
Abstract
Background
Manipulation of the amylose-amylopectin ratio in cereal starch has been identified as a major target for the production of starches with novel functional properties. In wheat, silencing of starch branching enzyme genes by a transgenic approach reportedly caused an increase of amylose content up to 70% of total starch, exhibiting novel and interesting nutritional characteristics.
In this work, the functionality of starch branching enzyme IIa (SBEIIa) has been targeted in bread wheat by TILLING. An EMS-mutagenised wheat population has been screened using High Resolution Melting of PCR products to identify functional SNPs in the three homoeologous genes encoding the target enzyme in the hexaploid genome.
Results
This analysis resulted in the identification of 56, 14 and 53 new allelic variants respectively for SBEIIa-A, SBEIIa-B and SBEIIa-D. The effects of the mutations on protein structure and functionality were evaluated by a bioinformatic approach. Two putative null alleles containing non-sense or splice site mutations were identified for each of the three homoeologous SBEIIa genes; qRT-PCR analysis showed a significant decrease of their gene expression and resulted in increased amylose content. Pyramiding of different single null homoeologous allowed to isolate double null mutants showing an increase of amylose content up to 21% compared to the control.
Conclusion
TILLING has successfully been used to generate novel alleles for SBEIIa genes known to control amylose content in wheat. Single and double null SBEIIa genotypes have been found to show a significant increase in amylose content.
Similar content being viewed by others

Background
Reserve starch represents the main component of wheat flour constituting roughly 60-70% of the wheat kernel and is chemically composed of a mixture of two glucan polymers known as amylose and amylopectin, representing 20-30% and 80-70% of total starch, respectively. The two glucan polymers differ in their degree of polymerization and of branching: amylose is essentially linear (DP < 104) and amylopectin is highly branched (DP 105-106). The two glucan polymers contribute differently to the functional properties of starch and the modulation of amylose/amylopectin ratio has been identified as a major target in order to develop starches with novel physical-chemical properties. In particular, high amylose starch is more and more in demand because of its unique nutritional properties and also for its technological characteristics that are opening new applications both in food as well as in non-food sectors [1–5].
Nutritionists and food industries are paying increasing attention to cereals with high amylose starch as derived foods have an increased amount of resistant starch, which has a role similar to dietary fibre inside the intestine, protecting against important diet related diseases [4]. An increased knowledge of starch biosynthesis is a necessary prerequisite for the determination of effective approaches to modify the amount of amylose in starch. Several starch enzymes have been identified as key factors in the modulation of the amylose/amylopectin ratio.
The two starch polymers are synthesized from a common substrate, ADP-glucose, by different pathways. Amylose biosynthesis involves a single enzyme, GBSSI (granule bound starch synthase I), known as waxy protein. In contrast, the branched structure of amylopectin is the result of a more complex biosynthetic mechanism involving several classes of enzymes: different types of starch synthases (SSs) promote the elongation of glucan chains by catalyzing the formation of α-1,4 glucosidic bonds; starch branching enzymes (SBEs) introduce α-1,6 links into the glucan backbone; debranching enzymes (DBEs) remove excess branches from glucan chains contributing to optimal packing of the semi-crystalline structure of the starch granule [6, 7].
Approaches to manipulate starch composition in wheat have involved both classical and biotechnological strategies. The silencing of genes encoding SSIIa (also known as Starch Granule Protein-1, SGP-1) and SBEIIa are currently two successful strategies for increasing amylose content. As starch granule proteins are easily detected by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), it has been possible to identify several mutant lines missing one of the three possible SGP-1 isoforms by screening natural germplasm and mutant populations [8, 9]. The absence of SSIIa has been found to cause a significant increase in amylose both in bread [10] (up to 35%) and durum wheat [11] (up to 45%). In wheat two classes of SBE, SBEI and SBEII, exist; the latter comprises two isoforms, SBEIIa and SBEIIb. The loss of SBEI has been reported not noticeably to affect starch composition [12]. SBEIIa and SBEIIb genes have been characterized and found to be located on the long arm of the homoeologous group 2 chromosomes [13]. SBEIIa has been shown to be the most abundant isoform and is found mainly in the soluble fraction of endosperm extracts, while SBEIIb is more highly represented in starch granules [14].
The ability to silence all copies of targeted genes through the use of RNA interference (RNAi) has permitted the elucidation of the role and functionality of the two different SBEII isoforms. Silencing of the SBEIIa and SBEIIb homoeologous gene families in bread wheat showed that only the loss of SBEIIa isoform was associated with a highly increased proportion of amylose in the transgenic lines (up to 70% of total starch) [15]. Although RNAi has now been shown to be effective in the production of high amylose lines in both bread and durum wheat [15, 16], the application of transgenic technology to crop improvement is still not completely accepted, encountering resistance from the general public and from governments.
Classical mutagenesis has been widely used in crop breeding over the past 60 years and is lately re-emerging as an efficient alternative to exploit and modify functionality of genes controlling important traits in crops. Chemical mutagenic treatment provides an efficient tool to generate high density mutations in the genome of the target organism, although in polyploids the presence of multiple copies of a gene has represented a major limitation in the detection of interesting phenotypes for valuable traits by forward genetics approaches. However, recent developments in sequence-level detection of mutations, coupled with the increased availability of both genomic and EST sequence data, have resulted in the development of a novel strategy of reverse genetics known as TILLING (Targeting Induced Local Lesions In Genomes) [17]. This technology was developed in Arabidopsis but has now been successfully applied to several crop species, including wheat, in which traits related to starch properties have been successfully targeted. Slade et al. [18] identified a total of 246 novel waxy (GBSSI) alleles in durum and bread wheat and crossed null mutants in different homoeologues to produce a waxy phenotype. Similarly, Sestili et al. [9] identified increased allelic variation present in the three homoeoloci of the SSIIa gene by analyzing a mutagenised population of the bread wheat cultivar Cadenza, using a combination of forward genetics and TILLING. More recently, Uauy et al. [19] using a modified TILLING approaches detected novel allelic variants of SBEIIa and SBEIIb genes in tetraploid and hexaploid wheats.
The most established method for the detection of DNA polymorphisms used in TILLING is a heteroduplex mismatch cleavage assay based on the endonuclease Cel1 [17]. An alternative technology, High Resolution Melting™(HRM), deriving from the combination of existing techniques of DNA melting analysis with a new generation of fluorescent dsDNA dyes [20] could also be used. This method is sensitive and specific for the detection of mutations in PCR products from genomic DNA and has recently been successfully applied in TILLING [21, 22].
In this work TILLING has been used to target genes encoding SBEIIa enzymes with the aim of developing non-transgenic wheat genotypes characterized by high amylose content and novel starch functionality.
Results
Selection of optimal genomic regions for TILLING
TILLING in polyploid species is complicated by the requirement for homoeoallele specific PCR for optimal sensitivity in SNP detection. As the three SBEIIa homoeoalleles share high similarity in their coding sequences, the intronic regions of the three genes were compared to identify sequence polymorphisms to facilitate the design of allele specific PCR primers. PCR amplicons for TILLING were also chosen to fulfill certain conditions. As our main objective was to identify functional mutations in the targeted genes, the exon density of potential amplicons was evaluated in order to select fragments that were as rich as possible in coding sequence. A further criteria used for the selection of TILLING fragments was the probability of finding deleterious SNPs (mutations affecting splicing sites or introducing stop codons) considering the types of transition mutation generally induced by EMS treatment (G → A; C → T).
Genomic regions selected for TILLING analysis are shown in Figure 1b. The amplicons vary in length between 1700 and 2200 bp. Three distinct regions of the gene were selected for the SBEIIa-A homoeoallele, two for SBEIIa-D and one for SBEIIa-B. Genome-specific primer pairs were designed for each target and validated for specificity using D-genome disomic substitution lines of the homoeologous group 2 chromosomes, produced in the durum wheat cultivar Langdon by Joppa and Williams [23] (Figure 1a).

Design and testing of primers for first round PCR. a) Electrophoretic profile of the PCR products obtained from Langdon (1), Langdon 2D(2A) (2), Langdon 2D(2B) (3) by using homoeoallele specific primer pairs. b) Graphical representation of the first round PCR amplicons. For SBEIIa-A the selected regions are: fragment from exon II to V (A(II-V) ); from exon VI to IX (A(VI-IX)); from exon X to XIII (A(X-XIII)). For SBEIIa-B: from exon IV to IX (B(IV-IX)). For SBEIIa-D: from exon II to VI (D(II-VI)); from exon X to XIII (D(X-XIII) ). Red, green and blue arrows represent PCR primers specific for genome A, B and D, respectively.
Detection of SNPs by HRM
The EMS-mutagenized population of bread wheat has been described elsewhere [24]. Briefly, this was derived from seeds of the UK spring wheat cultivar Cadenza treated with either 0.6% or 0.9% EMS solution overnight followed by growth to maturity. Single ears were harvested from each of the M1 plants and one grain from each ear sown to generate an M2 population of ~4,500 unique lines. Genomic DNA was isolated from the leaves of individual M2 plants and M3 seeds were harvested and archived. The M2 DNA samples were pooled two-fold and screened for mutations in the targeted regions (A(II-V), A(VI-IX), A(X-XIII); B(IV-IX); D(II-VI) e D(X-XIII) of SBEIIa (Figure 1b).
HRM was selected as the most suitable method for the detection of SNPs in the target genes considering their peculiar genomic structure. SBEIIa genes each contain 22 exons with sizes ranging between 40 bp and 240 bp spanning a region of 10 kb; moreover each exon is separated by introns of up to 1 kbp in size. In order to limit the number of mutations detected in introns and noting that HRM is most sensitive for the analysis of smaller fragments (100-400 bp), we chose to produce amplicons for HRM each covering the region of a single exon. As it was difficult to design homoeoallele-specific primers for each exon, amplicons with optimal sizes for HRM analysis were produced by nested PCR. First round, homoeoallele specific PCR fragments, as described above, were used as templates in 2nd round PCR using primer pairs targeting each included exon. The 2nd round primers were designed in the introns flanking each target exon and positioned approximately 5-20 nucleotides from the splice sites, resulting in PCR amplicons for HRM ranging in size from 100 bp to 350 bp.
Optimization of HRM analysis
The principle of the HRM technique is based on the change in fluorescence of a dsDNA-specific intercalating dye during temperature-induced denaturation of the DNA duplex. The HRM instrument allows the monitoring of fluorescence changes in real time as the temperature of the samples is slowly increased. While detection of SNPs in homoduplex DNA is possible, instability created by the presence of mismatched bases in heteroduplex DNA increases sensitivity, producing a melt curve usually characterized by a loss of fluorescence at a lower temperature than wild type homoduplex DNA [20]. For TILLING assays, heteroduplexes are derived from the melting and re-annealing of wild type and mutant amplicons, generated by two-fold pooling of genomic samples before PCR.
For each second round primer pair, optimization of the conditions for PCR and the subsequent HRM step were carried out, noting that the presence of the LCgreen Plus dye increased the primer Tm and thus raised the optimum annealing temperature of the PCR reaction. Analysis of the melt curve of the amplicon also allowed the specificity of the PCR to be confirmed. Although the presence of the mutation has been detected comparing the ΔF/T curves (Figure 2, panel d), produced by the HRM software, the observation of dF/dT curves (Figure 2, panel b) has proved useful for further confirmation of the mutations. In fact, heteroduplexes show a dF/dT curve visibly shifted at lower temperature in comparison with normal amplicons. All the amplicons have been analyzed in the temperature range between 75°C- 95°C; the two amplicons covering exon II and exon V have been further analyzed at higher temperatures to optimize the analysis of their GC rich domains (data not shown).

High Resolution Melting analysis of second round PCR products of 96 2-fold pooled samples. The figure shows the analysis of the amplicon correspondent to exon VI of the SBEIIa-B gene. a) Total fluorescence (F) vs temperature (T) curves; b) comparison of dF/T curves between normal and heteroduplex (indicated by arrows) DNA amplicons; c) normalized and temperature-shifted curves of fluorescence vs temperature showing wild types (grey) and mutants (red); d) ΔF/T difference curves with variants highlighted in red.
Novel allelic variants for SBEIIa-A, SBEIIa-B and SBEIIa-Dhomoeoalleles
Screening of genomic DNA from the TILLING library was conducted on two fold pools in consideration of the high mutation density associated with this hexaploid wheat EMS-mutagenised population. In Table 1 the numbers of plants analyzed and mutants identified for each of the three genes SBEIIa-A, SBEIIa-B and SBEIIa-D are reported. The mutation density has been calculated as follows: (total size of amplicons) × (total number of screened lines)/(number of identified mutations). Of the 53 novel alleles (plus three duplicated mutations) of SBEIIa-A that were characterized, 36 were mis-sense, 15 silent and two truncation mutations. 50 novel alleles (plus three duplicated mutations) were identified for the SBEIIa-D gene of which 34 were mis-sense, 14 silent, 1 on the splice junction and 1 non sense mutation. Of the 14 novel SBEIIa-B alleles 10 were mis-sense, 1 truncation and 1 splice junction mutation (Table 2, 3). The 18 putative mutants identified in the amplicon A(X-XIII) were not characterized by sequencing with the exception of one nonsense allele localized in exon XII.
We estimated an overall mutation density of 1 mutation per 40 kb screened. All mutations identified were shown to be transitions of the type C→T or G→A as expected for treatment with EMS, which acts via alkylation of G residues. The knock-out genotypes (C2907T and G5158A) identified for SBEIIa-A allele, respectively in exon IX and XII, will be referred to as SBEIIa-A-1 and SBEIIa-A-2 ; the two null genotypes for SBEIIa-B are named as SBEIIa-B-1 (G1948A, non sense mutation in exon VI) and SBEIIa-B-2 (G1916A, 3' splice site of intron V); the mutants C3693T (non sense mutation in exon X) and G5335A (5' splice site of intron XIII) of D genome allele are respectively named SBEIIa-D-1 and SBEIIa-D-2 .
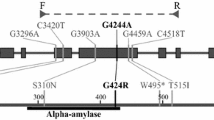
Non-synonymous SNPs result in an amino acid change in the protein that can affect protein functionality to varying extents. In order to evaluate the effect of mis-sense mutations identified, the web based program PARSESNP http://www.proweb.org/parsesnp/ has been used (Table 3; Figure 3). PARSESNP utilizes two different bioinformatic tools, PSMM (Position-Specific Scoring Matrix) and SIFT (Sorting Intolerant from Tolerant ) which predict whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids [25]. PARSESNP analysis of the non-synonymous mutations found in SBEIIa-A, SBEIIa-B and SBEIIa-D resulted in the identification of 4, 1 and 8 mis-sense mutations, respectively, that are predicted to have severe effects on protein functionality. For the four protein variants SBEIIa-A (P206S), SBEIIa-A (A208V), SBEIIa-B(A205V) and SBEIIa-D(A201T) the amino acid change induced by the EMS treatment is located in the region of the N-terminal domain of the glycogen branching enzyme family, reported to be essential for the size of the glucan chains transferred and also for the catalytic activity of BE [26]. The amino acids changes H362Y, G374R, G390S, V398I and D462N, identified for the SBEIIa-D protein, are all localized in the (α/β)8 barrel catalytic domain of related enzymes belonging to the α-amylase family. Secondary structures and catalytic residues were identified in the three SBEIIa proteins through homology with the crystallographic structure of glycogen branching enzyme of E. coli, the model protein for branching enzyme family [27]. On the basis of these information it has been determined that the amino acid changes G390S and D462N are localized in the two strands β3 and β4 respectively of the (α/β)8 barrel domain; H362Y is adjacent to the residue Tyr361 known to be involved in catalysis, while V398I is located between Asp396 and His401 also directly involved in enzymatic activity.

Representation of the allelic variants identified in SBEIIa genes by TILLING as obtained by PARSESNP. Red, black and violet triangles represent deleterious (non-sense and splicing junction), mis-sense and silent mutations, respectively.
In order to study more in detail the new SBEIIa variants described above, the amino acid sequences were submitted to the program i-Tasser http://zhanglab.ccmb.med.umich.edu/I-TASSER/[28] which predicts the 3D structures and functionality of the proteins. The comparison between the simulated 3D structures of non mutated and mutated SBEIIa proteins, in most cases, highlighted differences in the pattern of substrate binding sites and in the protein secondary structure. In Figure 4 we show as an example the case of SBEIIa-D(V398I): while in wild type protein residue 398 was involved in the β3 strand of the (α/β)8 domain, in the mutated protein it is in a coil structure. Moreover the program predicted a different pattern of substrate binding sites for normal and mutated protein: of the seven binding sites predicted for the normal SBEIIa-D, in SBEIIa-D(V398I) six residues were conserved and two new residues resulted involved in substrate binding (Figure 4). On the contrary in SBEIIa-D(D462N) the mutation caused the loss of two of the seven amino acids involved in the binding and catalytic activity in normal SBEIIa protein, respectively Arg465 and Asp467.

3D Structures of normal and mutated SBEIIa-D protein. Secondary (above) and 3D (bottom) structures as elaborated by I-TASSER for wild type and mutant forms of SBEIIa-D protein (V398I and D462N). The ligand is depicted in magenta colored ball & stick, the predicted binding site residues interacting with the ligand are shown as transparent green spheres, while the N and C terminus in the model are marked by blue and red spheres respectively.
Analysis of SBEIIa-transcripts in the knock out mutants
Expression of the three SBEIIa genes was evaluated in homozygous lines of the five putative knock out mutants, SBEIIa-A-1 , SBEIIa-A-2, SBEIIa-B-1 , SBEIIa-B-2 and SBEIIa-D-1 . All of these alleles are non-sense mutants with the exception of SBEIIa-B-2 , which is a splice-site mutation. Allele-specific qRT-PCR primer pairs were designed by comparing coding regions of the three SBEIIa genes. In some cases specificity was provided by the presence of small indels between the three genes; otherwise primers were designed based on sequence polymorphism in their 3' terminal ends. The specificity of the primers was validated by PCR on genomic DNA of the Langdon D-genome disomic substitution lines. Semi-quantitative and real time qRT-PCR experiments were performed on total RNA isolated from immature seeds (18 dpa) of homozygous mutant lines to investigate whether the expression levels of SBEIIa genes were affected by the presence of the putative knock-out mutations in the SBEIIa single null genotypes.
Figure 5a clearly shows a drastic decrease of SBEIIa-A transcript in the two non-sense mutant lines SBEIIa-A-1 and SBEIIa-A-2 compared to the wild type genotype. A similar effect was found in the SBEIIa-B-2 and SBEIIa-D-1 genotypes, showing a severe reduction in transcript level due to both the splicing and non sense mutations, respectively, on the expression of the genes. In one case, SBEIIa-B-1 , the presence of premature stop codon in the gene sequence has not resulted in a strong reduction of its transcript. Each mutant genotype was also investigated for the expression of the two remaining wild-type homoeologous copies of SBEIIa. No appreciable difference was detected in this case with respect to the wild type plant.

Semiquantitative and quantitative RT-PCR of SBEIIa transcripts. a) Semiquantitative RT-PCR of SBEIIa genes in SBEIIa homozygous single mutant genotypes: 1) SBEIIa-A-1; 2) SBEIIa-A-2; 3) SBEIIa-D-1; 4) SBEIIa-B-1; 5) SBEIIa-B-2; 6) wild-type Cadenza. b) Relative expression of SBEIIa homoeologs in single null genotypes as determined by Real Time quantitative PCR analysis: W.T.= wild type Cadenza; A-(1)= SBEIIa-A-1; A-(2)= SBEIIa-A-2; B-(1)= SBEIIa-B-1; B-(2)= SBEIIa-B-2; D-(1)= SBEIIa-D-1. Vertical bars indicate standard error.
The extent of gene silencing in the five putative knock out mutants was quantified by Real Time RT-PCR (Figure 5b). We registered the strongest effect on gene expression in the two SBEIIa-A null lines, SBEIIa-A-1 and SBEIIa-A-2 : transcripts of the target alleles were found to be reduced to 1.7% and 3.3%, respectively, of the level in the wild-type control. Weaker effects were identified in the other null genotypes: the B alleles, SBEIIa-B-1 (non-sense) and SBEIIa-B-2 (splice site), were found to be expressed at 20% and 12%, respectively, of wild-type levels and SBEIIa-D allele was found 8.5 fold reduced in the SBEIIa-D-1 genotype.
In order to investigate the effect of splice junction (S.J.) mutation (3' S.J. of intron V) on gene transcription, primers spanning exons II to IX were used to isolate transcripts from the SBEIIa-B-2 mutant. PCR amplification resulted in two bands of different size: the larger product showed the inclusion of the intron V, whereas the smaller one was found to contain a deletion of the first seven nucleotides of exon VI. The presence of the intron V in the longer transcript showed that mutation at 3' splice site of intron V caused an incorrect splicing of SBEIIa-B. The deletion in exon VI, found in the shorter fragment, is probably due to the selection of an alternative splice junction site, positioned 5 nucleotides downstream the normal S.J. site. This last mechanism has been previously found in plants [29, 30] and explained by the local scanning of the spliceosome that may select the best intron 3' splice site on the basis of sequence context [31]. Splicing of the immature mRNA at this junction would result in a frame-shift mutation leading to the production of a premature stop codon.
Estimation of amylose content, total starch and seed weight
In order to detect the phenotypic effect of null mutations in SBEIIa genes, amylose content was measured in the three single mutants SBEIIa-A-1, SBEIIa-B-1 and SBEIIa-D-1 (Table 4). Our results showed an increase of amylose content in the three genotypes between 6% and 12% in respect to the normal genotype.
Double null lines SBEIIa (SBEIIa-A-1B-1, SBEIIa-A-1D-1, SBEIIa-B-1D-1 ) have been produced by crossing single null genotypes and selecting the F2 progeny as described in Material and Methods. Pyramiding of two null homoeoalleles results correlated with an increase in amylose content included between 17%- 21% compared to the wild type (Table 4). In addition, comparison of 100 seed weights did not highlight significant differences among the single and double null genotypes compared to the control, although total starch content resulted decreased between 2% and 8% in the single and double null genotypes (Table 4).
Discussion
In the last twenty years, modification of starch has been highlighted by food scientists as a primary target to confer added value on cereal products for both nutritional and industrial uses [7]. Naturally occurring variation has been exploited in wheat to generate starch with novel properties [8, 32]. In polyploids the effect of mutations in single homoeologues is often masked by inherent genetic redundancy; therefore forward genetic screening for mutations requires extensive screening based on effective isoenzymatic or molecular markers. In addition, the shortage of mutations for most target loci in natural population makes the identification of the desired genotypes a slow process [32]. Both for Waxy and SGP-1, the availability of assays able to distinguish the individual protein products of the three homoeologous genes led to the identification of complete sets of single null mutants that were used to alter starch functionality in wheat [10, 32, 33]. However, a negative aspect of breeding programs based on natural genetic variation is the phenomena known as linkage drag. Extensive backcrossing is therefore required to remove undesirable characters inherited from exotic parental material making the breeding program time consuming.
In this work TILLING has been employed as a tool to identify novel genetic variability in the SBEIIa loci. In TILLING the desired variability is generated within a commercial variety selected by the breeder or researcher thus reducing genetic drag, although backcrossing is still required to remove excess mutations that may affect other characters. One disadvantage of TILLING in polyploid crops, compared to other reverse genetics approaches such as RNAi, is the need to combine mutations in all functional copies of the gene encoding the target protein. Pyramiding of the three null alleles is currently being carried out including backcrosses with Cadenza and we aim to complete this task within two years. On the other hand, mutants identified by TILLING are not considered to involve genetic manipulation and are relatively free of public and legislative concerns and, unlike RNAi which requires the production of transgenic plants, can be immediately introduced into breeding programs and tested in the field. If in diploid species chemical mutagenesis gives the opportunity to easily detect phenotypic changes linked to mutations in key genes, polyploids possess a higher tolerance of mutations resulting in a higher density in the population. This offers the possibility of identifying a wide variety of mutations in the target genes by screening a realistic number of mutagenised individuals.
TILLING in SBEIIa genes resulted in the production of large allelic series representing a valuable resource not only for starch modification but also to study structure-function relationship in the targeted enzyme. SBEs are found to contain three domains: an amino-terminal domain, a carboxyl-terminal domain and a central catalytic domain [27, 34]. The N-terminal region is important for specifying the chain length and is required for maximum enzyme activity [26, 35]. In this work protein variants characterized by mutations in functional domains of SBE enzyme have been identified and analyzed by bioinformatic tools able to predict the effect of the amino acid substitution on protein structure and functionality.
Although several mis-sense mutations have been found that potentially affect enzyme activity, the polyploidy nature of wheat prevents the immediate assessment of those allelic variants on phenotype. Thus, in a crop breeding perspective, the mutations of interest are those one known to prevent complete gene expression such as non-sense and splicing site located polymorphisms. To increase the frequency of the detection of knock-out mutants, a careful selection of gene regions rich in codons CAA, TGG, CAG and CGA was performed. The CODDLE application http://www.proweb.org/coddle/ is useful to evaluate truncation mutations frequency in the gene sequence; however we found that a more accurate selection of the fragments can be performed by manual sequence analysis. Moreover we finally selected gene fragments whose size is larger than that limited by CODDLE (up to1500 bp).
In general an efficient detection of SNPs in a gene is dependent upon the production of specific PCR products thus requiring the development of homoeoallele specific primers. In wheat obtaining full sequence data for target genes can be a significant challenge, although this is likely to be eased considerably in the next few years as shotgun and fully assembled sequence data is made available. We were able to design homoeoallele-specific primer pairs by identifying polymorphisms that exist among the three SBEIIa genes. In some cases oligonucleotides were designed corresponding to indel polymorphisms; however, it was also possible to develop specific primer pairs using a 3' terminal SNP in both the forward and reverse primers. Alternatively, a recent work suggests that it may be possible to use non-homoeoallele specific PCR to detect mutation in polyploids [21], although in our hands this resulted in reduced sensitivity.
High Resolution Melting has been recently applied to TILLING in plant species including tomato and wheat [21, 22, 36]. It is a closed tube PCR-based assay requiring no further processing of PCR amplicons; this results in significant advantages both in terms of costs and time saving in respect to other TILLING methods such as Cel1 digestion [37]. In our work the choice of HRM was strongly suggested by the consideration of the structure of SBEIIa genes, which contain many small exons (43-242 bp) interrupted by sizeable introns. As HRM is most suitable for the analysis of fragments up to 400 bp [38], this allowed us to target individual exons within the SBEIIa genes. Although traditional TILLING, based on Cel1 digestion, permits the analysis of larger amplicons (up to 1500 bp), this has as consequence the detection of mutations in the intronic regions that, excluding those in intron splice sites, do not impact on protein function [18].
HRM permitted an efficient detection of SNPs in two-fold pools of genomic DNA. The high mutation frequency of the wheat population used in the present work did not require deep pooling to increase the throughput of the screening. Our finding of a mutation density of 1 SNP for each 40 kb is in agreement with a previous report [36] that cited similar results for the same wheat population screened by traditional Cel1-based TILLING.
Hofinger et al. [37] have recently reported that HRM is less efficient in the detection of mutations localized at a distance of less than 20 nt from the PCR primers. Our data are in agreement with this hypothesis; in fact in some cases PCR primers were designed at a distance of less than 10 nucleotides from 5' and 3' ends of the exons as suggested by HRM software for primer design supplied by the manufacturer and this condition could have limited the number of mutations detected in the splicing sites of the exons analyzed. Suggestive of this we detected only two mutation in the splicing sites and in both cases primers had been designed at a distance of at least 20 nt from the ends of the exons.
The four non-sense genotypes SBEIIa-A-1, SBEIIa-A-2, SBEIIa-B-1 and SBEIIa-D-1 present a premature stop codon localized in the first twelve exons of the SBEIIa genes that prevents the production of a protein containing a functional (α/β)8 barrel catalytic domain essential for the enzyme activity. Also the two genotypes SBEIIa-B-2 and SBEIIa-D-2 present splice junction mutations, respectively localized at 5' end of exon VI and at 3' end of exon XIII, that would prevent a correct translation of the catalytic domain of SBEIIa enzyme by the introduction of premature stop codons.
The study of the effect of non-sense mutations on gene expression in plants is a poorly-explored topic [39, 40]. We found that non-sense mutations in the gene sequence were associated with a detectable decrease in transcript levels in respect to the control genotype. Moreover the splicing junction mutation in SBEIIa-B-2 also has been associated to a significant reduction of the gene expression. For each mutant genotype we tested the expression level of all the three homoeologous SBEIIa copies finding that just the gene with non sense mutation (or mutation in the splicing site) presented drastic decrease in the level of expression. Saito and Nakamura [41] reported similar results for a Wx-A1- mutant characterized by a premature stop codon in the gene sequence. Patron et al. [42] reported the characterization of a barley waxy mutant, derived by mutagenesis, in which a premature stop codon was associated to the absence of the protein product; in this case the transcript level of the mutant allele was found similar to that of wild type. Similar results were found by Zhu et al. [43] for the wheat mutant, obtained by chemical mutagenesis, lacking the high molecular weight glutenin subunit Bx14 due to the presence of a premature stop codon. The reduction of transcript level detected in our knockout mutants suggests an intervention of a mechanism of quality control preventing accumulation of non functional or deleterious truncated protein, which has been described previously and is known as Nonsense Mediated mRNA Decay (NMD) [44]. Although this mechanism has been extensively characterized in mammals, little is known about its mode of action in plants. NMD in mammals takes place in intron-containing genes when the premature stop codon is positioned 55 nucleotides or more upstream of the last exon-exon junction [45]. In plants NMD has been reported to act also in case of intronless genes [46] thus showing that different rules govern this mechanism in respect to mammals; however several genes containing a premature stop codon positioned 55 nucleotides upstream of the last exon-exon junction have been reported to be subjected to NMD in plants [41, 47–49].
All our knock out mutant genotypes present the premature stop codon at 55 nucleotides upstream of the last exon-exon junction thus following the consensus of NMD in mammals. Although reduction in transcript levels of the mutated genes has been detected in all our genotypes, the extent of the decrement varied among the 5 genotypes. In particular the mutant SBEIIa-B-1 did not show drastic decrease in transcript level of the mutated allele. Similar examples have been reported in literature [42, 43] indicating that NMD is a complex mechanism and further elucidation is needed to understand its mode of action in plants.
Amylose content was estimated in the control, the three non sense genotypes, for which seeds were available and double null mutants derived from their crossing. The modest increase of amylose content in single null mutants is presumable due to the compensation exerted by gene redundancy in polyploids, similarly to what reported by Miura and Sugawara [50] and Konik-Rose et al. [51] for other genes involved in starch biosynthesis. Further increase in amylose content was also observed for the three double null lines obtained from the cross of the three single null mutants. In addition, our results showed a modest decrease in starch content in the set of single and double null SBEIIa genotypes not correlated to a loss of seed weight. The discrepancy could be due to the limitation of the method to estimate total starch in high amylose cereals as reported by McClearly et al. [52].
Concluding, as previously found for the other genes controlling amylose content in wheat [10, 53], it has to be expected a much higher increment of amylose content in triple null SBEIIa wheat.
Conclusions
Novel allelic variants have been identified for the three SBEIIa homoeologs in bread wheat that represent a valuable resource both for functional genomics studies and for wheat improvement. In particular a complete set of single null SBEIIa wheat lines have been identified and characterized both at molecular and phenotypic level. Genic expression of null alleles resulted deeply reduced showing the intervention of NMD mechanism to prevent the production of a non functional protein. The set of the three single and double null genotypes showed an increase in amylose content which can further be increased when triple null lines will be available. The complete null lines will be used in breeding activities aimed to increase the level of resistant starch in wheat end products.
Methods
Plant material
Production of the EMS-mutagenised population of the spring bread wheat cv Cadenza has been described previously [9, 24].
Primer design
Alignment of the three gene sequences were performed by ClustalW http://www.ebi.ac.uk/clustalw. Gene- and homoeoallele-specific primers for TILLING were designed using the PRIMER 3 program. PCR primers for TILLING analysis were validated using D-genome disomic substitution lines of homoeologous group 2 chromosomes of the durum wheat cultivar Langdon [23]. Genomic DNA was extracted from 0.2 g of green tissue as reported in Tai and Tanksley [54]. Primers pairs are reported in Table 5.
PCR reactions for primer evaluation were carried out in 50 μl final volume using 50-100 ng of genomic DNA, 1× Red Taq ReadyMix PCR reaction mix (1.5 U Taq DNA Polymerase, 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 0.001% gelatine, 0.2 mM dNTPs) and 0.5 μM of each of the two primers. Amplification conditions for testing primers included an initial denaturation step at 94°C for 5 min, followed by 35 cycles at 94°C for 1 min, 62-67°C for 1 min and 72°C for 1 min, followed by a final incubation at 72°C for 5 min.
Screening of the TILLING library
Amplicons analyzed in TILLING were produced by a nested PCR strategy. 1st round PCR was carried out in a 10 μl volume using 10 ng of two-fold pooled genomic wheat DNA, 5 μl of Hot Shot™Mastermix (Cadama Medical Ltd), 0.5 μM primers. The PCR program was: 97°C, 5 min; (97°C, 30 s; 62-67, 30 s; 72°C for 1.5-2 min)x 38 cycles; 72°C, 10 min. 96 well plates were used for the screening.
For HRM, the 1st round PCR reaction was diluted 60 fold and 1 μl was used as template in the 2nd round PCR. The 2nd round PCR reaction was prepared as follows: 1 μl of diluted DNA template (1:60); 5 μl of Hot Shot™Mastermix (Cadama Medical Ltd); 1 μl of LCGreen Plus; 0.5 μM primers (Table 6). The PCR program used was: 97°C, 5 min; (97°C, 30 s; 60°C, 20 s; 72°C, 20-30 s)x 39 cycles; 72°C, 10 min. After the final extension step, PCR amplicons were denatured at 95°C for 30 s and reannealed at 25°C for 1 min. Both 1st and 2nd round PCR reaction were overlaid with 10 μl of mineral oil (Sigma-Aldrich M5904) to prevent sample evaporation. 2nd round PCRs were run in 96 well Frame-Star plates (4titude Ltd, Surrey, UK).
High Resolution Melting by LightScanner
The 96 well plates (2nd PCR) were used for HRM using the LightScanner instrument (Idaho Technology, Inc). Samples were normally heated using a temperature range from 75°C to 95°C. For amplicons containing high GC regions a further analysis was conducted in a temperature range from 85°C to 98°C to guarantee optimal resolution in SNP detection.
The data obtained were analyzed by LightScanner software analysis provided with the instrument. Melting curves were normalized according to the manufacturer's instructions. The results obtained by HRM were visualized as differential curves ΔF/T displaying the relative difference in fluorescence of a respective sample in respect to a reference sample. F/T normalized curves show the decrement in fluorescence of each sample during the denaturation of the PCR amplicon as the temperature increases. As stated by the manufacturer's instructions, ΔF > 0.05 was considered significant; furthermore the shape of the melting curve and position (along temperature axis) along the dF/T curve were observed and used as criteria to distinguish false positive from real mutants. Samples identified as putative mutants were selected and the amplicon re-amplified from each individual in the pool for sequencing. DNA sequence analyses were conducted by a commercial sequencing service (Eurofins MWG Operon, Ebersberg, Germany). The PARSESNP http://www.proweb.org/parsesnp/ application was used for the evaluation of protein variants coded by mutated alleles.
Semi-quantitative reverse transcriptase-polymerase chain reaction
Total RNA was extracted from immature seeds (18 DPA) as reported in Laudencia-Chingcuanco et al. [55] with some modifications. The starting material was 0.1 g and all volumes of buffers and solutions were diluted 1 to 10. For reverse transcriptase-mediated PCR studies, cDNA was synthesized from 1 μg of total RNA using an oligo(dT) primer and Superscript Reverse Transcriptase III (Invitrogen). One of twentieth volume of each cDNA was used as a template for PCR amplification. PCR reactions were carried out in 20 μl final volume using 1 units of Ex-Taq (Takara), 1× buffer, 0.2 mM of each dNTPs, 0.5 μM of each primer. Amplification conditions included an initial denaturation step at 98°C, followed by 35 cycles at 98°C for 10 sec., 58°C for 1 min. and 72°C for 1 min, followed by a final extension at 72°C for 5 min. The following gene-specific primers were designed for: SBEIIa-A [EMBL:HE591389] (5'accagtatgtttcacggaaacac3'; 5'caccttgtacttcccaggcc3'), SBEIIa-B [EMBL: FM865435] (5'atatcgtggtatgcaagagttcgac3'; 5'caagaaagagcgcggccta3'), SBEIIa-D [GenBank: AF338431] (5'gaggaagataaggtgatcatcctca3'; 5'caaagagtgcatcgtcagagtcc3'). Amplification of the wheat actin gene [GenBank: AB181991] was used as reference for transcript amplification and the primers used have the following sequence: (TaACTINF) 5'-aagagtcggtgaaggggact-3' and (TaACTINR) 5-ttcatacagcaggcaagcac- 3'.
Isolation of the SBEIIa-BmRNA sequences of the splice junction mutant
Gene transcripts were isolated from cDNA by using the homoeoallele specific primer 5'gacttggcggccactcca 3' and the gene specific primer 5'ctctggtcgtttaggttgaggatg 3'.
Real-Time RT-PCR (qRT-PCR)
One microlitre of the cDNA prepared above was used for real-time PCR in a 20 μL volume. For each sample three technical replicates were used for PCR amplification. The PCR reaction consisted of 10 μL of iQ™ SYBR Green Supermix 2× (BIO-RAD), which contained buffer, dNTPs and SYBR Green I. Concentrations of the forward and reverse oligodeoxynucleotide primers in the reaction were 500 nM for all the genes of interest. qRT-PCR experiments were performed using the iCycler iQ (Bio-Rad Laboratories, Hercules, CA1, USA). Amplification conditions were as follows: initial 95°C for 15 min and 40 cycles of 95°C for 30 s, 60°C for 1 min and 72°C for 1 min each.
Relative expression analysis was determined by using the 2-ΔΔCT method [56] (Applied Biosystems User Bulletin No. 2-P/N 4303859). Calculation and statistical analyses were performed by Gene Expression Macro™ Version 1.1 (Bio-Rad Laboratories, Hercules, CA, USA). The efficiencies of target and housekeeping genes were determined by qRT-PCR on serial dilutions of RNA template over a 100-fold range [57], with similar results (data not shown). Amplified products were checked by gel electrophoresis and sequencing to verify primer specificity. The relative expression of each gene is reported as the fold increase of the transcript level at each time point, compared to the lowest transcript level. As in semi-quantitative RT-PCR, actin was used as the housekeeping gene.
Selection of double null SBEIIamutants
Double null SBEIIa lines were obtained by crossing SBEIIa-A1- , SBEIIa-B1- and SBEIIa-D1-. Double null homozygous lines of the F2 progenies were selected by PCR using CAPS or dCAPS primers followed by a specific restriction enzyme reaction. dCAPs primers have been designed by dCAPs Finder application [57]. Digested PCR amplicons were run on agarose gels (2%) stained with ethidium bromide for band visualization.
The following primer pairs and restriction enzymes were used: SBEIIa-A1- (Fw taaatcctcagtgactctggtcgtttaggttgaggattc, Rv aagtgacatatgcattaattcaccttctaa; Xba); SBEIIa-B1 - (Fw ctggagcgcatgtacgtcttaac, Rv caccataatcatcctgaaaagatcg; MfeI); SBEIIa-D1- (Fw gaggcagtgggcatgtgaaagtc, Rv ccaaagcttgcatagtatgaatgctcctggattgccattgtcg; SalI)
Determination of amylose and total starch content
Amylose content (percentage of total starch) was determined by a iodometric assay as reported in Chrastil [58] using starch extracted from whole flour by the "dough ball" method [59]. Seeds were obtained from plants grown in the field. Three biological and six technical replicates have been used for all materials.
A standard curve was used using mixtures of potato amylose (Fluka 10130) and amylopectin isolated from waxy wheat. Total starch content of kernels was determined by Megazymes Total Starch Assay Kit (AA/AMG, Megazyme Pty Ltd., Wicklow, Ireland).
References
Soh HN, Sisson MJ, Turner M: Effect of starch granule size distribution and elevated amylose content on durum dough rheology and spaghetti cooking quality. Cereal Chem. 2006, 83: 513-519. 10.1094/CC-83-0513.
Waring S: Functionality of resistant starch in food applications. 2005, [http://eu.foodinnovation.com/pdfs/functresist.pdf]
Chanvrier H, Appelqvist IAM, Bird AR, Gilbert E, Htoon A, Li Z, Lillford PJ, Lopez-Rubi A, Morell MK, Topping DL: Processing of novel elevated amylose wheats: functional properties and starch digestibility of extruded products. J Agric Food Chem. 2007, 55: 10248-10257. 10.1021/jf0718650.
Sajilata MG, Singhal RS, Kulkarni PR: Resistant starch: a review. Compr Rev Food Sci Food Safety. 2006, 5: 5-17.
Freire A, Podczeck F, Veiga F, Sousa J: Starch-based coatings for colon-specific delivery. Part II: Physicochemical properties and in vitro drug release from high amylose maize starch films. Eur J Pharm Biopharm. 2009, 72: 587-594. 10.1016/j.ejpb.2009.02.010.
Ball SG, Morell MK: From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Ann Rev Plant Biol. 2003, 54: 207-233. 10.1146/annurev.arplant.54.031902.134927.
Stone B, Morell MK: Carbohydrate. In Wheat, Chemistry and Technology.. 4edition. Edited by: Khan K, Shewry PR. AACC International St. Paul MN;2009:299-362.
Yamamori M, Endo TR: Variation of starch granule proteins and chromosome mapping of their coding genes in common wheat. Theor Appl Genet. 1996, 93: 275-281. 10.1007/BF00225757.
Sestili F, Botticella E, Bedo Z, Phillips A, Lafiandra D: Production of novel allelic variation for genes involved in starch biosynthesis through mutagenesis. Mol Breeding. 2010, 25: 145-154. 10.1007/s11032-009-9314-7.
Yamamori M, Fujita S, Hayakawa K, Matsuki J, Yasui T: Genetic elimination of a starch granule protein, SGP-1, of wheat generates an altered starch with apparent high amylose. Theor App Genet. 2000, 101: 21-29. 10.1007/s001220051444.
Lafiandra D, Sestili F, D'Ovidio R, Janni M, Botticella E, Ferrazzano G, Silvestri M, Ranieri R, DeAmbrogio E: Approaches for the modification of starch composition in durum wheat. Cereal Chem. 2010, 87 (1): 28-34. 10.1094/CCHEM-87-1-0028.
Regina A, Kosar-Hashemi B, Li Z, Rampling L, Cmiel M, Gianibelli MC, Konik-Rose C, Larroque O, Rahman S, Morell MK: Multiple isoforms of starch branching enzyme-I in wheat: lack of the major Sbe-I isoform does not alter starch phenotype. Funct Plant Biol. 2004, 31: 591-601. 10.1071/FP03193.
Rahman S, Regina A, Li Z, Mukai Y, Yamamoto M, Kosar-Hashemi B, Abrahams S, Morell MK: Comparison of starch-branching enzyme genes reveals evolutionary relationships among isoforms. Characterization of a gene for starch-branching enzyme IIa from the wheat genome donor. Aegilops tauschii Plant Physiol. 2001, 125: 1314-24.
Regina A, Kosar-Hashemi B, Li Z, Pedler A, Mukai Y, Yamamoto M, Gale K, Sharp PJ, Morell MK, Rahman S: Starch branching enzyme IIb in wheat is expressed at low levels in. the endosperm compared to other cereals and encoded at a non-syntenic locus. Planta. 2005, 222: 899-909. 10.1007/s00425-005-0032-z.
Regina A, Bird A, Topping D, Freeman S, Barsby T, Kosar-Hashemi B, Rahman S, Morell MK: High-amylose wheats generated by RNA interference improves indices of large-bowel health in rats. Proc Nat Acad Sci USA. 2006, 103: 3546-3551. 10.1073/pnas.0510737103.
Sestili F, Janni M, Doherty A, Botticella E, D'Ovidio R, Masci S, Jones H, Lafiandra D: Increasing the amylose content of durum wheat through silencing of the SBEIIa genes. BMC Plant Biol. 2010, 10: 144-10.1186/1471-2229-10-144.
McCallum CM, Comai L, Greene EA, Henikoff S: Targeting induced local lesions in genomes (TILLING) for plant functional genomics. Plant Physiol. 2000, 123: 439-442. 10.1104/pp.123.2.439.
Slade AJ, Fuerstenberg SI, Loeffler D, Steine MN, Facciotti D: A reverse genetic, non transgenic approach to wheat crop improvement by TILLING. Nat Biotechnol. 2005, 23: 75-81. 10.1038/nbt1043.
Uauy C, Paraiso F, Colasuonno P, Tran RK, Tsai H, Berardi S, Comai L, Dubcovsky J: A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheats. BMC Plant Biol. 2009, 9: 115-10.1186/1471-2229-9-115.
Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ: High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003, 49: 853-860. 10.1373/49.6.853.
Dong C, Vincent K, Sharp S: Simultaneous mutation detection of three homoeologous genes in wheat by High Resolution Melting analysis and Mutation Surveyor. BMC Plant Biol. 2009, 9: 143-10.1186/1471-2229-9-143.
Gady ALF, Hermans FWK, Van de Wal MHBJ, Van Loo EN, Visser RGF, Bachem CWB: Implementation of two high through-put techniques in a novel application, detecting point mutations in large EMS mutated plant populations. Plant Methods. 2009, 5: 13-10.1186/1746-4811-5-13.
Joppa LR, Williams ND: Langdon durum disomic substitution lines and aneuploid analysis in tetraploid wheat. Genome. 1988, 30: 222-228. 10.1139/g88-038.
Rakszegi M, Kisgyorgy BN, Tearall K, Shewry PR, Lang L, Phillips A, Bedo Z: Diversity of agronomic and morphological traits in a mutant population of bread wheat studied in the Healthgrain program. Euphytica. 2010, 174: 409-421. 10.1007/s10681-010-0149-4.
Ng PC, Henikoff S: SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31: 3812-3814. 10.1093/nar/gkg509.
Kuriki T, Stewart DC, Preiss J: Construction of chimeric enzymes out of maize endosperm branching enzymes I and II activity and properties. J Biol Chem. 1997, 272: 2899-2904.
Abad MC, Binderup K, Rios-Steiner J, Arni RK, Preiss J, Jeiger JH: The x-ray crystallographic structure of Escherichia coli branching enzyme. J Biol Chem. 2002, 277: 42164-42170. 10.1074/jbc.M205746200.
Zhang Y: I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008, 9: 40-10.1186/1471-2105-9-40.
Lal S, Choi J, Curtis Hannah L: The AG dinucleotide terminating introns is important but not always required for pre-mRNA splicing in the maize endosperm. Plant Physiol. 1999, 120 (1): 65-72. 10.1104/pp.120.1.65.
Brown JWS: Arabidopsis intron mutations and pre-mRNA splicing. Plant J. 1996, 10: 771-780. 10.1046/j.1365-313X.1996.10050771.x.
Smith CJW, Chu TT, Nadal-Ginard B: Scanning and competition between AGs are involved in 39 splice site selection in mammalian introns. Mol Cell Biol. 1993, 13: 4939-4952.
Yamamori M, Nakamura T, Kuroda A: Variations in the content of starch-granule bound protein among several Japanese cultivars of common wheat (Triticum aestivum L). Euphytica. 1992, 64: 215-219. 10.1007/BF00046051.
Urbano M, Margiotta B, Colaprico G, Lafiandra D: Waxy protein in diploid, tetraploid and hexaploid wheats. Plant Breeding. 2002, 121: 1-5. 10.1046/j.1439-0523.2002.00662.x.
Svensson B: Protein engineering in the alpha-amylase family: catalytic mechanism, substrate specificity and stability. Plant Mol Biol. 25: 141-157.
Hong S, Mikkelsen R, Preiss J: Analysis of the amino terminus of maize branching enzyme II by polymerase chain reaction random mutagenesis. Arch Biochem Biophys. 2001, 386: 62-68. 10.1006/abbi.2000.2179.
Parry MAJ, Madgwick PJ, Bayon C, Tearall K, Lopez AH, Baudo M, Rakszegi M, Hamada W, Al-Yassin A, Ouabbou H, Labhilili H, Phillips A: Mutation discovery for crop improvement. J Exp Bot. 2009, 60: 2817-2825. 10.1093/jxb/erp189.
Hofinger BJ, Jing HC, Hammond KE, Kanyuka K: High-resolution melting analysis of cDNA-derived PCR amplicons for rapid and cost-effective identification of novel alleles in barley. Theor Appl Genet. 2009, 119: 851-865. 10.1007/s00122-009-1094-2.
Reed GH, Wittwer CT: Sensitivity and specificity of single nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem. 2004, 50: 1748-1754. 10.1373/clinchem.2003.029751.
Cai XL, Wang ZY, Zhang JL, Hong MM: Aberrant splicing of intron 1 leads to the heterogeneous 5' UTR and decreased expression of waxy gene in rice cultivars of intermediate amylose content. Plant J. 1998, 14: 459-465. 10.1046/j.1365-313X.1998.00126.x.
Henikoff S, Comai L: Single-nucleotide mutations for plant functional genomics. Ann Rev Plant Biol. 2003, 54: 375-401. 10.1146/annurev.arplant.54.031902.135009.
Saito M, Nakamura T: Two point mutations identified in emmer wheat generate null Wx-A1 alleles. Theor Appl Genet. 2005, 110: 276-282. 10.1007/s00122-004-1830-6.
Patron NJ, Smith AM, Fahy BF, Hylton CM, Naldrett MJ, Rossnagel BG, Denyer K: The altered pattern of amylose accumulation in the endosperm of low-amylose barley cultivars is attributable to a single mutant allele of granule-bound starch synthase I with a deletion in the 5'-non-coding region. Plant Physiol. 2002, 130: 190-198. 10.1104/pp.005454.
Zhu YF, Li YW, Chen Y: Generation and characterization of a high molecular weight glutenin 1Bx14-deficient mutant in common wheat. Plant Breeding. 2005, 124: 421-427. 10.1111/j.1439-0523.2005.01145.x.
Frischmeyer PA, Dietz HC: Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1998, 8: 1893-1900.
Maquat LE: Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004, 5: 89-99.
Jofuku KD, Schipper RD, Goldberg RB: A frameshift mutation prevents Kunitz trypsin inhibitor mRNA accumulation in soybean embryos. Plant Cell. 1989, 1: 427-435.
Dehesh K, Franci C, Parks BM, Seeley KA, Short TW, Tepperman JM, Quail PH: Arabidopsis HY8 locus encodes phytochrome A. Plant Cell. 1993, 5: 1081-1088.
Marchant A, Bennett MJ: The Arabidopsis AUX1 gene: a model system to study mRNA processing in plants. Plant Mol Biol. 1998, 36: 463-471. 10.1023/A:1005961303167.
Isshiki M, Yamamoto Y, Satoh H, Shimamoto K: Nonsense-mediated decay of mutant waxy mRNA in rice. Plant Physiol. 2001, 125: 1388-1395. 10.1104/pp.125.3.1388.
Miura H, Sugawara A: Dosage effect of Waxy genes on amylose synthesis in wheat. Theor Appl Genet. 1996, 93 (7): 1066-1070. 10.1007/BF00230126.
Konik-Rose C, Thistleton J, Chanvrier H, Tan I, Halley P, Gidley M, Kosar-Hashemi B, Wang H, Larroque O, Ikea J, McMaugh S, Regina A, Rahman S, Morell M, Li Z: Effects of starch synthase IIa gene dosage on grain, protein and starch in endosperm of wheat. Theor Appl Genet. 2007, 115: 1053-1065. 10.1007/s00122-007-0631-0.
McClearly BV, Solah V, Gibson TS: Quantitative measurement of total starch in cereal flours and products. J Cereal Sci. 1994, 20: 51-58. 10.1006/jcrs.1994.1044.
Nakamura T, Yamamori M, Hirano H, Hidaka S, Nagamine T: Production of waxy (amylose-free) wheats. Mol Gen Genet. 1995, 248: 253-259. 10.1007/BF02191591.
Tai TH, Tanksley SD: A rapid and inexpensive method for isolation of total DNA from dehydrated plant tissue. Plant Mol Biol Rep. 1991, 8: 297-303.
Laudencia-Chingcuanco DL, Stamova BS, You FM, Lazo GR, Beckles DM, Anderson OD: Transcriptional profiling of wheat caryopsis development using cDNA microarrays. Plant Mol Biol. 2006, 63: 651-668.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Neff MM, Neff JD, Chory J, Pepper AE: dCAPS, a simple technique for the genetic analysis of single nucleotide polym.orphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 1998, 14: 387-392. 10.1046/j.1365-313X.1998.00124.x.
Chrastil J: Improved colorimetric determination of amylose in starches or flours. Carbohydr Res. 1987, 159: 154-158. 10.1016/S0008-6215(00)90013-2.
Vansteelandt J, Delcour JA: Physical behaviour of durum wheat starch (Triticum durum) during industrial pasta processing. J Agric Food Chem. 1988, 46: 2499-2503.
Acknowledgements
This study is partially financially supported by the European Commission in the Communities 6th Framework Programme, Project HEALTHGRAIN (Food- CT-2005-514008). It reflects the authors' views and the Community is not liable for any use that may be made of the information contained in this publication and by AGER in the From Seed to Pasta section.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
EB carried out the TILLING analysis, molecular and bioinformatic characterization of mutants and drafted the paper with FS and DL. AHL collaborated to the optimization of HRM analysis. AP provided the EMS wheat population and the HRM TILLING platform. EB, FS, AP and DL edited the manuscript. DL coordinated the work. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Botticella, E., Sestili, F., Hernandez-Lopez, A. et al. High resolution melting analysis for the detection of EMS induced mutations in wheat Sbella genes. BMC Plant Biol 11, 156 (2011). https://doi.org/10.1186/1471-2229-11-156
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-11-156