
Abstract
Background
Vegetative buds provide plants in temperate environments the possibility for growth and reproduction when environmental conditions are favorable. In grapevine, crucial developmental events take place within buds during two growing seasons in consecutive years. The first season, the shoot apical meristem within the bud differentiates all the basic elements of the shoot including flowering transition in lateral primordia and development of inflorescence primordia. These events practically end with bud dormancy. The second season, buds resume shoot growth associated to flower formation and development. Gene expression has been previously monitored at specific stages of bud development but has never been followed along the two growing seasons.
Results
Gene expression changes were analyzed along the bud annual cycle at eight different time points. Principal Components Analysis (PCA) revealed that the main factors explaining the global gene expression differences were the processes of bud dormancy and active growth as well as stress responses. Accordingly, non dormant buds showed an enrichment in functional categories typical of actively proliferating and growing cells together with the over abundance of transcripts belonging to stress response pathways. Differential expression analyses performed between consecutive time points indicated that major transcriptional changes were associated to para/endodormancy, endo/ecodormancy and ecodormancy/bud break transitions. Transcripts encoding key regulators of reproductive development were grouped in three major expression clusters corresponding to: (i) transcripts associated to flowering induction, (ii) transcripts associated to flower meristem specification and initiation and (iii) transcripts putatively involved in dormancy. Within this cluster, a MADS-box gene (VvFLC2) and other transcripts with similar expression patterns could participate in dormancy regulation.
Conclusions
This work provides a global view of major transcriptional changes taking place along bud development in grapevine, highlighting those molecular and biological functions involved in the main events of bud development. As reported in other woody species, the results suggest that genes regulating flowering could also be involved in dormancy regulatory pathways in grapevine.
Similar content being viewed by others

Background
Woody perennial plant species have specific morphological and physiological constraints when compared with annual plants, leading to different reproductive and somatic developmental strategies. Polycarpic woody plants develop terminal or axillary buds with embryonic shoots from which complete branches can develop after specific signals [1]. To elude unfavorable environmental conditions, these buds become dormant providing the possibility to resume growth under viable conditions. Three dormancy states have been distinguished in buds: (i) Paradormancy, induced by distal organs of the plant; (ii) Endodormancy, due to signals internal to the bud itself and (iii) ecodormancy, when bud growth is prevented by environmental factors such as low temperatures [2, 3].
Bud para/endodormancy transition (also known as endormancy onset) is generally triggered by environmental factors. Among them, day length and temperature are stable annual cues regulating this process in many plant species in temperate regions. Endo/ecodormancy transition (or endodormancy release) requires the completion of a chilling period that leaves the bud in an ecodormant stage, susceptible to initiate bud break upon a period of favorable temperatures [4–7]. Regulation of endodormancy onset and release involves plant hormones such as auxins, ethylene, abscisic acid and gibberellins which could also interact with sugars signaling [4, 6]. Epigenetic regulation throughout chromatin modification has also been proposed to be involved in dormancy regulatory processes based on differential expression of several chromatin modifying proteins [5].
In woody species that set dormant terminal buds, cessation of vegetative growth precedes the onset of dormancy and the terminal bud differentiates directly from the shoot apical meristem (SAM). Thus, bud formation is concomitant with dormancy onset [6–8]. In other woody species, including grapevine, axillary buds become dormant when perceiving the environmental signals triggering the dormancy onset, whereas SAM ceases growth when environmental conditions become unfavorable [9, 10].
Little is known about the molecular events underlying bud dormancy in woody species. Photoperiodic regulation of dormancy could involve Phytochromes (PHY) and the circadian clock since over-expression of PHYA prevents short day (SD)-photoperiod endodormancy induction in Populus[11, 12]. In addition, it has been proposed that signaling pathways regulating dormancy onset and release may share genetic components with flowering regulation, for example members of the Flowering Locus T (FT)/ Terminal Flower1 (TFL1) and the MADS-box gene families [5]. Both FT and TFL1 homologs are repressed in Populus and leafy spurge by environmental factors inducing dormancy [5, 11, 12]. Moreover, over-expression of PHYA in transgenic Populus prevents FT and TFL1 homologs repression and the onset of dormancy [12]. Finally, both genes are up regulated by the chilling temperatures causing endodormancy release [13].
Some members of the MADS-box transcription factors family, such as Flowering Locus C (FLC) and Short Vegetative Phase (SVP), involved in the temperature regulation of flowering response in Arabidopsis [14, 15], could participate in the regulation of bud dormancy in woody perennial species. FLC homologs are up regulated during endodormancy and their expression decreases after dormancy release both in Poncirus and leafy spurge [16, 17]. Similarly, SVP homologs, known as Dormancy Associated MADS-box (DAM) genes, have also been involved in growth cessation and terminal bud formation in woody perennial plants such as peach, raspberry, kiwi, apricot, leafy spurge, Poncirus or Populus[18–24]. The peach evergrowing (evg) mutant, has been shown to carry a deletion of six SVP-like genes (PpDAM1–6) resulting in a complete lack of dormancy of the terminal shoot meristems [18]. Considering these evidences, it has been proposed that FLC and SVP-like genes could act by repressing FT expression, as they do during flowering in Arabidopsis [15]. This would provoke growth cessation and/or endodormancy [5, 17].
In grapevine, winter dormant buds develop from basal axillary buds of lateral shoots. These shoots initiate from prompt buds in the same growing season. Dormant buds are complex bract-protected organs constituted by a primary bud and one or two additional secondary buds [9, 10, 25]. Growth and development of these buds are initially prevented by paradormancy signals from the apex. However, these latent buds maintain active processes of cell division and differentiation until perceiving (at the end of the summer) the SD photoperiods and temperatures drop that would trigger the onset of dormancy in grapevine [2, 25]. Endodormancy has been shown to end when buds have already experienced enough chilling [25, 26]. However, buds remain ecodormant until temperature is permissive. When ecodormancy is released, buds swell and the shoot apical meristems (SAMs) in the primary bud follows a program of organ differentiation and growth that gives rise to the new season shoots [10, 27]. In Northern hemisphere, the para/endodormancy transition starts in August and the dormant state is released during November [28]. Flowering induction in grapevine takes place in latent buds during the first growing season, whereas flower meristems and flower organs will develop during the second growing season in the consecutive year after bud break [10, 29, 30]. Processes of inflorescences and flowers initiation and development have been widely reviewed in grapevine [31–33].
Recent works have studied transcriptional characterization of bud responses to chilling [34], photoperiod [35] and dormancy breaking treatments [36, 37]. Their results have led to the identification of candidate genes with a dual role in flowering and dormancy in Vitis riparia[34, 35] and to propose of a role for oxidative stress as part of the dormancy releasing mechanisms [36–38].
In this study we have followed the grapevine bud transcriptome along the complete annual cycle which includes two growing seasons. Bud transcriptome analyses identified three major phases of transcriptome change, mainly associated to dormancy. Furthermore, the expression of members of the MIKC-type MADS-box gene family [39], the SPL (Squamosa Promoter Binding Protein-Like) family, the FT-TFL1 family [31] and the VFL gene (the grapevine Floricaula/Leafy ortholog) [40], was also analyzed during bud development.
Results and discussion
Bud transcriptome variation along the annual cycle
Grapevine bud development is modulated by environmental factors such as temperature and day length [25]. In our experimental conditions, Tempranillo cv. latent buds are formed during the first growing season in the young sprouting stems between April and May (APR-MAY) and experience active developmental processes involved in the set up of the vegetative and reproductive growth until the end of the summer of the following year (Figure 1) [32]. Flowering induction takes place within latent buds around the middle of June (JUN) and inflorescence primordia differentiate from lateral meristems developed by the shoot apex. Inflorescence meristems proliferate to generate inflorescence branch meristems in complex inflorescence primordia along July (JUL) and August (AUG) [40]. In our growing conditions, Tempranillo buds are endodormant in the second half of September (SEP) and endodormancy is released by the end of November (NOV), although buds remain in an ecodormant stage from DEC to MAR [41]. Once winter is over, ecodormancy is released and inflorescence branch meristems proliferate to produce flower meristems in April (APR) swelling buds that initiate the second growing season [40].

Bud developmental evolution along the year. A) Environmental change in temperature and day length. B) Pictures depicting grapevine axillary buds along the year, corresponding to May, June and July, dormant buds from September to March, and during bud break in April. Temperatures are shown as monthly minimum average (blue line), monthly maximum average (orange line) and monthly average (yellow line). Day length (grey line) is shown as light hours per day. Assignments of bud stages were based on previous data on cv. Tempranillo [40, 41].
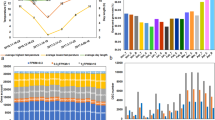
High throughput transcriptional analysis was performed along bud development on bud samples collected at 8 different time points during the annual cycle (see Methods). Principal Components Analysis (PCA) was performed on the whole expression dataset (Additional file 1) to verify correlation among different biological replicates and to identify main sources of gene expression variation. The results of the PCA plot showed consistency across biological replicates, as shown in Figure 2. The first two principal components (PC1 and PC2) explained 77.5 percent of the total variability in gene expression (62.2 percent and 15.3 percent respectively). PC1 seems to represent the time course evolution of bud developmental stages and appears to be reset to the original status with bud swelling, since APR bud samples of the second season were neighboring to MAY bud samples of the first season (in the same quadrant), revealing a high transcriptome similarity between those two stages. PC2 highlights major transcriptome differences between JUN, JUL and SEP samples and the remaining time points.

Bi-dimensional loading score plot of sample replicates resulting from PCA analysis. X axis represents PC1 that explains 62.2% and Y axis represents PC2 that explains 15.3% of the total variability for gene expression. Samples belonging to the same time-point are represented by the same color.
To investigate the biological basis of the principal components, transcripts with the highest contribution to each component in the analysis were identified according to the absolute value of their component score (CS) for PC1 and PC2 (Additional files 2 and 3). Figure 3A shows the expression profiles of transcripts mostly contributing to PC1. Transcripts with negative CS values (547 transcripts, blue color) are up-regulated in non-dormant buds and show declining expression during endodormancy and the lowest expression during ecodormancy. Transcripts with positive CS values (204 transcripts, orange color) follow the opposite trend (Figure 3A). Functional enrichment analyses indicated that the transcripts up-regulated in non-dormant buds were characteristic of actively proliferating and growing cells (Figure 3B). Among them, transcripts related with cell division, cell growth and differentiation (cell-cycle regulation, microtubule-driven movement, chromatin assembly, cell growth, peptidase-mediated proteolysis and cell wall metabolism) and those related with primary and secondary metabolism (photosynthesis, fatty acid biosynthesis, flavonoid biosynthesis and aromatic compounds glycosidation) showed high CS value.

Top-scored transcripts for Principal Component 1. A) Expression profiles of the transcripts with positive component score values (orange lines) and negative component score values (blue lines). Each single line represents the average of mean-centered expression values for an individual transcript. B) Functional categories over-represented in each cluster. Color code is the same as in A. Absolute values of the log10 transformed P-values were used for the bar diagram representing statistical signification, only categories with P-values < 0.05 were shown. BF, bud formation; FI, flowering induction; END, endodormancy; ECD, ecodormancy; GR, growth resumption.
On the other hand, the most significant functional categories contributing to PC1 and enriched in dormant buds (orange color) were those related to stress responses (stilbenoid biosynthesis, "HSP-mediated protein folding", temperature stress response, as well as CCAAT transcription factor family) [42]. These functions could be related to bud responses to dehydration and temperature changes that take place together with dormancy, as previously reported in Populus[24]. Furthermore, the observed up-regulation of genes involved in ABA catabolism could be related to the decay of this hormone previous to dormancy release [5]. Finally, an increase in the expression of starch catabolism genes together with a down-regulation of genes encoding photosynthetic proteins, also observed in Populus[24], is in agreement with the physiological state of dormant buds.
Regarding PC2, the enriched functional categories contributing to its negative values were those related to stress responses characteristic of JUN, JUL and SEP samples, whereas the functional categories associated with its positive values were those related to cell proliferation that show higher expression in non-dormant buds (Additional file 4). Therefore, PC2 could represent the transcriptional effects of the stress experienced by buds from JUN to SEP with respect to the remaining stages.
In summary, our results suggest that active growth, dormancy and stress responses are major contributors to the gene expression variability observed along the bud annual cycle. Processes characteristic of actively proliferating and growing cells are up-regulated in non-dormant buds and decline during bud dormancy together with the up-regulation of stress response pathways.
Transcriptome changes in bud developmental transitions
In order to identify the developmental stages representing major transcriptome changes during the annual cycle of the bud, we performed a pair wise differential expression analysis between consecutive time points. The number of differentially expressed genes varied strikingly among sample pair comparisons (Figure 4). Major changes were observed between JUL and SEP (involving 3139 transcripts), SEP and NOV (involving 2002 transcripts) and MAR and APR (involving 5658 transcripts). Interestingly, these transitions could be associated, respectively, to the proposed timing for para/endodormancy, endo/ecodormancy and ecodormancy/bud break transitions [28, 41]. These results supported the conclusions of the PCA experiment, suggesting that the transitions between bud dormancy and active growth explains most of the variation in bud gene expression profiles.

Differential expressions between consecutive bud time points. The histogram represents the number of differentially expressed transcripts between each consecutive sample pair, sense of the bars indicates whether transcripts were differentially expressed in the earlier versus later stage (on the left side in blue) or in later stage versus the earlier one (on the right side in red). Numbers correspond to the number of non redundant transcripts in each class.
In order to identify biological functions involved in the three major bud transcriptional changes we performed studies of functional categories enrichment (Figure 5). The para/endodormancy transition (JUL to SEP) (Figure 5A) was characterized by a major reduction in enriched functional categories that mostly contributed to PC1 in Figure 3. Among them, categories related to cell proliferation (including regulation of cell cycle, chromatin assembly and microtubule organization and biogenesis) and cell growth and death, were down regulated from JUL to SEP. These results are consistent with the shutdown of those processes during bud dormancy that has been described in other systems [24, 43]. Cell wall organization and biogenesis, also linked to cellular processes, was significantly reduced with endodormancy. Modification of the cell wall, mainly xyloglucans metabolism and cell wall proteins, was an enriched category in JUL samples but not in SEP. However, cell wall biosynthesis (mainly including cellulose biosynthesis transcripts) appeared as a significant category in SEP. Metabolism of carbohydrates was in some aspects related to cell wall metabolism, thus 1,3-β-glucan catabolism predominated in JUL while in SEP carbohydrate metabolism was mainly represented by starch and sucrose metabolism. These results could be related to the required sealing of plasmodesmata with callose to lower their size exclusion limit, a process dependent on 1,3-β-glucansynthase and 1,3-β-glucanase activities. Additionally, cell wall composition must be modified to reduce water and molecules movement among cells during this stage [44].

Functional categories over-represented in the pair wise comparisons. Bar chart summarizes the significantly enriched functional categories between stages flanking these transitions. The sense of the bars indicate whether transcripts were differentially expressed in the earlier versus later stage (on the left side) or in later stage versus the earlier one (on the right side). Absolute values of the log10 transformed P-values (of the enrichment analysis) were used for plotting, only categories with P-values lower than threshold (0.05) were shown.
Representation of photosynthesis-related functional categories also decreased during para/endodormancy transition, in agreement with results reported in Populus[24]. Lipid metabolism (including fatty acid biosynthesis, glycerolipds metabolism and oxylipins) was also under-represented in this transition. This change could be related to the formation of lipid bodies (LBs) which store triacylglicerols and improve freezing tolerance previous to endodormancy, as has been reported in other systems [44]. However, as far as we know, no cytological evidence on the formation of LBs exists in grapevine buds.
Other significant functional categories that were under-represented from JUL to SEP were those related to stress responses (mainly abiotic and temperature), chaperone-mediated protein folding and aquaporins (mainly TIP). In Populus, genes responsible for adaptation to dehydration and low temperatures have been shown to be expressed in response to SD even in the absence of those stresses [24]. In leafy spurge, genes responsive to cold stress were also up-regulated in fall and winter [43]. Within the signaling pathways functional category, 129 transcripts were down-regulated between JUL and SEP. Among them, salicylic acid-mediated signaling was significantly enriched, what could be related to stress responses as well as elicitation of phytoallexin biosynthesis [45].
The transcription factor functional category was also significantly enriched among transcripts down-regulated in this comparison. Among them, a significant enrichment was also proven for the ERF subfamily (9 transcripts), within the AP2-like [46] transcription factor family, most of them related to ethylene regulated responses. In Populus, the temporal expression of some ERF-like transcripts at the beginning of SD photoperiods has suggested a role for ethylene in the regulation of dormancy [24]. In fact, Populus, leafy spurge and potato have a transient peak in ethylene or ethylene perception associated with endodormancy induction [5].
Only a few functional categories were significantly enriched among transcripts showing up-regulation from JUL to SEP. Apart from those concerning cell wall organization and biogenesis and carbohydrate metabolism that were previously described, functional categories related to metabolism of nucleotides and nucleic acids and protein metabolism were also significant. Among them, enrichment of RNA metabolism and translation initiation could reveal the existence of mechanisms relying on stored mRNAs transcripts ready to be translated, as described in dry angiosperm seeds [47].
The endo/ecodormancy transition (SEP to NOV) (Figure 5B) was characterized by a further decline in transcripts participating in cellular processes as well as primary and secondary metabolism functional categories, similar to what was observed in the para/endodormancy transition. Functional categories related to cell wall metabolism and biogenesis (mainly based on biosynthesis of cellulose, catabolism of pectins and modification of pectins and xyloglucans) were still relevant in SEP and further down-regulated in NOV. Moreover, carbohydrate metabolism-related transcripts that were down-regulated in SEP versus NOV corresponded now to oligosaccharides metabolism and glucans catabolism. A parallel situation has been reported in Vitis riparia during the chilling period required for endodormancy release [34]. Stress responses were also under-represented from SEP to NOV, especially categories related to oxidative stress. An activation of the oxidative stress response machinery preceding endodormancy release would be in agreement with previous reports showing that oxidative stress affecting mitochondrial function could participate in endodormancy release in grapevine [36, 37].
On the other hand, the SEP to NOV transition was marked by an enrichment of ABA catabolism, in agreement with the role of ABA in endodormancy and its decay during endo/ecodormancy transition [5]. ABA levels correlate with bud dormancy in several species and decay throughout the transition to ecodormancy. Both in Populus and leafy spurge, ABA content peaked after few weeks of SD and decayed later [24, 43]. Moreover, an ABA-related transcript has also been reported to be down-regulated during the chilling period required for endodormancy release in grapevine [34]. Buds of NOV also showed an enrichment of stilbenoid biosynthesis that in grapevine usually responds to biotic and abiotic elicitors [48]. Finally, enrichment of nicotinate and nicotinamide metabolism in NOV as well as nucleotide and amino acid transport might suggest the initiation of certain metabolic activity paralleling the endo/ecodormancy transition. Interestingly, the ERF subfamily of transcription factors that was significantly enriched in JUL versus SEP was also found in NOV versus SEP. Moreover, five of these transcripts were common in JUL and NOV, suggesting common functions before para/endodormancy and after endo/ecodormancy transitions.
The ecodormancy/budbreak transition (MAR to APR) (Figure 5C) was characterized by down-regulation of specific functional categories involved in starch and sugar catabolism or signaling and stress responses mainly related to previous periods. Similar underrepresentations were also observed in transcription factor families known to be involved in stress adaptive responses such as HSF [49], NAC [50] and WRKY [51] families, as has been reported in other species [19, 43]. In contrast, bud break in APR was characterized by the presence of most functional categories that were enriched in JUL before bud para/endodormancy transition. Those categories were related to cellular processes required for cell proliferation (chromatin assembly, microtubule organization and biogenesis, cell cycle regulation) and cell growth (cell growth and death, cell wall organization and biogenesis, photosynthesis and primary and secondary metabolism). Up-regulation of cell wall organization and biogenesis and carbohydrate metabolism categories (mono, oligo and polysaccharides and more specifically glucans and 1,3-β-glucan catabolism), could also be related to restoring cell wall properties and cell communication throughout callose hydrolysis at plasmodesmata. Induction of 1,3-β-glucanase after the application of dormancy-release agents has also been reported in grapevine [38]. Secondary metabolism categories included enrichment of flavonoid biosynthesis (mainly anthocyanins) as well as aromatic compound and shikimate metabolism as probable flavonoid precursors. Interestingly, the significantly enriched hormone signaling functional categories were those related to auxin and salicylic acid, that likely have a role in cell proliferation and expansion [52] as well as in the response to biotic stress [45].
Expression profiles of key regulators of reproductive development
Flowering induction in grapevine takes place in latent buds at the beginning of the summer whereas flower meristem differentiation and flower development take place in the second growing season [10, 30–33]. To identify putative genes involved in flowering induction and flower development, we examined in detail the expression profiles of reproductive development key genes such as VFL, the MIKC-type MADS-box, the SPL and the FT-TFL1 gene families. Hierarchical clustering based on bud expression values of these transcripts along the annual cycle were represented in Figure 6. Consistently with the main bud transcriptional profiles described in the previous section, expression analysis of these transcripts identified three major distinct clusters. The first cluster grouped transcripts up-regulated in non-dormant buds and down-regulated during dormancy. The second cluster contained transcripts with highest expression level during bud break. The third cluster grouped transcripts up-regulated in dormant buds with an opposite expression to those of cluster 1.

Hierarchical clustering of expression profiles of key regulators of reproductive development along bud development. MADS-box gene family (blue boxes), SPL gene family (green boxes), FT-TFL1 gene family (orange boxes) and the VFL gene (red box). Color scale (on top) represents mean-centered expression values.
Cluster 1 showed transcripts expressed in latent buds when inflorescence primordia are initiated and proliferate. A well characterized gene within this cluster is the MADS-box gene VvSOC1.1[39], which expression pattern suggests that it could play a crucial role in flowering induction, as does SOC1, its Arabidopsis homolog [53]. Other MADS-box genes in cluster 1 were members of the FLC (VvFLC1) and SVP subfamilies (VvSVP1, VvSVP3 and VvSVP5) [39]. VvFLC1 showed high expression during the first season, decreased during dormancy and increased again during the second growing season (Additional file 5). Expression patterns of the three grapevine SVP homologs were distinct than that observed for SVP in Arabidopsis [54]. Their expression levels were high at flowering induction, reduced during dormancy and increased again at flower meristem formation. No significant expression changes of SVP homologs were observed along grapevine bud dormancy, what would not justify their role in that process, as has been reported in other species [18–24]. Cluster 1 also included VvMFT1[31], a member of the FT-TFL1 family of transcriptional regulators [55, 56], with highest expression during the first season. In addition, detection within this cluster of APETALA3.2, PISTILLATA and AGAMOUS homologs suggests that these genes, involved in the specification of flower organs identity, could already be expressed in the inflorescence meristems of first season buds in preparation for the flower organ specification that takes place during the second growing season. Finally, cluster 1 included many homologs of A. thaliana SPL genes (SPL2, SPL3, SPL4, SPL5, SPL8 and SPL9) what will be discussed later when considering the expression patterns observed within this gene family.
Cluster 2 grouped transcripts showing their highest expression during bud break in the second growing season and likely associated with the events of flower meristems and flower organs differentiation. Consistently with the developmental processes taking place in bud break, cluster 2 contained both VvFT and VFL, which have homologs in Arabidopsis that are required for flower induction and flower meristem specification. VvFT was over-expressed during flowering induction, decreased during bud dormancy and increased again during the second season, which is compatible with the roles proposed for its homologs in Populus and other woody plants [7, 11, 13, 43]. Although the putative TFL1 homolog (VvTFL1A) was not present in the Grapegen GeneChip®, our previous analysis of this gene family [31] showed that it is down-regulated during dormancy and up-regulated with dormancy release. This expression pattern parallels what has been reported for TFL1 homologs in Populus[13]. VvSPL13-L and VvAG3 on one side and VvFUL-L, VvAGL6.2 and VvSEP4 on the other, showed a similar expression pattern and could also participate in the processes of flower meristem and flower organ specification.
Cluster 3 included transcripts showing their highest expression during dormancy. Interestingly, this cluster included two MADS-box genes, VvFLC2 and VvAGL15.1, which Arabidopsis homologs have been involved in flowering repression. In addition, it contains several SPL genes (VvSPL6-L, VvSPL12-L and VvSPL14-L). The remaining genes also showed some expression in non-dormant stages. The two FLC homologs found in grapevine (VvFLC1 and VvFLC2) [39] showed an opposite expression pattern in buds (Additional file 5). VvFLC1 behaved as PEP1, the Arabis alpina FLC homolog. PEP1 expression is reduced during the winter and increases when growth is resumed after the cold period [57]. Fluctuations in the FLC transcript levels in this perennial species would allow some meristems to undergo flowering transition while others maintain the vegetative growth of the plant. A similar role for VvFLC1 in maintaining vegetative growth of the young meristems that will give rise to the new latent buds could be proposed in grapevine. In contrast, the opposite expression pattern of VvFLC2, resembles expression reported for FLC-like genes in Poncirus[16] and leafy spurge [17], where they could be involved in the regulation of dormancy. Similarly, a role as repressor of flower meristem initiation has been proposed for AGL15 and AGL18 in Arabidopsis [58]. Interestingly, their putative grapevine homolog, VvAGL15.1, has the same expression pattern as VvFLC2. The search of FLC gene homologs with opposite expression pattern in other polycarpic plant species could help to elucidate their role in these processes. In addition, two members of the FT/TFL1 gene family (VvTFLC1 and VvMFT2) [31] were also found in cluster 3, opening the possibility that their function could be related with the control of dormancy in grapevine. In agreement, high expression of a member of the FT/TFL1 family (PaFT4) in Norway spruce (Picea abies L.) correlates with growth cessation and bud set [59], in contrast to that observed for the PtFT1 gene of Populus[11]. Indeed, comparative sequence and expression analysis of the FT/TFL1 family in gymnosperm and angiosperm species lead to speculate that the original function of this gene family could be related to the regulation of growth arrest and/or dormancy [60].
Different members of the SPL family of transcription factors were found within the three clusters. This family of transcription factors is known to participate in the regulation of diverse plant developmental processes such as plant phase transition, flower and fruit development and plant architecture [61–64]. Ten of the 16 Arabipdopsis SPL genes are post-transcriptionally regulated by miR156, which incorporates endogenous age/development signals into vegetative phase transition and flowering [61, 62]. This vegetative phase regulatory mechanism is also conserved in woody perennials [65]. Interestingly, cluster 1 contains transcripts homologous to SPL3, 4, 5 and 9, all belonging to the miR156/7-targeted SPL subfamily, which act as positive regulators of juvenile-to-adult phase change transition and flowering in Arabidopsis [63, 64, 66] and are regulated by SOC1[67]. Cluster 1 also included two SPL2 and one SPL8 homologs. Arabidopsis SPL2 is also miR156 targeted and seems to be involved in lateral organ development within the reproductive phase [68]. The miR156/7 non-targeted SPL8 gene is involved in pollen sac development [69] and required for male fertility [70]. Finally, several members of this gene family were found in Cluster 3 displaying an expression pattern more restricted to the dormancy period (VvSPL1-L, VvSPL6-L, VvSPL7-L, VvSPL12-L, VvSPL14-L and VvSPL13-L2). Most Arabidopsis counterparts of these genes belong to the miR156/7 non-targeted SPL subfamily (SPL1, 7, 12, 14 and 16) that comprises the larger proteins in the family [70]. Little is known about the functions of these putative transcriptional regulators with the exception of SPL14, which seems to regulate plant architecture and the length of vegetative phase. An Arabidopsis mutant with reduced SPL14 expression, had elongated petioles, serrated leaf margins and accelerated vegetative phase change [71], suggesting that this gene could play a role as a negative regulator of phase transition and flowering [72], having antagonistic function to other SPL proteins that promote vegetative phase change. Interestingly, both VvSPL14-L and VvSPL12-L showed an expression pattern very similar to VvFLC2 and VvAGL15.1 which could also suggest a role for these SPL genes in dormancy maintenance. Little is known about the role of SPL-like genes in woody species. Two genes (SPL-like 3 and 6) were detected during dormancy in Populus[24] with SPL6-like increasing and SPL3-like decreasing along dormancy. In addition, an SPL-2 homolog has also been found in grapevine which seems to be regulated by photoperiod [35]. Further studies will be needed to elucidate the possible role of the SPL gene family in bud dormancy.
Additional mechanisms involving transcriptional repressors could be required during the dormancy period to prevent premature flower meristems formation from the inflorescence meristems. The establishment of annual and perennial life has independently arisen several times in flowering plants [73], so it is likely that mechanisms involved in the control of bud dormancy or repression of flower meristem formation have recruited different regulatory genes in different botanical families. Complementary experiments will be needed to assess the biological function of the members of these transcriptional regulators families in such processes.
Conclusions
Transcriptional analyses along bud development have shown that principal components explaining the observed expression variability are determined by genes involved in active cell growth and proliferation, dormancy regulation and stress responses, indicating that these are the most active events in bud development. Major transcriptional changes were detected between samples collected in July and September (para/endodormancy transition), September and November (endo/ecodormancy transition) and March and April (ecodormancy/bud break transition). The functional categories enriched in these transitions are in agreement with the results of the Principal Component analysis.
Expression profiles of key regulators of reproductive development were assigned to three major transcriptional clusters corresponding to (i) transcripts associated only to flowering induction; (ii) transcripts associated to flowering induction and flower meristem initiation; and (iii) transcripts putatively involved in dormancy. Those results suggest that VvFLC2 and other transcripts with similar expression patterns such as VvAGL15.1 or VvSPL14-L could have a role in bud dormancy regulation in grapevine whereas no evidence for a participation of VvSVP genes in this process could be observed.
Methods
Plant materials
Grapevine (Vitis vinifera L. cultivar Tempranillo) buds were obtained from an experimental vineyard at the Instituto Madrileño de Investigación y Desarrollo Rural, Agrario y Alimentario (IMIDRA, Alcalá de Henares, Madrid). Samples were collected from triplicate blocks in the same vineyard during two consecutive years. Plants and buds developmental stages were classified following the developmental series of Baggiolini (1952) [74] and modified E-L system [75]. Buds were collected at equivalent stem positions from the base and always at the same time of the day. Bud samples were frozen in liquid nitrogen and stored at -80°C before RNA extraction. Samples corresponding to May (MAY), June (JUN), July (JUL), September (SEP), November (NOV), January (JAN), March (MAR) and April (APR) buds were analyzed. Meteorological data were obtained from a station at Finca El Encin (IMIDRA, Alcalá de Henares).
RNA extraction
Total RNA was extracted from frozen bud samples according to Reid et al., 2006 [76]. RNA purification was performed using the RNeasy Mini Kit (QIAGEN) according to manufacturer's protocols. To remove DNA traces in RNA samples, DNase I digestion was carried out with the RNase-Free DNase Set (QIAGEN). RNA integrity and quantity were assessed by Agilent’s Bioanalyzer 2100. Microarray hybridizations were performed at the Genomics Unit of the National Centre for Biotechnology (CNB-CSIC, Madrid). Raw microarray data from the reported experiments are publicly available at the Plant Gene Expression Database (PlexDB) [77] and labelled as “VV36: Time course of grapevine bud development”.
Microarray data processing and analysis
Monitoring of bud transcriptional activity was performed in three biological replicates for each time point using Affymetrix Grapegen GeneChip®. Raw Affymetrix CEL files were imported to Robin software suite [78] to perform data normalization using the RMA method. Principal component analysis was performed using Acuity software [79] (Molecular Devices, LLC, CA, US). The score matrix was used to select probe-sets that best fit the first principal component (PC1) and those with PC1 scores greater than 7 or lower than -7 were chosen. Likewise, probe-sets that best fitted PC2 were those with component score greater than 3 or lower than -3.
Differential expression analyses were performed in Multi Experiment Viewer [80] using LIMMA, applying a 0.01 cut-off for P-value and log2 fold ratio greater than 1 and lower than -1. P values were corrected using the Benjamini-Hochberg test.
To identify the biological functions over-represented within selected probe sets functional enrichment analyses were performed using FatiGO [81] (P-value< 0.05). Functional categories were based on manual annotation of the custom made GrapeGen GeneChip®, based on 12X v1 grape genome assembly, described in Grimplet et al., 2012 [82].
Key regulators of reproductive development were selected according to their functional annotation [82]. Expression values were extracted from the whole experiment normalized data matrix (averaged from the triplicates per sample and probe set). When more than one probe set matched a single gene transcript, only one was selected. Hierarchical clustering was performed using MultiExperiment Viewer [80] based on Pearson's correlation and using the complete linkage option.
References
Horvath DP, Anderson JV, Chao WS, Foley ME: Knowing when to grow: signals regulating bud dormancy. Trends Plant Sci. 2003, 8: 534-540. 10.1016/j.tplants.2003.09.013.
Lang G, Early J, Martin G, Darnell R: Endodormancy, paradormancy, and ecodormancy - physiological terminology and classification for dormancy research. HortScience. 1987, 22: 371-377.
Rohde A, Bhalerao RP: Plant dormancy in the perennial context. Trends Plant Sci. 2007, 12: 217-223. 10.1016/j.tplants.2007.03.012.
Chao WS, Foley ME, Horvath DP, Anderson JV: Signals regulating dormancy in vegetative buds. Intl J Plant Dev Biol. 2007, 1: 49-56.
Horvath D: Common mechanisms regulate flowering and dormancy. Plant Sci. 2009, 177: 523-531. 10.1016/j.plantsci.2009.09.002.
Anderson J, Horvath D, Chao W, Foley M: Bud dormancy in perennial plants: a mechanism for survival. In Dormancy and Resistance in Harsh Environments. 2010, Berlin, 69-90. Lubzens, Esther and Cerda, Joan and Clark, Melody (Series Editor): Topics in Current Genetics, vol 21.
Olsen JE: Light and temperature sensing and signaling in induction of bud dormancy in woody plants. Plant Mol Biol. 2010, 73: 37-47. 10.1007/s11103-010-9620-9.
Jansson S, Douglas CJ: Populus: a model system for plant biology. Annu Rev Plant Biol. 2007, 58: 435-458. 10.1146/annurev.arplant.58.032806.103956.
Morrison J: Bud development in Vitis vinifera L. Bot Gaz. 1991, 152: 304-315. 10.1086/337894.
Mullins MG, Bouquet A, Williams LE: Biology of the Grapevine. Cambridge: Cambridge University Press 1992.
Böhlenius H, Huang T, Charbonnel-Campaa L, Brunner AM, Jansson S, Strauss SH, Nilsson O: CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science. 2006, 312: 1040-1043. 10.1126/science.1126038.
Ruonala R, Rinne PLH, Kangasjärvi J, van der Schoot C: CENL1 expression in the rib meristem affects stem elongation and the transition to dormancy in Populus. Plant Cell. 2008, 20: 59-74. 10.1105/tpc.107.056721.
Rinne PLH, Welling A, Vahala J, Ripel L, Ruonala R, Kangasjärvi J, van der Schoot C: Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-beta-glucanases to reopen signal conduits and release dormancy in Populus. Plant Cell. 2011, 23: 130-146. 10.1105/tpc.110.081307.
Hemming MN, Trevaskis B: Make hay when the sun shines: the role of MADS-box genes in temperature-dependant seasonal flowering responses. Plant Sci. 2011, 180: 447-453. 10.1016/j.plantsci.2010.12.001.
Lee JH, Yoo SJ, Park SH, Hwang I, Lee JS, Ahn JH: Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21: 397-402. 10.1101/gad.1518407.
Zhang J, Li Z, Mei L, Yao J, Hu C: PtFLC homolog from trifoliate orange (Poncirus trifoliata) is regulated by alternative splicing and experiences seasonal fluctuation in expression level. Planta. 2009, 229: 847-859. 10.1007/s00425-008-0885-z.
Doğramaci M, Horvath DP, Chao WS, Foley ME, Christoffers MJ, Anderson JV: Low temperatures impact dormancy status, flowering competence, and transcript profiles in crown buds of leafy spurge. Plant Mol Biol. 2010, 73: 207-226. 10.1007/s11103-010-9621-8.
Bielenberg DG, Wang Y, Li Z, Zhebentyayeva T, Fan S, Reighard GL, Scorza R, Abbott AG: Sequencing and annotation of the evergrowing locus in peach (Prunus persica (L.) Batsch) reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet Genomes. 2008, 4: 495-507. 10.1007/s11295-007-0126-9.
Mazzitelli L, Hancock RD, Haupt S, Walker PG, Pont SDA, McNicol J, Cardle L, Morris J, Viola R, Brennan R, Hedley PE, Taylor MA: Co-ordinated gene expression during phases of dormancy release in raspberry (Rubus idaeus L.) buds. J Exp Bot. 2007, 58: 1035-1045. 10.1093/jxb/erl266.
Wu R, Walton EF, Richardson AC, Wood M, Hellens RP, Varkonyi-Gasic E: Conservation and divergence of four kiwifruit SVP-like MADS-box genes suggest distinct roles in kiwifruit bud dormancy and flowering. J Exp Bot. 2012, 63: 797-807. 10.1093/jxb/err304.
Yamane H, Kashiwa Y, Ooka T, Tao R, Yonemori K: Suppression subtractive hybridization and differential screening reveals endodormancy-associated expression of an SVP/AGL24-type MADS-box gene in lateral vegetative buds of japanese apricot. J Amer SocHort Sci. 2008, 133: 708-716.
Horvath DP, Sung S, Kim D, Chao W, Anderson J: Characterization, expression and function of DORMANCY ASSOCIATED MADS-BOX genes from leafy spurge. Plant Mol Biol. 2010, 73: 169-179. 10.1007/s11103-009-9596-5.
Li Z, Zhang J, Mei L, Deng X, Hu C, Yao J: PtSVP, an SVP homolog from trifoliate orange (Poncirus trifoliata L. Raf.), shows seasonal periodicity of meristem determination and affects flower development in transgenic Arabidopsis and tobacco plants. Plant Mol Biol. 2010, 74: 129-142. 10.1007/s11103-010-9660-1.
Ruttink T, Arend M, Morreel K, Storme V, Rombauts S, Fromm J, Bhalerao RP, Boerjan W, Rohde A: A molecular timetable for apical bud formation and dormancy induction in poplar. Plant Cell. 2007, 19: 2370-2390. 10.1105/tpc.107.052811.
Lavee S, May P: Dormancy of grapevine buds - facts and speculation. Aust J Grape Wine Res. 1997, 3: 31-46. 10.1111/j.1755-0238.1997.tb00114.x.
Dokoozlian N: Chilling temperature and duration interact on the budbreak of 'Perlette' grapevine cuttings. HortScience. 1999, 34: 1054-1056.
Gerrath J, Posluszny U: Morphological and anatomical development in the Vitaceae. 2. Vegetative development in Vitis riparia. Can J Bot. 1988, 66: 209-224. 10.1139/b88-037.
Pouget R: Considérations générales sur le rythme végétatif et la dormance des bourgeons de la Vigne. Vitis. 1972, 11: 198-217.
Gerrath J, Posluszny U: Morphological and anatomical development in the Vitaceae. 2. Floral development in Vitis riparia. Can J Bot. 1988, 66: 1334-1351. 10.1139/b88-188.
Gerrath JM: Developmental morphology and anatomy of grape flowers. Hortic Rev. 1993, 13: 315-337.
Carmona MJ, Calonje M, Martínez-Zapater JM: The FT/TFL1 gene family in grapevine. Plant Mol Biol. 2007, 63: 637-650. 10.1007/s11103-006-9113-z.
Carmona MJ, Chaïb J, Martínez-Zapater JM, Thomas MR: A molecular genetic perspective of reproductive development in grapevine. J Exp Bot. 2008, 59: 2579-2596. 10.1093/jxb/ern160.
Lebon G, Wojnarowiez G, Holzapfel B, Fontaine F, Vaillant-Gaveau N, Clement C: Sugars and flowering in the grapevine (Vitis vinifera L.). J Exp Bot. 2008, 59: 2565-2578. 10.1093/jxb/ern135.
Mathiason K, He D, Grimplet J, Venkateswari J, Galbraith DW, Or E, Fennell A: Transcript profiling in Vitis riparia during chilling requirement fulfillment reveals coordination of gene expression patterns with optimized bud break. Funct Integr Genomics. 2009, 9: 81-96. 10.1007/s10142-008-0090-y.
Sreekantan L, Mathiason K, Grimplet J, Schlauch K, Dickerson JA, Fennell AY: Differential floral development and gene expression in grapevines during long and short photoperiods suggests a role for floral genes in dormancy transitioning. Plant Mol Biol. 2010, 73: 191-205. 10.1007/s11103-010-9611-x.
Halaly T, Pang X, Batikoff T, Crane O, Keren A, Venkateswari J, Ogrodovitch A, Sadka A, Lavee S, Or E: Similar mechanisms might be triggered by alternative external stimuli that induce dormancy release in grape buds. Planta. 2008, 228: 79-88. 10.1007/s00425-008-0720-6.
Perez FJ, Kuhn N, Ormeno-Nunez J, Rubio S: The expression of VvPHYA and VvPHYB transcripts is differently regulated by photoperiod in leaves and buds of grapevines. Plant Signal Behav. 2009, 4: 614-616. 10.4161/psb.4.7.8907.
Pacey-Miller T, Scott K, Ablett E, Tingey S, Ching A, Henry R: Genes associated with the end of dormancy in grapes. Funct Integr Genomics. 2003, 3: 144-152. 10.1007/s10142-003-0094-6.
Díaz-Riquelme J, Lijavetzky D, Martínez-Zapater JM, Carmona MJ: Genome-wide analysis of MIKCC-type MADS-box genes in grapevine. Plant Physiol. 2009, 149: 354-369. 10.1104/pp.108.131052.
Carmona MJ, Cubas P, Martínez-Zapater JM: VFL, the grapevine FLORICAULA/LEAFY ortholog, is expressed in meristematic regions independently of their fate. Plant Physiol. 2002, 130: 68-77. 10.1104/pp.002428.
Martínez de Toda F: Biología de la vid. Fundamentos biológicos de la viticultura. Madrid: Mundi-Prensa 1991.
Nilsson L, Müller R, Nielsen TH: Dissecting the plant transcriptome and the regulatory responses to phosphate deprivation. Physiol Plant. 2010, 139: 129-143. 10.1111/j.1399-3054.2010.01356.x.
Horvath DP, Chao WS, Suttle JC, Thimmapuram J, Anderson JV: Transcriptome analysis identifies novel responses and potential regulatory genes involved in seasonal dormancy transitions of leafy spurge (Euphorbia esula L.). BMC Genomics. 2008, 9: 536. 10.1186/1471-2164-9-536.
van der Schoot C, Rinne PLH: Dormancy cycling at the shoot apical meristem: transitioning between self-organization and self-arrest. Plant Sci. 2011, 180: 120-131. 10.1016/j.plantsci.2010.08.009.
Li X, Zheng X, Yan S, Li S: Effects of salicylic acid (SA), ultraviolet radiation (UV-B and UV-C) on trans-resveratrol inducement in the skin of harvested grape berries. Front Agric China. 2008, 2: 77-81. 10.1007/s11703-008-0014-6.
Dietz K, Vogel MO, Viehhauser A: AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma. 2010, 245: 3-14. 10.1007/s00709-010-0142-8.
Kimura M, Nambara E: Stored and neosynthesized mRNA in Arabidopsis seeds: effects of cycloheximide and controlled deterioration treatment on the resumption of transcription during imbibition. Plant Mol Biol. 2010, 73: 119-129. 10.1007/s11103-010-9603-x.
Bavaresco L, Fregoni C, Van-Zeller-de-Macedo-Basto-Gonçalves MI, Vezzulli S: Physiology & Molecular Biology of Grapevine Stilbenes: An Update. In Grapevine molecular physiology and biotechnology. 2009, Edited by Roubelakis-Angelakis KA, Dordrecht, Heidelberg, London, New York: Springer, 341-364.
Scharf K, Berberich T, Ebersberger I, Nover L: The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta. 2012, 1819: 104-119. 10.1016/j.bbagrm.2011.10.002.
Olsen AN, Ernst HA, Leggio LL, Skriver K: NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10: 79-87. 10.1016/j.tplants.2004.12.010.
Rushton PJ, Somssich IE, Ringler P, Shen QJ: WRKY transcription factors. Trends Plant Sci. 2010, 15: 247-258. 10.1016/j.tplants.2010.02.006.
Perrot-Rechenmann C: Cellular responses to auxin: division versus expansion. Cold Spring Harb Perspect Biol. 2010, 2: a001446. 10.1101/cshperspect.a001446.
Lee J, Lee I: Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot. 2010, 61: 2247-2254. 10.1093/jxb/erq098.
Hartmann U, Höhmann S, Nettesheim K, Wisman E, Saedler H, Huijser P: Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant J. 2000, 21: 351-360. 10.1046/j.1365-313x.2000.00682.x.
Yoo SY, Kardailsky I, Lee JS, Weigel D, Ahn JH: Acceleration of flowering by overexpression of MFT (MOTHER OF FT AND TFL1). Mol Cells. 2004, 17: 95-101.
Xi W, Liu C, Hou X, Yu H: MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Plant Cell. 2010, 22: 1733-1748. 10.1105/tpc.109.073072.
Wang R, Farrona S, Vincent C, Joecker A, Schoof H, Turck F, Alonso-Blanco C, Coupland G, Albani MC: PEP1 regulates perennial flowering in Arabis alpina. Nature. 2009, 459: 423-427. 10.1038/nature07988.
Adamczyk BJ, Lehti-Shiu MD, Fernandez DE: The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J. 2007, 50: 1007-1019. 10.1111/j.1365-313X.2007.03105.x.
Gyllenstrand N, Clapham D, Källman T, Lagercrantz U: A Norway spruce FLOWERING LOCUS T homolog is implicated in control of growth rhythm in conifers. Plant Physiol. 2007, 144: 248-257. 10.1104/pp.107.095802.
Karlgren A, Gyllenstrand N, Källman T, Sundström JF, Moore D, Lascoux M, Lagercrantz U: Evolution of the PEBP gene family in plants: functional diversification in seed plant evolution. Plant Physiol. 2011, 156: 1967-1977. 10.1104/pp.111.176206.
Guo A, Zhu Q, Gu X, Ge S, Yang J, Luo J: Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family. Gene. 2008, 418: 1-8. 10.1016/j.gene.2008.03.016.
Wu G, Poethig RS: Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development. 2006, 133: 3539-3547. 10.1242/dev.02521.
Wu G, Park MY, Conway SR, Wang J, Weigel D, Poethig RS: The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009, 138: 750-759. 10.1016/j.cell.2009.06.031.
Amasino R: Seasonal and developmental timing of flowering. Plant J. 2010, 61: 1001-1013. 10.1111/j.1365-313X.2010.04148.x.
Wang JW, Park MY, Wang LJ, Koo Y, Chen XY, Weigel D, Poethig RS: MiRNA Control of Vegetative Phase Change in Trees. PLoS Genet. 2011, 7 (2): e1002012. 10.1371/journal.pgen.1002012.
Yamaguchi A, Wu M, Yang L, Wu G, Poethig RS, Wagner D: The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev Cell. 2009, 17: 268-278. 10.1016/j.devcel.2009.06.007.
Jung J, Ju Y, Seo PJ, Lee J, Park C: The SOC1-SPL module integrates photoperiod and gibberellic acid signals to control flowering time in Arabidopsis. Plant J. 2012, 69: 577-588. 10.1111/j.1365-313X.2011.04813.x.
Shikata M, Koyama T, Mitsuda N, Ohme-Takagi M: Arabidopsis SBP-box genes SPL10, SPL11 and SPL2 control morphological change in association with shoot maturation in the reproductive phase. Plant Cell Physiol. 2009, 50: 2133-2145. 10.1093/pcp/pcp148.
Unte US, Sorensen A, Pesaresi P, Gandikota M, Leister D, Saedler H, Huijser P: SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell. 2003, 15: 1009-1019. 10.1105/tpc.010678.
Xing S, Salinas M, Höhmann S, Berndtgen R, Huijser P: miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell. 2010, 22: 3935-3950. 10.1105/tpc.110.079343.
Stone JM, Liang X, Nekl ER, Stiers JJ: Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to fumonisin B1. Plant J. 2005, 41: 744-754. 10.1111/j.1365-313X.2005.02334.x.
Usami T, Horiguchi G, Yano S, Tsukaya H: The more and smaller cells mutants of Arabidopsis thaliana identify novel roles for SQUAMOSA PROMOTER BINDING PROTEIN-LIKE genes in the control of heteroblasty. Development. 2009, 136: 955-964. 10.1242/dev.028613.
Thomas H, Thomas HM, Ougham H: Annuality, perenniality and cell death. J Exp Bot. 2000, 51: 1781-1788. 10.1093/jexbot/51.352.1781.
Baggiolini M: Les stades repères dans le developpement annuel de la vigne et leur utilisation pratique. Rev Romande Agric Vitic Arbor. 1952, 8: 4-6.
Coombe BG: Growth Stages of the Grapevine: Adoption of a system for identifying grapevine growth stages. Aust J Grape Wine Res. 1995, 1: 104-110. 10.1111/j.1755-0238.1995.tb00086.x.
Reid KE, Olsson N, Schlosser J, Peng F, Lund ST: An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006, 6: 27. 10.1186/1471-2229-6-27.
Dash S, Van Hemert J, Hong L, Wise RP, Dickerson JA: PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Res. 2012, 40: D1194-201. 10.1093/nar/gkr938.
Lohse M, Nunes-Nesi A, Krüger P, Nagel A, Hannemann J, Giorgi FM, Childs L, Osorio S, Walther D, Selbig J, Sreenivasulu N, Stitt M, Fernie AR, Usadel B: Robin: an intuitive wizard application for R-based expression microarray quality assessment and analysis. Plant Physiol. 2010, 153: 642-651. 10.1104/pp.109.152553.
Acuity software. [http://www.moleculardevices.com/Products/Software/Acuity.html].
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34: 374-378.
Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tárraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, García F, Marbà M, Montaner D, Dopazo J: Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010, 38: W210-W213. 10.1093/nar/gkq388.
Grimplet J, Van Hemert J, Carbonell-Bejerano P, Diaz-Riquelme J, Fennell A, Pezzotti M, Martinez-Zapater JM: Comparative analysis of grapevine whole-genome predictions, functional annotation and categorization of the predicted gene sequences. BMC Research notes. 2012, 5: 213. 10.1186/1756-0500-5-213.
Acknowledgements
This study was funded by Project BIO2008-03892 and a predoctoral fellowship to J.D.-R. from the Spanish Ministry of Science and Innovation (MICINN). JG was supported by the JAE-Doc program from Agencia Estatal CSIC, co-funded by the European Social Fund. The authors want to thank Félix Cabello and the Instituto Madrileño de Investigación y Desarrollo Rural, Agrario y Alimentario (IMIDRA, Alcalá de Henares, Madrid) for providing access to the grapevine experimental plots at El Encin and Javier Ibañez (ICVV-CSIC) who kindly facilitate us the meteorological data from the station at Finca El Encin. We are also very grateful to Diego Lijavetzky, for discussion on the experimental approach and to Pablo Carbonell-Bejerano and Rafael Torres-Pérez for their work generating functional annotation files and Marisela Velez for editing the manuscript. We wish to thank the Genomics service at CNB-CSIC for the microarray hybridizations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Authors declare that they have no competing interests.
Authors’ contributions
JDR and MJC performed the sample collection and RNA extractions. JDR together with JG performed the transcriptomic analysis. MJC drafted the manuscript. MJC and JMZ conceived the study, partook in its design and coordination. All authors worked in developing the final manuscript and read and approved it.
Electronic supplementary material
12870_2012_1155_MOESM1_ESM.xls
Additional file 1: Table containing the RMA normalized expression values for all GrapeGen GeneChip® probesets in the analyzed samples together with their correspondences to genes in the 12X V1 version of grapevine reference genome and their annotations. This file also includes the component scores for each probe-set in the first three principal components. (XLS 13 MB)
12870_2012_1155_MOESM2_ESM.xls
Additional file 2: Table containing the RMA normalized expression values for the probesets selected by PC1 component score (both positive and negative values) in the analyzed samples.(XLS 409 KB)
12870_2012_1155_MOESM3_ESM.xls
Additional file 3: Table containing the RMA normalized expression values for the probesets selected by PC2 component score (both positive and negative values) in the analyzed samples. (XLS 667 KB)
12870_2012_1155_MOESM4_ESM.pdf
Additional file 4: Functional categories significantly enriched in the clusters selected by PC2 component score (both positive and negative values). Categories enriched in the positive cluster are depicted in green while those enriched in the negative one are in pink. Absolute values of the log10 transformed P-values were used for the bar diagram representing statistical signification, only categories with P-values < 0.05 were shown. (PDF 22 KB)
12870_2012_1155_MOESM5_ESM.pdf
Additional file 5: Expression pattern of the two grapevine FLC homologs. Average expression values for each time-point are shown. (PDF 19 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Díaz-Riquelme, J., Grimplet, J., Martínez-Zapater, J.M. et al. Transcriptome variation along bud development in grapevine (Vitis viniferaL.). BMC Plant Biol 12, 181 (2012). https://doi.org/10.1186/1471-2229-12-181
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-181