
Abstract
Background
Transcription factors (TFs) are proteins that have played a central role both in evolution and in domestication, and are major regulators of development in living organisms. Plant genome sequences reveal that approximately 7% of all genes encode putative TFs. The DOF (DNA binding with One Finger) TF family has been associated with vital processes exclusive to higher plants and to their close ancestors (algae, mosses and ferns). These are seed maturation and germination, light-mediated regulation, phytohormone and plant responses to biotic and abiotic stresses, etc. In Hordeum vulgare and Oryza sativa, 26 and 30 different Dof genes, respectively, have been annotated. Brachypodium distachyon has been the first Pooideae grass to be sequenced and, due to its genomic, morphological and physiological characteristics, has emerged as the model system for temperate cereals, such as wheat and barley.
Results
Through searches in the B. distachyon genome, 27 Dof genes have been identified and a phylogenetic comparison with the Oryza sativa and the Hordeum vulgare DOFs has been performed. To explore the evolutionary relationship among these DOF proteins, a combined phylogenetic tree has been constructed with the Brachypodium DOFs and those from rice and barley. This phylogenetic analysis has classified the DOF proteins into four Major Cluster of Orthologous Groups (MCOGs). Using RT-qPCR analysis the expression profiles of the annotated BdDof genes across four organs (leaves, roots, spikes and seeds) has been investigated. These results have led to a classification of the BdDof genes into two groups, according to their expression levels. The genes highly or preferentially expressed in seeds have been subjected to a more detailed expression analysis (maturation, dry stage and germination).
Conclusions
Comparison of the expression profiles of the Brachypodium Dof genes with the published functions of closely related DOF sequences from the cereal species considered here, deduced from the phylogenetic analysis, indicates that although the expression profile has been conserved in many of the putative orthologs, in some cases duplication followed by subsequent divergence may have occurred (neo-functionalization).
Similar content being viewed by others


Background
Transcriptional control is the single most important regulatory mechanism in all organisms. It ultimately depends on transcription factors (TFs) that recognise cis-regulatory elements in the promoters of their target genes. It has become evident that TFs are essential components in the regulation of many traits in plants, including some of agronomic importance such as yield or response to biotic or abiotic stresses and to hormones. TFs have played also a central role in crop domestication and in the evolution of plants [1, 2]. Molecular genetic studies have so far identified major domestication genes in plants, and many of them encode TFs [3], including the rice shattering genes sh4 and qSH[4, 5], the Teosinte Branched1 (Tbr1) gene, first described in maize, which affects plant architecture [6], and the AP2-like wheat gene Q, which is involved in the inflorescence structure [7].
The availability of plant genome sequences has made a great impact on plant biology, defining the protein-coding content of several species and illustrating how these have increased in complexity during the evolution of higher plants [8–10]. Complex organisms contain a large number of TFs. In plants, ≈ 7% of all genes encode putative TFs [11]; by genome-wide identification, the Arabidopsis and the rice genomes have been predicted to contain at least 1,600 TF genes [12, 13] and over 2,000 TF genes in the Brachypodium distachyon genome [10].
The TF proteins are composed of at least two discrete domains: a DNA binding domain, which interacts with cis-regulatory elements in the promoters of their target genes, and an activation or repression domain. TFs operate in complex networks based on protein-protein interactions to regulate many physiological and biochemical processes by modulating the rate of transcriptional initiation. This combinatorial control, involving both transcriptional activators and repressors, integrates signals and results in diverse outcomes of gene expression. TFs are classified into families, mainly on the basis of their conserved DNA binding domains, and both, the number of families, as well as, the number of members in each family have increased in the course of evolution [14].
The DOF (DNA binding with One Finger) transcription factor family belongs to the class of zinc finger domains and it is characterized by a binding domain of 52 amino acid residues that is structured as a Cys2Cys2 (C2/C2) zinc finger [15] that binds specifically to cis-regulatory elements containing the common core 5’-T/AAAAG-3’ [16]. The family has evolved from a common ancestor in Chlamydomonas reinharditii, where only one Dof gene has been found, and expanded into the different taxonomic groups from ferns and mosses to vascular plants [17]. DOF TFs are not found in other eukaryotes such are yeast, Drosophila, Caenorhabditis or humans. The number of Dof genes varies depending on the species; bioinformatic analysis of the Arabidopsis and rice genome predicts 36 and 30 Dof genes respectively [18], while 26 have been described in barley [17], 31 in wheat [19], and 28 in sorghum [20].
Since the identification of the first DOF protein, ZmDOF1, from maize, that acts as a transcriptional activator of the light regulated C4 Phospho-Enol-Pyruvate-Carboxylase (PEPC) gene [21, 22], Dof genes have been extensively studied from various plant species. Generally, DOF proteins are reported to participate as transcriptional regulators in many plant-specific biological processes, such is the case of CDF1, 2, 3 and 5 DOF proteins from A. thaliana that regulate the photoperiodic flowering time by repressing the CONSTANS gene [23–25]. HPPBF3, COG1 and OBP3 are three DOF proteins that participate in the signalling pathways mediated by phytochrome A and B and Cryptochrome 4 [26–28]. The OBP binding proteins (OBP 1–4) interact with the ocs stress-response element in plant promoters [29, 30]. Other DOF transcription factors such as AtDOF5.6/HCA2 regulates inter-fascicular cambium and vascular tissue formation [31], and AtDOF4.7 is expressed in the abscission zone of flowers and participates in the expression of cell wall hydrolytic enzymes [32]. In Arabidospsis, the DOF proteins DAG1 and DAG2 influence, with opposite effects, seed germination [33, 34], and DOF6 has been recently characterized as a negative regulator of seed germination that interacts with TCP14 [35].
The Nicotiana tabacum NtBBF, is induced by auxin and acts as a regulator of the expression of the oncogene rolB[36]. In potato StDOF1 regulates guard cells specific gene expression [37] and StSRF1 modulates the carbohydrate metabolism in the storage roots [38]. PsDOF7 from Pisum sativum activates the expression of the chloroplast thioredoxins f and m that are linked to short-term changes in the sugar and thiol status in plants [39]. In the higher plant ancestor Physcomitrella patens the PpDof1 acts as transcriptional repressor, controlling nutrient-dependent filament growth [40].
In cereal seeds, DOF transcription factors have been shown to regulate gene expression both during seed maturation and upon germination. In maize, PBF (Prolamin-Binding-Factor) and its orthologs from barley and wheat, HvDof24-BPBF and WPBF, are important activators of genes encoding reserve proteins during endosperm development, and its presence is associated with crude protein content and starch content of barley seeds [41–43]. Besides, BPBF is a transcriptional repressor of gibberellin-responsive hydrolase genes induced in the aleurone layers upon seed germination [44]. Another barley DOF, HvDOF23-SAD, has been shown to activate gene expression both during seed maturation and upon germination [45, 46]. Other DOF proteins from barley, HvDOF19 and HvDOF17, mediate the ABA-repression of hydrolase genes in germinating aleurone cells [47]. These DOF regulators interact physically with other proteins belonging to different TF families such as bZIP, MYBR2R3, MYBR1 and WRKY [42, 45, 47–50]. Other seed DOFs, like GmDOF4 and GmDOF11 from soybean, have been implicated in lipid metabolism and their over-expression in A. thaliana increase seed lipid content by activating genes associated with fatty acid biosynthesis [51].
Brachypodium distachyon has been the first member to be sequenced within the Triticeae tribe [10], that includes important crops like wheat, barley and oats [52]. Due to its small genome size, short life cycle, and easy transformation, Brachypodium has become a model system for functional genomic studies in temperate cereals.
This paper reports genome wide in silico identification of the Dof gene family of B. distachyon. Phylogenetic comparison with closely related DOF proteins from rice and barley has been done, as well as, an expression analysis with special emphasis during seed maturation and germination.
Results
In silico identification of Brachypodium distachyon DOFencoding genes
To identify the DOF proteins encoded by the B. distachyon genome, the consensus amino acid sequence of the DNA binding domain of DOF proteins, previously annotated from barley [17], has been used to perform a BLAST against the whole Brachypodium genome database (http://blast.brachypodium.org/) [10]. Twenty seven non-redundant Dof transcription factor genes have been identified (Additional file 1) numbered from BdDof1 to BdDof27, according to their homology to the corresponding barley DOFs. All of them have a typical binding domain of 52 residues spanning a single C2/C2 zinc finger structure (DOF domain). Their schematic distribution along the five Brachypodium chromosomes appears in Figure 1A and their exon-intron structure is shown in Figure 1B. According to their predicted structures, sixteen of the BdDof genes have no introns whereas ten of them contain one intron, generally placed up-stream of the DNA binding domain, with the exception of BdDof7 with an intron down-stream of it, and of BdDof18, which has two introns. The highly conserved 52 residues of the binding domain of B. distachyon DOFs, has been used to produce a phylogenetic tree (Figure 2) where these proteins are grouped into four Major Clusters A; B; C and D. Clusters A and C comprise the majority of BdDOF family members (22 in total), whereas the Clusters B and D contain only 3 and 2 members, respectively. A high bootstrap value, as well as, the comparative analyses of the deduced amino acid sequences of the BdDOF proteins by the MEME software supports this phylogenetic tree [53, 54]. The schematic representation of the different conserved motifs can be found in Additional file 2, and the amino acid consensus sequence of each motif in Additional file 3.

Chromosomal localization and intron-exon structure of the 27 BdDof genes annotated along the five chromosomes of Brachypodium. (A) The chromosome numbers and their size (Mb) are indicated at the bottom of each bar. (B) The intron-exon structure is that predicted by the Brachypodium Genome Annotation Project. Exons are represented by bars: black bars indicate the open reading frames and white bars the non-translated regions. Introns are represented by connecting lines. The Dof domain is represented by a grey circle. The size of exons and introns can be estimated using the horizontal scales.

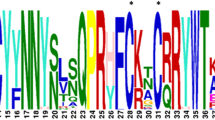
Deduced amino-acids sequences of the BdDof DNA-binding domains and phylogenetic analysis. The phylogenetic tree has been constructed from a complete alignment of the 52 amino acid residues spanning the classical DOF-binding domain of the predicted 27 DOF proteins in B. distachyon. Bootstraping values are indicated as percentages (when > 50%) along the branches. The resulting four gene clusters are indicated in different colours: A = green, B = red, C = blue, D = orange. Strongly conserved residues are indicated below the alignment: asterisks indicate a perfectly conserved residue; dots indicate similar residues and double dots indicate that residue variation occurs within weaker conserved residue groups. The four cysteine residues (C) putatively responsible of the zinc-finger structure are indicated above.
Phylogenetic comparison of DOF proteins from Brachypodium, barley and rice
In order to gain insight into the evolutionary relationships among the DOF proteins from Brachypodium, rice and barley, a combined phylogenetic tree has been constructed (Figure 3) where these proteins are grouped into four Major Clusters of Orthologous Groups (MCOG A; B; C and D) subdivided into subfamilies. Several putative orthologs (i.e. BdDOF12/HvDOF12/OsDOF7 or BdDOF15/HvDOF15/OsDOF15) and paralogs (BdDOF2/BdDOF23 or BdDOF11/BdDOF20/ BdDOF16) have been identified. This phylogenetic tree is further supported by the comparative analyses of the deduced amino acid sequences of the whole DOF proteins by the MEME software (Figure 4 and Table 1). The majority of the conserved motifs are found in a given subfamily, with the exception of motif 3 that is found both in subfamilies A1 and A2 of MCOG A. The D family members have only in common the DOF binding domain.

Joined phylogenetic tree of the Brachypodium, barley and rice DOF transcription factors. An unrooted tree is shown for a homologous region of 52 amino acids that spans the predicted DOF domain sequences of the 81 Brachypodium, barley and rice Dof genes. Bootstraping values are indicated as percentages (when >50%) along the branches. The resulting Major Clusters of Orthologous Genes (MCOG) are shown in different colours: A = green, B = red, C = blue, D = orange. The scale bar corresponds to 0.05 estimated amino acids substitution per site.

Distribution of the conserved motifs along the DOF transcription factors clustered in the MCOGs. Motifs have been identified by means of the MEME software using the deduced amino acid sequences of the 81 Brachypodium, rice and barley Dof genes listed in Figure 3. Position of the identified motifs is relative to the Dof domain. Multilevel consensus sequences for the MEME defined motifs are listed in Table 1.
Expression of the BdDof genes in the major organs of B. distachyon
To study the expression of the annotated BdDof genes, RT-qPCR analyses have been done in several organs: leaves (L: a mixture of young and mature leaves, 12 and 40-day-old), roots (R: 12-day-old), pre-anthesis spikes (S), developing seeds (DS: a mixture from 2 to 12 days after pollination, dap), dry embryos (E), and germinating seeds (a mixture from 8 to 96 hours after imbibition, hai). In this latter case, germinating embryo (GE) and the de-embryonated seed containing the germinating aleurone (GA) have been processed separately. For this purpose, a set of gene-specific primers from the 3’-non coding regions of the BdDof genes have been designed; to ensure reliability of the data, primer pairs that displayed PCRs efficiencies between 2 ± 0.1 have been selected (Additional file 4) and the expression values compared to that of the constitutive BdGAPDH gene (Glyceraldehyde 3-Phosphate Dehydrogenase) [55]. A colored schematic representation has been done (Figure 5) using the TM4 software [56]; in Additional file 5 the expression values of the BdDof genes (mean ± standard error) can be found. According to their expression profiles, the genes have been divided into two groups: the 9 genes with the highest seed expression values are grouped in Figure 5A; in Figure 5B, the global expression of the 18 remaining BdDof genes are included.

Expression levels of the BdDof genes in various organs of the Brachypodium distachyon plants. Expression levels are measured by RT-qPCR in leaves (L), roots (R), spikes (S), developing seeds (DS), dry embryos (E), germinating embryos (GE) and de-embryonated germinating seeds containing the aleurones (GA). (A) Expression pattern of the 9 most abundant Dof genes in seeds; (B) expression pattern of the 17 less abundant Dof genes in seeds. The number inside the square indicates the relative expression levels standardized to the constitutive BdGAPDH gene expression (%). Values are the mean of at least three independent experiments.
Among the genes preferentially expressed in seeds (BdDof4, BdDof17 and BdDof24), BdDof4 presents their maximum transcript levels in the developing seeds, but it is also abundantly expressed in dry embryos and can be detected, although moderately, in the de-embryonated germinating seeds (GA). Transcripts of BdDof17 are especially abundant in dry embryos and faintly detected during seed maturation. The most abundant transcript in germinating seeds is that of BdDof24, both in the embryos and in the rest of the seeds. BdDof3 is ubiquitously expressed and its transcripts are abundant in leaves, pre-anthesis spikes and in the maturation phase of seeds. Genes BdDof2, BdDof8, BdDof16, BdDof22 and BdDof23 are expressed preferentially in leaves, and BdDof2, BdDof16 and BdDof23 are also abundant in roots.
The BdDof genes appearing in Figure 5B are poorly expressed in seeds. However twelve of them are present in leaves, and four of these: BdDof5, BdDof6, BdDof14 and BdDof25 are also found in roots (>20% of the BdGAPDH expression). The transcripts of the BdDof7, BdDof9, BdDof10, BdDof13, BdDof15 and BdDof19 are faintly detectable in all the organs analyzed.
Expression profiles of the BdDofgenes during seed development
A more detailed expression profile for the genes included in Figure 5A has been further investigated in the two phases of seed development: maturation and germination. Seeds have been collected at different maturation stages (2, 4, 6, 8, 10 and 12 dap), and in germinating seeds at different hours after imbibition (8, 16, 24, 48, 72 and 96 hai); in this case the embryos have been processed separately from the rest of the seeds. The expression profile for the BdDof genes analysed is shown in Figures 6 and 7.

Expression kinetic profiles of the most abundant BdDof genes during seed maturation. Seeds at different days after pollination (2, 4, 6, 8, 10 and 12 dap). Relative transcript levels are standardized to the constitutive BdGAPDH gene expression (%). Values are the means ± SE of at least three independent experiments. The scale on the Y axis in (A) or (B) varies depending of the expression level of the genes analysed.

Expression kinetic profiles of the most abundant BdDof genes expressed in germinating seeds. (A) De-embryonated seeds containing the aleurone and (B) embryos from germinating seeds at different hours after imbibition (8, 16, 24, 48, 72 and 96 hai). Relative transcript levels are standardized to the constitutive BdGAPDH gene expression (%). Values are the means ± SE of at least three independent experiments. The scale on the Y axis in (A) or (B) varies depending of the expression level of the genes analysed.
The most abundant transcripts in seeds during maturation are those of BdDof3, with a sharp peak of expression at 10 dap (600% that of GAPDH). Similarly, the expression of the genes BdDof8, BdDof4 and BdDof17 increases during development, whereas BdDof24 presents a high expression level at the beginning and at the end of the maturation period considered (2 dap and 12 dap), decreasing to low levels at the middle stage (Figure 6A). The less abundant transcripts of this group are those of genes BdDof23, BdDof22, BdDof2, BdDof19 and BdDof16, their expression moderately increasing with seed maturation with the exception of BdDof19 that has a peak at 10 dap and decreases thereafter (Figure 6B).
In Figure 7, the expression kinetics in the embryos and in the de-embrionated (containing the aleurones) germinating seeds of BdDof24, BdDof17, BdDof3, BdDof22, and BdDof23 are presented. The most abundant transcripts in both cases are those of BdDof24, but whereas in de-embrionated germinating seeds (GA), the transcript levels increase progressively with time of imbibition reaching a plateau value ( ~300% that of GAPDH) from 48 hai to 96 hai, in germinating embryos an expression peak is reached at 24 hai that sharply decreases after 48 hai. In germinating seeds, the transcripts of BdDof2, BdDof4, BdDof8, BdDof16 and BdDof19 are barely detected (data not shown).
Discussion
Transcriptional regulation is an important mechanism underlying gene expression; the number, position and interaction between different cis-elements and the TFs at a given gene promoter determine the gene expression pattern. These TFs can be classified into gene families according to the presence of a particular DNA-binding domain. In this study, we have conducted a comprehensive search to identify the family of BdDof genes in the Brachypodium distachyon genome database. A complete overview of this gene family in Brachypodium is presented, including a multiple sequence alignment, the intron-exon structures, a phylogenetic comparison with closely related cereal species and its expression in different organs.
The overall intron-exon structure indicates that, the majority of BdDof genes are intronless, as observed in other species, or have one intron, as occurs with five members of Cluster A; exceptionally BdDof18 in Cluster C have two introns [17, 18]. Multiple sequence alignment of these BdDOF proteins with the 52 amino acid residues spanning the DOF binding domain have defined four groups of paralogous genes in Brachypodium, that share common proteins motifs outside the DOF domain, detected in the MEME analysis. The comparative phylogenetic analysis of these BdDOF proteins with those from rice and barley, have defined four MCOGs.
RT-qPCR analysis has shown that the majority of BdDof genes are expressed in leaves, although nine of them are highly (or exclusively) expressed in seeds. No similarities in expression patterns are generally observed within members of the same Cluster of paralogous genes, although BdDof2 and BdDof23 in Cluster C, that share at least five motifs outside of the binding domain, are also quite similar in their expression patterns, indicating that probably they arose by a recent gene duplication, followed by a translocation event since they are located in Chromosomes 5 and 3 respectively. Genes preferentially expressed in leaves (approximately ten times more than in the rest of the organs analysed) are those of BdDof11, BdDof27, BdDof1, BdDof18, BdDof12 and BdDof20. A similar expression pattern has been found in some of their orthologous partners in rice, using the tool Rice Gene Expression Anatomy Viewer from the Rice Genome Annotation Project [57]. This is the case of BdDof12 and its orthologous OsDof7, and BdDof1 and OsDof21. Preferentially expressed in leaves are genes BdDof5, BdDof6, BdDof14, BdDof25 and BdDof26, although their transcripts are ubiquitous. Genes BdDof7, BdDof9, BdDof10, BdDof13, BdDof15 and BdDof19 are barely detected in the organs assayed.
Since several studies support a role for DOF TFs in the regulation of genes encoding seed storage proteins during seed maturation as well as of genes encoding hydrolases involved in the mobilization of reserves upon seed germination, we have selected for a more thorough analysis the genes highly expressed in seeds. These are BdDof2, BdDof3, BdDof4, BdDof8, BdDof16, BdDof17, BdDof22, BdDof23 and BdDof24. BdDof19 is also included in the seed time-course study, because it is closely related in sequence to HvDof19, an important regulator of hydrolase gene expression upon germination in barley seeds [47]. BdDof4, BdDof17 and BdDof24 are preferentially expressed in seeds, with very low levels of expression in the vegetative organs analysed. The rice ortholog of BdDof4 (OsDof4) that is expressed in developing seeds, has been described as having a role during the grain filling process [58]. BdDof24 is highly expressed in seeds, both in the maturation phase and upon germination. Its putative orthologs from barley (HvDof24 BPBF) and rice (OsDof10 RPBF), have similar expression patterns and they are transcriptional activators of genes encoding endosperm-specific storage proteins [59, 60] and transcriptional repressors of hydrolase genes in the aleurone upon seed germination [44]. Other DOF TFs reported to be important in the regulation of barley hydrolase genes upon germination are those encoding transcriptional repressors HvDof17 and HvDof19[47]. The expression of their putative Brachypodium orthologs shows that BdDof17 has a predominant expression in dry embryos, as well as in de-embryonated germinating seeds, peaking at the first two stages analysed (8 and 16 hai) and decreasing thereafter, and it is also expressed during maturation. The same analyses for BdDof19 indicate that although its expression is ten times lower than that of BdDof17 during seed maturation, its pattern is compatible with a possible role in the regulation of storage protein genes; however, no transcripts are detected in the samples of germinating seeds.
BdDof8, BdDof2 and BdDof23, have been included in the group of genes expressed abundantly in seeds. Transcript levels of BdDof8 increase as maturation progresses and an expression peak is detected in germinating seeds (data not shown). BdDof2 and BdDof23 are expressed in the maturation phase of seed development, and only the transcripts of BdDof23 are detected in germinating embryos. These data suggest that BdDof23 may have a similar role during the maturation process as its orthologous in barley HvDof23 SAD[45], and its absence in de-embryonated germinating seeds probably indicates that it does not control the expression of hydrolase genes in the aleurone as opposed to its ortholog in barley [46].
Although not preferentially expressed in seeds, the time course during seed maturation and germination has been studied for the three remaining genes that presented high levels in seeds: BdDof3, BdDof16 and BdDof22. The BdDof3 transcripts are the most abundant in the maturation phase of seed development, peaking at 10 dap and decreasing drastically thereafter, and can also be detected in germinating seeds; however its orthologous gene in barley has not been found among the ESTs and other sequences presently available.
Conclusions
The sequences of the 27 members belonging to the DOF gene family have been compiled from the Brachypodium distachyon genome database, and its inclusion into four clusters is supported by the sequence of the DNA-binding domain, the conservation of different domains outside the DOF domain and their intron-exon structure. A phylogenetic comparison with the barley and rice DOF proteins and a detailed expression profile study in different organs together with comparison with published functions of the barley and rice DOFs proteins, suggest that there is a strong sequence conservation between the DOFs of these monocot species, although some of their functions may have diverged in the course of evolution (see Additional file 6). This works opens the possibility of a further more focused investigation of the functional role of these DOF regulatory proteins.
Methods
Database searches for the identification of Dof family members in Brachypodium distachyon
A non-redundant compilation of the Hordeum vulgare and Oryza sativa Dof genes have been collected from two different databases: NCBI (http://www.ncbi.nlm.nih.gov) and TIGR (http:/blast.jcvi.org/euk-blast/), respectively. The amino acid consensus sequence of the DOF domain from barley family members has been used to search for potential Dof genes in the genome of Brachypodium distachyon through BLASTP at the “Brachypodium Database” (http://www.brachypodium.org). The putative DOF protein sequences of B. distachyon are analysed with the Interpro program using the PFAM database (http://pfam.sanger.ac.uk[61]) and their DOF domains deduced.
Dof protein alignment and phylogenetic analysis
The identification of a homologous region in all the DOF proteins sequences (from barley, rice and Brachypodium) that spans the classical DOF-binding domain has been done through a multiple alignment using CLUSTALW [62]. Since the DOF domains of HvDof11 and HvDof26 genes found in the databases are truncated, these genes have not been included in the subsequent analysis. From the deduced amino acid sequences identified, phylogenies have been computed using the Phylogeny.fr platform [63], which uses MUSCLE for multiple alignment with Gblocks for alignment curation and the maximum likelihood PhyML method for tree building using the MEGA software 4.0 [64].
Identification of conserved motifs
The deduced protein sequences of the 81 DOF genes from O.sativa, H.vulgare and B.distachyon have been further analyzed by means of the MEME program [65], (http://meme.sdsc.edu/meme4_6_0/intro.html). To identify conserved motifs in these sequences, the selection of maximum number of motifs was set to 50 with minimum width of 8 amino acid residues. Conserved motifs identified by MEME have been scanned using PSORT server (http://www.psort.org/) to find subcellular localization signals.
Plant material and RNA extraction for expression analysis of the BdDOFgenes
Brachypodium distachyon strain Bd21, a community standard diploid inbred line [66], kindly provided by Prof. Garvin (University of Minesota), has been used in this study. Seed have been germinated in the dark at 22°C for one week and then the seedlings transferred to pots in a controlled-environment growth chamber at 22°C and under a 16h day/8h night photoperiod. Samples from young leaves (12-day-old), old leaves (40-day-old), roots (12-day-old), spikes and seeds, at 2, 4, 6, 8, 10 and 12 days after pollination (dap), and dry embryos have been harvested and used for RNA extraction. After-ripened seeds (storage at 22°C and 30% relative humidity in the dark for 3 months) have been surface sterilized, washed and germinated in water imbibed filter paper at 22°C in the dark and samples collected at different times (8, 16, 24, 48, 72 and 96 hours after imbibition, hai) and used for RNA extraction as described above.
Total RNA from vegetative tissues (leaves and roots), spikes and dry embryos has been isolated using the phenol/chloroform method and precipitation with 2M LiCl [67]. For the isolation of RNA from seeds the standard protocol for Arabidopsis seed RNA isolation has been followed [68]. Genomic DNA in the RNA preparations has been eliminated after a DNAse treatment using the DNAse I, RNAse-free from Roche Diagnostics. First strand cDNA has been synthesized with random hexameres using the High-Capacity cDNA Reverse Transcription Kit according to the manufacture’s recommendations (Applied Biosystems).
RT-qPCR analysis
The transcript levels of 27 BdDof genes have been quantified by RT-qPCR with a 7300-Real Time PCR System (Applied Biosystems) using SYBR-green as the intercalating dye. The analysis has been done using three different biological replicates. The primer pairs used for the RT-qPCR analysis (Additional file 4) have been designed on the 3’-non coding region according to the parameters established on the Primer3Plus program (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi); the primer gene specificity of the BdDof primer pairs has been checked by blasting primer sequences in the Brachypodium data base (http://www.brachybase.org) and confirmed by a single peak in the melting temperature curve of the RT-qPCR analyses. To ensure reliability of the results, PCR efficiency has been determined [69]. To this end, four serial ten-fold dilutions of pooled cDNAs have been made starting from 10 to 0.01 ng/μl; when the expression level was too low, genomic DNA has been used. The raw Ct values have been plotted against log-transformed concentrations of the serial dilutions, the corresponding PCR efficiency (E) of one cycle in the exponential phase calculated according to the equation: E= 10(−1/slope). The investigated transcripts have showed high PCR efficiency rates (Additional file 4). Quantification has been standardized to the expression of the BdGAPDH (glyceraldehyde 3-phosphate dehydrogenase) gene, that was validated as a suitable reference gene across all the plant samples examined (Additional file 7), calculated using the 2-∆CT method and referred as percentage [55, 69].
The expression data from leaves (a mixture of young and adult leaves, 12 and 40-day-old respectively), roots (12 day-old), spikes, developing seeds (a mixture from 2 to 12 dap), dry embryos and germinating seeds (a mixture from 8 to 96 hai) have been analyzed using the TM4 software [56].
References
Doebley J, Lukens L: Transcriptional regulators and the evolution of plant form. Plant Cell. 1998, 10 (7): 1075-1082.
Purugganan MD, Fuller DQ: The nature of selection during plant domestication. Nature. 2009, 457 (7231): 843-848. 10.1038/nature07895.
Doebley JF, Gaut BS, Smith BD: The molecular genetics of crop domestication. Cell. 2006, 127 (7): 1309-1321. 10.1016/j.cell.2006.12.006.
Li C, Zhou A, Sang T: Rice domestication by reducing shattering. Science. 2006, 311 (5769): 1936-1939. 10.1126/science.1123604.
Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M: An SNP caused loss of seed shattering during rice domestication. Science. 2006, 312 (5778): 1392-1396. 10.1126/science.1126410.
Wang RL, Stec A, Hey J, Lukens L, Doebley J: The limits of selection during maize domestication. Nature. 1999, 398 (6724): 236-239. 10.1038/18435.
Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai YS, Gill BS, Faris JD: Molecular characterization of the major wheat domestication gene Q. Genetics. 2006, 172 (1): 547-555.
Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408 (6814): 796-815. 10.1038/35048692.
Tanaka T, Antonio BA, Kikuchi S, Matsumoto T, Nagamura Y, Numa H, Sakai H, Wu J, Itoh T, Sasaki T, Aono R, Fujii Y, Habara T, Harada E, Kanno M, Kawahara Y, Kawashima H, Kubooka H, Matsuya A, Nakaoka H, Saichi N, Sanbonmatsu R, Sato Y, Shinso Y, Suzuki M, Takeda J, Tanino M, Todokoro F, Yamaguchi K, Yamamoto N, et al: The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008, 36: D1028-D1033.
The International Brachypodium Initiative: Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010, 463 (7282): 763-768. 10.1038/nature08747.
Udvardi MK, Kakar K, Wandrey M, Montanari O, Murray J, Andriankaja A, Zhang JY, Benedito V, Hofer JM, Chueng F, Town CD: Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol. 2007, 144 (2): 538-549. 10.1104/pp.107.098061.
Riechmann JL, Ratcliffe OJ: A genomic perspective on plant transcription factors. Curr Opin Plant Biol. 2000, 3 (5): 423-434. 10.1016/S1369-5266(00)00107-2.
Xiong Y, Liu T, Tian C, Sun S, Li J, Chen M: Transcription factors in rice: a genome-wide comparative analysis between monocots and eudicots. Plant Mol Biol. 2005, 59 (1): 191-203. 10.1007/s11103-005-6503-6.
Pabo CO, Sauer RT: Transcription factors: structural families and principles of DNA recognition. Annu Rev Biochem. 1992, 61: 1053-1095. 10.1146/annurev.bi.61.070192.005201.
Yanagisawa S: A novel DNA-binding domain that may form a single zinc finger motif. Nucleic Acids Res. 1995, 23 (17): 3403-3410. 10.1093/nar/23.17.3403.
Yanagisawa S, Schmidt RJ: Diversity and similarity among recognition sequences of Dof transcription factors. Plant J. 1999, 17 (2): 209-214. 10.1046/j.1365-313X.1999.00363.x.
Moreno-Risueno MA, Martinez M, Vicente-Carbajosa J, Carbonero P: The family of DOF transcription factors: from green unicellular algae to vascular plants. Mol Genet Genomics. 2007, 277 (4): 379-390. 10.1007/s00438-006-0186-9.
Lijavetzky D, Carbonero P, Vicente-Carbajosa J: Genome-wide comparative phylogenetic analysis of the rice and Arabidopsis Dof gene families. BMC Evol Biol. 2003, 3: 17-10.1186/1471-2148-3-17.
Shaw LM, McIntyre CL, Gresshoff PM, Xue GP: Members of the Dof transcription factor family in Triticum aestivum are associated with light-mediated gene regulation. Funct Integr Genomics. 2009, 9 (4): 485-498. 10.1007/s10142-009-0130-2.
Kushwaha H, Gupta S, Singh VK, Rastogi S, Yadav D: Genome wide identification of Dof transcription factor gene family in sorghum and its comparative phylogenetic analysis with rice and Arabidopsis. Mol Biol Rep. 2011, 38 (8): 5037-5053. 10.1007/s11033-010-0650-9.
Yanagisawa S, Izui K: Molecular cloning of two DNA-binding proteins of maize that are structurally different but interact with the same sequence motif. J Biol Chem. 1993, 268 (21): 16028-16036.
Yanagisawa S, Sheen J: Involvement of maize Dof zinc finger proteins in tissue-specific and light-regulated gene expression. Plant Cell. 1998, 10 (1): 75-89.
Imaizumi T, Schultz TF, Harmon FG, Ho LA, Kay SA: FKF1 F-box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science. 2005, 309 (5732): 293-297. 10.1126/science.1110586.
Imaizumi T, Kay SA: Photoperiodic control of flowering: not only by coincidence. Trends Plant Sci. 2006, 11 (11): 550-558. 10.1016/j.tplants.2006.09.004.
Fornara F, Panigrahi KC, Gissot L, Sauerbrunn N, Ruhl M, Jarillo JA, Coupland G: Arabidopsis DOF transcription factors act redundantly to reduce constans expression and are essential for a photoperiodic flowering response. Dev Cell. 2009, 17 (1): 75-86. 10.1016/j.devcel.2009.06.015.
Tepperman JM, Zhu T, Chang HS, Wang X, Quail PH: Multiple transcription-factor genes are early targets of phytochrome A signaling. Proc Natl Acad Sci U S A. 2001, 98 (16): 9437-9442. 10.1073/pnas.161300998.
Park DH, Lim PO, Kim JS, Cho DS, Hong SH, Nam HG: The Arabidopsis COG1 gene encodes a Dof domain transcription factor and negatively regulates phytochrome signaling. Plant J. 2003, 34 (2): 161-171. 10.1046/j.1365-313X.2003.01710.x.
Ward JM, Cufr CA, Denzel MA, Neff MM: The Dof transcription factor OBP3 modulates phytochrome and cryptochrome signaling in Arabidopsis. Plant Cell. 2005, 17 (2): 475-485. 10.1105/tpc.104.027722.
Zhang B, Chen W, Foley RC, Buttner M, Singh KB: Interactions between distinct types of DNA binding proteins enhance binding to ocs element promoter sequences. Plant Cell. 1995, 7 (12): 2241-2252.
Kang HG, Singh KB: Characterization of salicylic acid-responsive, arabidopsis Dof domain proteins: overexpression of OBP3 leads to growth defects. Plant J. 2000, 21 (4): 329-339. 10.1046/j.1365-313x.2000.00678.x.
Guo Y, Qin G, Gu H, Qu LJ: Dof5.6/HCA2, a Dof transcription factor gene, regulates interfascicular cambium formation and vascular tissue development in Arabidopsis. Plant Cell. 2009, 21 (11): 3518-3534. 10.1105/tpc.108.064139.
Wei PC, Tan F, Gao XQ, Zhang XQ, Wang GQ, Xu H, Li LJ, Chen J, Wang XC: Overexpression of AtDOF4.7, an Arabidopsis DOF family transcription factor, induces floral organ abscission deficiency in Arabidopsis. Plant Physiol. 2010, 153 (3): 1031-1045. 10.1104/pp.110.153247.
Gualberti G, Papi M, Bellucci L, Ricci I, Bouchez D, Camilleri C, Costantino P, Vittorioso P: Mutations in the Dof zinc finger genes DAG2 and DAG1 influence with opposite effects the germination of Arabidopsis seeds. Plant Cell. 2002, 14 (6): 1253-1263. 10.1105/tpc.010491.
Gabriele S, Rizza A, Martone J, Circelli P, Costantino P, Vittorioso P: The Dof protein DAG1 mediates PIL5 activity on seed germination by negatively regulating GA biosynthetic gene AtGA3ox1. Plant J. 2010, 61 (2): 312-323.
Rueda-Romero P, Barrero-Sicilia C, Gomez-Cadenas A, Carbonero P, Onate-Sanchez L: Arabidopsis thaliana DOF6 negatively affects germination in non-after-ripened seeds and interacts with TCP14. J Exp Bot. 2012, 63 (5): 1937-1949. 10.1093/jxb/err388.
Baumann K, De Paolis A, Costantino P, Gualberti G: The DNA binding site of the Dof protein NtBBF1 is essential for tissue-specific and auxin-regulated expression of the rolB oncogene in plants. Plant Cell. 1999, 11 (3): 323-334.
Plesch G, Ehrhardt T, Mueller-Roeber B: Involvement of TAAAG elements suggests a role for Dof transcription factors in guard cell-specific gene expression. Plant J. 2001, 28 (4): 455-464.
Tanaka M, Takahata Y, Nakayama H, Nakatani M, Tahara M: Altered carbohydrate metabolism in the storage roots of sweet potato plants overexpressing the SRF1 gene, which encodes a Dof zinc finger transcription factor. Planta. 2009, 230 (4): 737-746. 10.1007/s00425-009-0979-2.
Barajas-Lopez Jde D, Tezycka J, Travaglia CN, Serrato AJ, Chueca A, Thormahlen I, Geigenberger P, Sahrawy M: Expression of the chloroplast thioredoxins f and m is linked to short-term changes in the sugar and thiol status in leaves of Pisum sativum. J Exp Bot. 2012, 63 (13): 4887-4900. 10.1093/jxb/ers163.
Sugiyama T, Ishida T, Tabei N, Shigyo M, Konishi M, Yoneyama T, Yanagisawa S: Involvement of PpDof1 transcriptional repressor in the nutrient condition-dependent growth control of protonemal filaments in Physcomitrella patens. J Exp Bot. 2012, 63 (8): 3185-3197. 10.1093/jxb/ers042.
Marzabal P, Gas E, Fontanet P, Vicente-Carbajosa J, Torrent M, Ludevid MD: The maize Dof protein PBF activates transcription of gamma-zein during maize seed development. Plant Mol Biol. 2008, 67 (5): 441-454. 10.1007/s11103-008-9325-5.
Vicente-Carbajosa J, Moose SP, Parsons RL, Schmidt RJ: A maize zinc-finger protein binds the prolamin box in zein gene promoters and interacts with the basic leucine zipper transcriptional activator Opaque2. Proc Natl Acad Sci U S A. 1997, 94 (14): 7685-7690. 10.1073/pnas.94.14.7685.
Haseneyer G, Stracke S, Piepho HP, Sauer S, Geiger HH, Graner A: DNA polymorphisms and haplotype patterns of transcription factors involved in barley endosperm development are associated with key agronomic traits. BMC Plant Biol. 2010, 10: 5-10.1186/1471-2229-10-5.
Mena M, Cejudo FJ, Isabel-Lamoneda I, Carbonero P: A role for the DOF transcription factor BPBF in the regulation of gibberellin-responsive genes in barley aleurone. Plant Physiol. 2002, 130 (1): 111-119. 10.1104/pp.005561.
Diaz I, Martinez M, Isabel-LaMoneda I, Rubio-Somoza I, Carbonero P: The DOF protein, SAD, interacts with GAMYB in plant nuclei and activates transcription of endosperm-specific genes during barley seed development. Plant J. 2005, 42 (5): 652-662. 10.1111/j.1365-313X.2005.02402.x.
Isabel-LaMoneda I, Diaz I, Martinez M, Mena M, Carbonero P: SAD: a new DOF protein from barley that activates transcription of a cathepsin B-like thiol protease gene in the aleurone of germinating seeds. Plant J. 2003, 33 (2): 329-340. 10.1046/j.1365-313X.2003.01628.x.
Moreno-Risueno MA, Diaz I, Carrillo L, Fuentes R, Carbonero P: The HvDOF19 transcription factor mediates the abscisic aciddependent repression of hydrolase genes in germinating barley aleurone. Plant J. 2007, 51 (3): 352-365. 10.1111/j.1365-313X.2007.03146.x.
Zou X, Neuman D, Shen QJ: Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol. 2008, 148 (1): 176-186. 10.1104/pp.108.123653.
Diaz I, Vicente-Carbajosa J, Abraham Z, Martinez M, Isabel-La Moneda I, Carbonero P: The GAMYB protein from barley interacts with the DOF transcription factor BPBF and activates endosperm-specific genes during seed development. Plant J. 2002, 29 (4): 453-464. 10.1046/j.0960-7412.2001.01230.x.
Rubio-Somoza I, Martinez M, Abraham Z, Diaz I, Carbonero P: Ternary complex formation between HvMYBS3 and other factors involved in transcriptional control in barley seeds. Plant J. 2006, 47 (2): 269-281. 10.1111/j.1365-313X.2006.02777.x.
Wang HW, Zhang B, Hao YJ, Huang J, Tian AG, Liao Y, Zhang JS, Chen SY: The soybean Dof-type transcription factor genes, GmDof4 and GmDof11, enhance lipid content in the seeds of transgenic Arabidopsis plants. Plant J. 2007, 52 (4): 716-729. 10.1111/j.1365-313X.2007.03268.x.
Feuillet C, Muehlbauer GJ: Genetics and genomics of the Triticeae. London: Springer 2009.
Graur D, Li W-H: Fundamentals of molecular evolution. 2nd edition. Sunderland: Sinauer Associates, 2000.
Thornton JW, DeSalle R: Gene family evolution and homology: genomics meets phylogenetics. Annu Rev Genomics Hum Genet. 2000, 1: 41-73. 10.1146/annurev.genom.1.1.41.
Hong SY, Seo PJ, Yang MS, Xiang F, Park CM: Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 2008, 8: 112-10.1186/1471-2229-8-112.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003, 34 (2): 374-378.
Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, Orvis J, Haas B, Wortman J, Buell CR: The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007, 35: D883-D887. 10.1093/nar/gkl976.
Gaur VS, Singh US, Kumar A: Transcriptional profiling and in silico analysis of Dof transcription factor gene family for understanding their regulation during seed development of rice Oryza sativa L. Mol Biol Rep. 2011, 38 (4): 2827-2848. 10.1007/s11033-010-0429-z.
Mena M, Vicente-Carbajosa J, Schmidt RJ, Carbonero P: An endosperm-specific DOF protein from barley, highly conserved in wheat, binds to and activates transcription from the prolamin-box of a native B-hordein promoter in barley endosperm. Plant J. 1998, 16 (1): 53-62. 10.1046/j.1365-313x.1998.00275.x.
Yamamoto MP, Onodera Y, Touno SM, Takaiwa F: Synergism between RPBF Dof and RISBZ1 bZIP activators in the regulation of rice seed expression genes. Plant Physiol. 2006, 141 (4): 1694-1707. 10.1104/pp.106.082826.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2002, 30 (1): 276-280. 10.1093/nar/30.1.276.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25 (24): 4876-4882. 10.1093/nar/25.24.4876.
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O: Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36: W465-469. 10.1093/nar/gkn180.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994, 2: 28-36.
Vogel JP, Gu YQ, Twigg P, Lazo GR, Laudencia-Chingcuanco D, Hayden DM, Donze TJ, Vivian LA, Stamova B, Coleman-Derr D: EST sequencing and phylogenetic analysis of the model grass Brachypodium distachyon. Theor Appl Genet. 2006, 113 (2): 186-195. 10.1007/s00122-006-0285-3.
Lagrimini LM, Burkhart W, Moyer M, Rothstein S: Molecular cloning of complementary DNA encoding the lignin-forming peroxidase from tobacco: Molecular analysis and tissue-specific expression. Proc Natl Acad Sci U S A. 1987, 84 (21): 7542-7546. 10.1073/pnas.84.21.7542.
Onate-Sanchez L, Vicente-Carbajosa J: DNA-free RNA isolation protocols for Arabidopsis thaliana, including seeds and siliques. BMC Res Notes. 2008, 1: 93-10.1186/1756-0500-1-93.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29 (9): e45-10.1093/nar/29.9.e45.
Acknowledgements
We wish to thank Dr. David F. Garvin from USDA-ARS for providing the seeds of the inbred diploid Brachypodium distachyon line Bd21. Financial support from Ministerio de Ciencia e Innovación, Spain (Project BFU2009-11809; principal investigator PC) is gratefully acknowledged. SHA and VGC are recipients of predoctoral fellowships from Ministerio de Educación y Ciencia and from Universidad Politécnica de Madrid, Spain, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SHA has annotated the BdDof genes, done the MEME analysis and performed the phylogenetic analysis; VGC has done the RT-qPCR expression analyses. PC and CBS conceived the study, participated in its design, coordination and interpretation of the data. CBS has written the manuscript and PC has financed the study and edited the final text. All authors have read and approved the final manuscript.
Sara Hernando-Amado, Virginia González-Calle contributed equally to this work.
Electronic supplementary material
12870_2012_1229_MOESM1_ESM.pdf
Additional file 1: Deduced amino acid sequences of all the Brachypodium Dof TFs annotated. The DOF domain sequences used for the alignment are highlighted in bold. (PDF 21 KB)
12870_2012_1229_MOESM2_ESM.pdf
Additional file 2: Distribution of the conserved motifs along the BdDOF transcription factors clustered in Figure2. Motifs identified by means of the MEME software using the deduced amino acid sequences of the 27 Brachypodium BdDof genes represented in Figure 2. Position of the identified motifs is relative to the DOF domain. Multilevel consensus sequences for the MEME defined motifs are listed in Additional file 3. (PDF 13 KB)
12870_2012_1229_MOESM3_ESM.pdf
Additional file 3: Group and sub-group of consensus specific conserved amino acid motifs. Numbers correspond to the motifs described in Additional file 2. Consensus sequences obtained from the analysis of the 27 Brachypodium DOF proteins with the MEME software. Motif 1 corresponds to the DOF DNA-binding domain. (PDF 11 KB)
12870_2012_1229_MOESM4_ESM.pdf
Additional file 4: Primer sequences used for RT-qPCR analyses, amplicon length and PCR efficiency. The corresponding RT-PCR efficiency (E) of one cycle in the exponential phase has been calculated according to the ecuation E=10(−1/slope). (PDF 48 KB)
12870_2012_1229_MOESM5_ESM.pdf
Additional file 5: Expression patterns of the BdDof genes in varios organs of the Brachypodium distachyon plants. The numbers indicate the relative expresion levels standardized to BdGAPDH (mean ± standard error). (PDF 48 KB)
12870_2012_1229_MOESM6_ESM.pdf
Additional file 6: Joined phylogenetic tree of the Brachypodium, barley, rice and the most important Dof genes functionally characterized.(PDF 64 KB)
12870_2012_1229_MOESM7_ESM.pdf
Additional file 7: BdGAPDH expression in different organs (A) and at developmental stage of maturating (B) and germinating (C) seed.(PDF 190 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hernando-Amado, S., González-Calle, V., Carbonero, P. et al. The family of DOF transcription factors in Brachypodium distachyon: phylogenetic comparison with rice and barley DOFs and expression profiling. BMC Plant Biol 12, 202 (2012). https://doi.org/10.1186/1471-2229-12-202
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-202