.1093/bioinformatics/bti425.
Wijaya E, Yiu S-M, Son NT, Kanagasabai R, Sung W-K: MotifVoter: a novel ensemble method for fine-grained integration of generic motif finders. Bioinformatics. 2008, 24 (20): 2288-2295. 10.1093/bioinformatics/btn420.
Sandve G, Abul O, Walseng V, Drablos F: Improved benchmarks for computational motif discovery. BMC Bioinformatics. 2007, 8 (1): 193. 10.1186/1471-2105-8-193.
Dooner HK, Robbins TP, Jorgensen RA: Genetic and developmental control of anthocyanin biosynthesis. Annu Rev Genet. 1991, 25 (1): 173-199. 10.1146/annurev.ge.25.120191.001133.
Grotewold E, Drummond BJ, Bowen B, Peterson T: The myb-homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell. 1994, 76 (3): 543-553. 10.1016/0092-8674(94)90117-1.
Lesnick ML, Chandler VL: Activation of the maize anthocyanin gene A2 is mediated by an element conserved in many anthocyanin promoters. Plant Physiol. 1998, 117 (2): 437-445. 10.1104/pp.117.2.437.
Tuerck JA, Fromm ME: Elements of the maize A1 promoter required for transactivation by the anthocyanin B/C1 or phlobaphene P regulatory genes. Plant Cell. 1994, 6 (11): 1655-1663.
Grotewold E, Sainz MB, Tagliani L, Hernandez JM, Bowen B, Chandler VL: Identification of the residues in the Myb domain of maize C1 that specify the interaction with the bHLH cofactor R. Proc Natl Acad Sci USA. 2000, 97 (25): 13579-13584. 10.1073/pnas.250379897.
Sainz MB, Grotewold E, Chandler VL: Evidence for direct activation of an anthocyanin promoter by the maize C1 protein and comparison of DNA binding by related Myb domain proteins. Plant Cell. 1997, 9 (4): 611-625.
Sekhon RS, Lin H, Childs KL, Hansey CN, Buell CR, de Leon N, Kaeppler SM: Genome-wide atlas of transcription during maize development. Plant J. 2011, 66 (4): 553-563. 10.1111/j.1365-313X.2011.04527.x.
Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ: The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32 (suppl 1): D493-D496.
Schmid CD, Bucher P: ChIP-Seq data reveal nucleosome architecture of human promoters. Cell. 2007, 131 (5): 831-832. 10.1016/j.cell.2007.11.017.
Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, Matasci N, Wang L, Hanlon M, Lenards A: The iPlant Collaborative: cyberinfrastructure for plant biology. Frontiers Plant Sci. 2011, 2: 34.
Grant CE, Bailey TL, Noble WS: FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011, 27 (7): 1017-1018. 10.1093/bioinformatics/btr064.
Zambelli F, Pesole G, Pavesi G: Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009, 37 (suppl 2): W247-W252.
Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z: Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004, 32 (4): 1372-1381. 10.1093/nar/gkh299.
Clarke ND, Granek JA: Rank order metrics for quantifying the association of sequence features with gene regulation. Bioinformatics. 2003, 19 (2): 212-218. 10.1093/bioinformatics/19.2.212.
Sequencing Project International Rice G: The map-based sequence of the rice genome. Nature. 2005, 436 (7052): 793-800. 10.1038/nature03895.
Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M: The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012, 40 (D1): D1202-D1210. 10.1093/nar/gkr1090.
Levine M, Tjian R: Transcription regulation and animal diversity. Nature. 2003, 424 (6945): 147-151. 10.1038/nature01763.
Zheng Z, Kawagoe Y, Xiao S, Li Z, Okita T, Hau TL, Lin A, Murai N: 5′ distal and proximal cis-acting regulator elements are required for developmental control of a rice seed storage protein glutelin gene. Plant J. 1993, 4 (2): 357-366. 10.1046/j.1365-313X.1993.04020357.x.
Crooks GE, Hon G, Chandonia J-M, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2004, 14 (6): 1188-1190. 10.1101/gr.849004.
Watson CG: Chart-Clicker. 2010, In: http://searchcpanorg/~gphat/Chart-Clicker-267/lib/Chart/Clickerpm. 2.67 edn: the CPAN
Mahony S, Benos PV: STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007, 35 (Web Server issue): W253-W258.
Kankainen M, Loytynoja A: MATLIGN: a motif clustering, comparison and matching tool. BMC Bioinformatics. 2007, 8 (1): 189. 10.1186/1471-2105-8-189.
Hartmann U, Valentine WJ, Christie JM, Hays J, Jenkins GI, Weisshaar B: Identification of UV/blue light-response elements in the Arabidopsis thaliana chalcone synthase promoter using a homologous protoplast transient expression system. Plant Mol Biol. 1998, 36 (5): 741-754. 10.1023/A:1005921914384.
Hatton D, Sablowski R, Yung MH, Smith C, Schuch W, Bevan M: Two classes of cis sequences contribute to tissue-specific expression of a PAL2 promoter in transgenic tobacco. Plant J. 1995, 7 (6): 859-876. 10.1046/j.1365-313X.1995.07060859.x.
Lam E, Chua NH: Tetramer of a 21-base pair synthetic element confers seed expression and transcriptional enhancement in response to water stress and abscisic acid. J Biol Chem. 1991, 266 (26): 17131-17135.
Chaubet N, Flenet M, Clement B, Brignon P, Gigot C: Identification of cis-elements regulating the expression of an Arabidopsis histone H4 gene. Plant J. 1996, 10 (3): 425-435. 10.1046/j.1365-313X.1996.10030425.x.
Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon J-M, Westerman RP, SanMiguel PJ, Bennetzen JL: Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 2009, 5 (11): e1000732. 10.1371/journal.pgen.1000732.
Kim E-Y, Kim S-Y, Ashlock D, Nam D: MULTI-K: accurate classification of microarray subtypes using ensemble k-means clustering. BMC Bioinformatics. 2009, 10 (1): 260. 10.1186/1471-2105-10-260.
McNicholas PD, Murphy TB: Model-based clustering of microarray expression data via latent Gaussian mixture models. Bioinformatics. 2010, 26 (21): 2705-2712. 10.1093/bioinformatics/btq498.
Carey CC, Strahle JT, Selinger DA, Chandler VL: Mutations in the pale aleurone color1 regulatory gene of the Zea mays anthocyanin pathway have distinct phenotypes relative to the functionally similar TRANSPARENT TESTA GLABRA1 gene in Arabidopsis thaliana. Plant Cell. 2004, 16 (2): 450-464. 10.1105/tpc.018796.
Bodeau JP, Walbot V: Structure and regulation of the maize Bronze2 promoter. Plant Mol Biol. 1996, 32 (4): 599-609. 10.1007/BF00020201.
Cone KC, Burr FA, Burr B: Molecular analysis of the maize anthocyanin regulatory locus C1. Proc Natl Acad Sci. 1986, 83 (24): 9631-9635. 10.1073/pnas.83.24.9631.
East EM: Inheritance of color in the aleurone cells of maize. Am Nat. 1912, 46 (546): 363-365. 10.1086/279285.
Styles ED, Ceska O: The genetic control of flavonoid synthesis in maize. Can J Genet Cytol. 1977, 19 (2): 289-302.
Acknowledgements
We thank Lewis Lukens and Gregory Downs (University of Guelph, Canada) for technical advice and use of server space; Mike Peppard, Paul Hobbs and Sean Yo (University of Guelph, Canada) for assistance in setting up server access; and Geir Kjetil Sandve and Kjetil Klepper (Norwegian University of Science and Technology, Norway) for assistance with their benchmark data set.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CLM developed and implemented Promzea software. CLM, MNR, DA, PDM, FF, MS, participated in the pipeline design. CLM and TL have tested and optimized Promzea Software. CLM and MNR wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2012_1284_MOESM1_ESM.pdf
Additional file 1: Supplemental materials and methods, and supplemental results. Supplementary materials and methods describing the details of the Promzea pipeline including the calculations and optimization of the parameters for filtering, ranking and visualizations. Additional File 1 also contains the supplementary results. (PDF 269 KB)
12870_2012_1284_MOESM2_ESM.pdf
Additional file 2: Table S1: Summary of promoters and GO annotated genes incorporated into Promzea from maize, Arabidopsis and rice. This table shows the compilation of numbers of promoters, GO annotations and GO-annotated genes retrieved for each plant genome. Table S2. Effectiveness of combining different motif discovery programs based on nucleotide sensitivity scores (nSn). Table S3. The effect of applying different MNCP score cut-offs. Table S4. List of input cDNAs and their corresponding genes from the maize anthocyanin and phlobaphene pathways used for Promzea motif searches. Identification of additional paralogs of genes associated with the maize anthocyanin and phlobaphene biosynthetic pathways. Homologous gene sequences were retrieved that also contained similar promoter motifs, following genome-wide searches by Promzea using the motifs as input. The cDNA sequences were retrieved from Genbank. This list shows corresponding genes from MaizeSequence.org (red text, true loci; blue text, closest paralogs) and additional functional paralogs (extreme right column). Table S5. Gene lists and annotations found in genome-wide searches for Promzea-predicted Motifs 1–5 from promoters of the maize anthocyanin and phlobaphene biosynthetic pathways. Table S6. List of the 127 genes in the maize genome with promoters containing all five of the anthocyanin/phlobaphene-related motifs predicted by Promzea. (PDF 481 KB)
12870_2012_1284_MOESM3_ESM.pdf
Additional file 3: Comparison of standalone motif discovery programs. Different motif discovery programs predicted motifs embedded in 125 sets of sequences belonging to the Sandve et al. (2007) benchmark data set. The benchmark software calculated the nucleotide Correlation Coefficient scores (nCC scores), a measure of the correlation between the known nucleotide positions and the predicted nucleotide positions. The nCC scores are compared for: (A) BioProspector and MEME, (B) Weeder and MEME, and (C) Weeder and BioProspector. The Spearman correlation (r) between the sets of nCC scores is indicated. (PDF 903 KB)
12870_2012_1284_MOESM4_ESM.pdf
Additional file 4: Effectiveness of combining different motif discovery programs. The output of each motif discovery program, applied to the Sandve et al. (2007) benchmark data set, was measured using the Nucleotide Correlation Coefficient (nCC) and the nucleotide Sensitivity (nSn). Shown are scores for the three data sets that comprise the Sandve data set: (A) synthetic (Algorithm Markov), (B) semi-synthetic (Algorithm Real), and (C) real promoters (Model Real). Shown are the scores of each standalone, unfiltered program, as well as the scores after combining the outputs of the three programs with filtering (combined). The error bars represent the 95% mean confidence interval. (PDF 937 KB)
12870_2012_1284_MOESM5_ESM.pdf
Additional file 5: Anthocyanin and phlobaphene pathway gene sequences. The sequences of the cDNAs encoding the enzymes involved in the maize anthocyanin and phlobaphene biosynthetic pathways. A subset of these cDNAs is known to contain experimentally defined cis-acting elements in their promoters that permit co-expression. (PDF 47 KB)
12870_2012_1284_MOESM6_ESM.pdf
Additional file 6: Promzea output for searches of the maize genome with the anthocyanin/phlobaphene-related motifs predicted by Promzea. Shown is the user output from the Promzea website or command line. (PDF 486 KB)
12870_2012_1284_MOESM7_ESM.zip
Additional file 7: Supplemental files for testing Promzea with data sets from the Maize Development Atlas. The zip folder contains 3 folders. The first contains the promoter input for Promzea for each maize tissue; the second folder has all the outputs from Promzea; the third folder contains the STAMP website outputs for comparisons of the predicted motifs with experimentally defined motifs. (ZIP 9 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liseron-Monfils, C., Lewis, T., Ashlock, D. et al. Promzea: a pipeline for discovery of co-regulatory motifs in maize and other plant species and its application to the anthocyanin and phlobaphene biosynthetic pathways and the Maize Development Atlas. BMC Plant Biol 13, 42 (2013). https://doi.org/10.1186/1471-2229-13-42
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-13-42