
Abstract
Protein kinases are critical modulators of a variety of cellular signal transduction pathways, and abnormal phosphorylation events can be a cause or contributor to disease progression in a variety of disorders. This has led to the emergence of protein kinases as an important new class of drug targets for small molecule therapeutics. A serine/threonine protein kinase, p38α mitogen-activated protein kinase (MAPK), is an established therapeutic target for peripheral inflammatory disorders because of its critical role in regulation of proinflammatory cytokine production. There is increasing evidence that p38α MAPK is also an important regulator of proinflammatory cytokine levels in the central nervous system, raising the possibility that the kinase may be a drug discovery target for central nervous system disorders where cytokine overproduction contributes to disease progression. Development of bioavailable, central nervous system-penetrant p38α MAPK inhibitors provides the required foundation for drug discovery campaigns targeting p38α MAPK in neurodegenerative disorders.
Similar content being viewed by others


Background
Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine protein kinases that play essential roles in eukaryotic cells by transducing environmental stress signals into altered gene expression. There are numerous human MAPKs, which are grouped into distinct families: the extracellular signal-regulated protein kinases (ERKs); the c-Jun N-terminal kinases (JNKs); and the p38 MAPKs (p38α, p38β, p38δ, p38γ). Different stressors, or combinations of stressors, result in differential activation of the discrete MAPK families, which can function in parallel in intracellular signal transduction cascades that alter cellular physiology. Signaling cross-talk among the individual MAPK cascades, as well as cross-talk with second messenger-mediated protein phosphorylation cascades, result in a high degree of biological selectivity in a tissue's response to stressors. Therefore, the presence of a given MAPK family member in a tissue or cell type does not provide a simple forecast of its physiological or pathophysiological role.
Various genetic and pharmacological inhibitors of individual protein kinases in stress-activated cells have provided causal linkages between the activation of a given kinase, or MAPK family pathway, and a particular cellular response endpoint, for example, increased production of proinflammatory cytokines. The p38 MAPK family of serine/threonine protein kinases was explicitly implicated in the regulation of key inflammatory responses in mammals, contributing to a large body of evidence that eventually established it as a therapeutic target for a range of diseases that have inflammation as a common disease progression mechanism. An isoform of the p38 MAPK family, p38α MAPK, was identified early as a drug discovery target and became the focus of intense investigations for over a decade. Currently, novel p38α MAPK inhibitors are in clinical development for peripheral tissue inflammatory disorders. On-going investigations continue to validate p38α MAPK as a therapeutic target for peripheral tissue disorders, displaying no target-related toxicities when appropriate compounds and dosing regimens are used. However, in vivo evidence supporting p38α MAPK as a central nervous system (CNS) therapeutic target has only recently become available. Here we provide a brief review of these emerging CNS data and highlight selected work that provided the firm foundation for considering bioavailable, blood brain barrier-penetrant, non-toxic p38α MAPK inhibitors as potential therapeutics for CNS disorders.
The p38 MAPK family as regulators of proinflammatory cytokine production
Proinflammatory cytokines are crucial components of physiological defense mechanisms, but chronic overproduction can lead to cellular dysfunction and damage [1]. One pathophysiology mechanism for peripheral tissue injury is the overproduction of proinflammatory cytokines, for example, tumor necrosis factor (TNF)α and interleukin (IL-1)β, which can lead to tissue barrier dysfunction and cell death. Current macromolecular therapeutics for peripheral tissue disorders used in the clinic target this increased cytokine activity [1]. Intracellular MAPK signal transduction cascades, especially the p38 MAPKs, are important regulators of proinflammatory cytokine biosynthesis [2–4]. p38 MAPK was first identified as a key regulator of IL-1β and TNFα production in human monocytes after lipopolysaccharide treatment [5, 6]. Later studies showed that activation of p38 MAPK regulates proinflammatory cytokine production at the transcriptional and post-transcriptional levels [7, 8], laying the foundation for exploration of p38 MAPK as a potential drug discovery target for attenuation of increased proinflammatory cytokine levels [3, 4].
Four isoforms of p38 MAPK have been identified, each the product of distinct genes: p38α, 38β, p38γ and p38δ [2, 9]. There are also several splice variants of these isoforms. p38α MAPK is widely expressed among tissues and is considered a crucial mediator of inflammatory responses activated by a variety of signaling mechanisms with a wide range of physiological endpoints [6, 10, 11]. Recently, O'Keefe et al. [12] demonstrated in an elegant approach using knock-in mice that the specific inhibition of the p38α isoform in vivo is sufficient and necessary for suppression of increased peripheral proinflammatory cytokine levels after lipopolysaccharide challenge. As with many intracellular signaling cascades mediated by serial protein phosphorylation steps, p38α MAPK is activated via transphosphorylations by upstream kinases [2]. The activation of p38α MAPK, in turn, allows it to efficiently phosphorylate its protein substrates [13]. The exact physiological outcomes from such integrated, complex networks are dependent on the type of stressor, cell type, tissue context of the cell, and previous stimulations.
In terms of the regulatory mechanisms of proinflammatory cytokine production, several of the p38 MAPK substrates are transcription factors, or other protein kinases, which in turn can phosphorylate regulatory proteins and thereby modulate function [13]. For example, p38α MAPK can phosphorylate a variety of transcription factors, for example, ATF2, ELK1, CREB, MEF2C, CHOP/GADD153, and C/EBPβ, leading to transcriptional stimulation of proinflammatory cytokines [14]. There are also a variety of p38α MAPK substrates that can regulate proinflammatory cytokine production through either transcriptional or translational mechanisms. One of the first endogenous substrates identified for p38α MAPK was MAP kinase-activated protein kinase-2 (MAPKAP K2 or MK2), which is critical for the biosynthesis of TNFα after lipopolysaccharide treatment [15–17]. This pathway has also been proposed to stabilize cytokine mRNA by mechanisms dependent on AU-rich elements in the untranslated regions of the cytokine genes [18, 19]. Another protein kinase substrate of p38α MAPK is mitogen and stress activated protein kinase 1 (MSK1), which is activated upon phosphorylation by p38α MAPK. Activated MSK1 can, in turn, stimulate transcription factors, allowing increased proinflammatory cytokine production [20]. MSK1 also appears to be involved in the expression of proinflammatory cytokine genes through phosphorylation of histone 3 (H3) and recruitment of NFκB [21–23]. The p38 MAPK/MSK1 pathway is also important for phosphorylation of CREB, a regulatory protein implicated in proinflammatory cytokine gene expression [24, 25]. Consistent with these proposed biological roles in cell function, p38 MAPKs are found in both the nucleus and cytoplasm [14, 26]. Thus, the wide variety of downstream substrates of p38 MAPKs, along with the spectrum of stressors and upstream activators that can converge on p38 MAPK activation, allow for fine control of proinflammatory cytokine production. The pivotal role of these pathways in the regulation of responses resulting in increased proinflammatory cytokine activity emphasizes the detrimental consequences dysregulation of these kinase cascades can have in disease.
Modulation of p38α MAPK as a therapeutic approach to peripheral inflammatory disorders
p38α MAPK is an established drug discovery target for peripheral inflammatory diseases, including rheumatoid arthritis and Crohn's disease, where increased levels of proinflammatory cytokines coincide with disease progression [27–30]. Treating animal models of these diseases with p38α MAPK inhibitors reduces the expression of proinflammatory cytokines, for example, IL-1β and TNFα, and alters disease-related pathology [31–33]. Early clinical development studies for the treatment of peripheral inflammatory diseases revealed toxicity issues related to the chemotype of the inhibitor and not to the target [34, 35]. This knowledge was used in the recursive development of later generation inhibitors based on structurally distinct chemotypes that showed improved safety, metabolic stability, and pharmacokinetic profiles while retaining or improving upon the affinity and selectivity for p38α MAPK [36–38].
Currently, p38α MAPK inhibitors based on a variety of chemotypes are in clinical development for the treatment of peripheral tissue diseases, for example, multiple myeloma, atherosclerosis, chronic obstructive pulmonary disease, rheumatoid arthritis, and pain. Compounds include, among others, SCIO 469 (Scios, Johnson and Johnson), VX-702 (Vertex), and SB 681323 (GlaxoSmithKline) [11]. These p38α MAPK inhibitors displayed concentration-dependent inhibition of kinase activity when tested in vitro, with IC50 values (concentration of inhibitor required for 50% inhibition) well below the desired 1 μM in vitro inhibition activity sought before moving to in vivo studies. This landmark value is based on the finding that the K m values (concentration of substrate that gives half-maximal activity) for the two physiological substrates of signaling kinases, ATP and the respective endogenous protein substrate, range from 1–20 μM [39], and the assumption that a bioavailable and comparatively stable inhibitor should exhibit in vivo function if the target is valid. Approved kinase inhibitor drugs provide a precedent consistent with this guideline. Imatinib (Gleevec), the first example of these agents, has IC50 values of 0.1–0.35 μM for its kinase targets [40]. The p38α MAPK inhibitors in clinical development also showed selectivity of inhibition when examined by in vitro kinase inhibition screens that include pathway- and structurally-related protein kinases. Generally, there is some inhibition of p38β MAPK due to the close structural similarity between the p38α and p38β isoforms. However, p38δ MAPK and p38γ MAPK have key differences in the site targeted by many inhibitors designed by structure-assisted methods, allowing good selectivity against them [12, 29, 35–38, 41, 42].
Contemporary approaches to the design of selective p38α MAPK inhibitors take advantage of the extensive information available from high-resolution structures of p38 MAPK isoforms and complexes with inhibitors. Many p38α MAPK inhibitors are designed to exploit the small 'gatekeeper' amino acid that controls access to a hydrophobic binding pocket in the enzyme active site [37, 38, 41]. p38α and p38β MAPKs both have a threonine at the gatekeeper residue, 106, whereas p38γ and p38δ MAPKs have a significantly larger methionine present at the corresponding position. This enables the design of small molecules that can dock bulky constituents into the large hydrophobic pocket of the active site of the kinases with the smaller gatekeeper residue, that is, p38α and p38β MAPKs. Exploitation of this gatekeeper and hydrophobic pocket feature is combined with other aspects of the structure to create affinity and selectivity for p38α MAPK/p38β MAPK versus the other MAPK isoforms as well as other kinases. A recent example is the combination of the gatekeeper discrimination with another p38α MAPK/p38β MAPK structural feature, the potential for hydrogen bonding with the nearby peptide backbone segment that is conserved in the two kinases [37, 38, 41, 42]. This is done by a vicinal arrangement of the two features in small molecules so that spacing and steric constraints within the targets are utilized.
p38α MAPK as a drug discovery target for CNS disorders
Evidence from both clinical studies and preclinical animal models suggests proinflammatory cytokine overproduction as a potential driving force for pathology progression in CNS disorders [1, 41, 43–46]. Although the importance of p38α MAPK in the regulation of proinflammatory cytokine production in peripheral inflammatory disorders is well established, much less is known about its role in CNS inflammatory disorders. Glia and neuron cell culture studies demonstrated the importance of p38α MAPK activation for up-regulated cytokine production by stressed glia and provided a link between p38α MAPK and stressor-induced neuronal dysfunction in vitro [47–53]. In addition to glial p38α MAPK and its linkage in cell culture to cytokine increases in response to stressors, p38α MAPK is also expressed in neurons [54, 55]. Neuronal p38α MAPK is considered a contributor to the phosphorylation and abnormal functioning of tau, a microtubule-associated protein found in neurons that correlates with the clinical pathology in AD and other dementias [56–59]. An interesting potential link between p38α MAPK activation in both glia and neurons comes from studies of co-cultures that showed microglia activation and release of IL-1β leading to increased neuronal tau phosphorylation [60]. Activation of p38α MAPK is also seen in brain tissue from AD transgenic mouse models [55, 61, 62]. Finally, clinical pathology results revealed the presence of activated p38α MAPK in brain samples from AD patients, with the activated p38α MAPK localized primarily to neurofibrillary tangles, neurons near neuritic amyloid plaques, and in glial cells [59, 63–65].
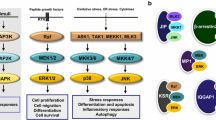
An indication of the potential complexity of p38 MAPK involvement in the physiology and pathophysiology of glia and neurons is shown in Figure 1. This pictorial representation of the integrated and redundant aspects of intracellular signal transduction, mediated by protein phosphorylation pathways, indicates the currently unpredictable nature of how a particular pathway may be quantitatively involved in the response to a given stressor. The tissue context of a given cell type will add another variable. Due to the inherent limitations of removing glia and neurons from their tissue environment, it is critical that in vivo studies test explicit hypotheses about the quantitative importance of p38 MAPKs in specific pathophysiological progression mechanisms. This knowledge can provide a foundation for future therapeutic development campaigns.

Diagrammatic outline of p38 mitogen-activated protein kinase (MAPK) pathways in glia and neurons. Disease-relevant stressors or stimuli can activate a variety of cross-talking and interacting signal transduction pathways, some of which can converge on activation of the p38 MAPK signaling cascades. For example, a typical p38 MAPK cascade consists of a three-tiered series of protein kinases: a MAPK (p38) and two upstream components (a MAPK kinase (MEK) and a MAPKK kinase (MEKK)) that activate the p38 MAPK by a series of activating phosphorylations. Activation of p38 MAPK and phosphorylation of its downstream substrates in activated glia (primarily microglia) can lead to up-regulation of proinflammatory cytokine production. Proinflammatory cytokines can act back on glia to stimulate multiple intracellular signaling pathways. Neurons can also respond to proinflammatory cytokine or other stressors/stimuli and activate neuronal p38 MAPK, culminating in neuron damage. The consequences of p38 MAPK activation in glia and neurons depend on the set of upstream signals, the isoform of p38 MAPK, the cell type, and the set of substrates that are stimulated.
An in vivo causative link between p38α MAPK and disease-relevant CNS pathophysiology has been provided by the use of small molecule p38 MAPK inhibitors in animal models of brain injury. For example, administration of a second-generation p38α MAPK inhibitor, SB239063, reduced infarct volume and attenuated neurological deficits in a rat model of focal ischemic brain injury [66, 67]. Oral administration of a CNS-penetrant p38α MAPK inhibitor, MW01-2-069A-SRM, suppressed increases in hippocampal proinflammatory cytokine levels and the mechanistically associated synaptic marker protein loss in a mouse model of amyloid-beta (Aβ)-induced injury [41]. Consistent with the compound's positive effect on hippocampal synaptic marker protein levels was an attenuation of hippocampal-dependent behavioral deficits [41]. Such results indicate the potential for targeting p38α MAPK as a therapeutic approach in some CNS disorders and provide causative links between p38α MAPK, increased proinflammatory cytokine levels, and synaptic dysfunction.
The in vivo results with small molecule p38α MAPK inhibitors raise the possibility that appropriate dosing with bioavailable, blood brain barrier-penetrant, and non-toxic p38α MAPK inhibitors could generate a desired therapeutic outcome for CNS disorders. The mechanism could include a combined effect of attenuation of up-regulated proinflammatory cytokine production by activated glia and a potential neuroprotective effect on neuronal dysfunction. In terms of chronic neurodegenerative disorders like AD, compounds such as MW01-2-069A-SRM (Figure 2) have a number of appealing features [41]. The compound has molecular properties that are associated with successful human CNS therapeutics, and has good oral bioavailability and brain penetrance in rodents. For example, after oral administration to mice, the compound exhibited a peak brain:blood concentration ratio of approximately 0.7. Oral administration of MW01-2-069A-SRM at a low dose (2.5 mg/kg) after the start of exposure to the stressor (for example, toxic forms of human Aβ1–42) resulted in hippocampal levels of the proinflammatory cytokines TNFα and IL-1β, and the presynaptic marker protein synaptophysin, being indistinguishable from control levels measured weeks later [41]. These data provide evidence that the p38α MAPK pathway is quantitatively important in the Aβ-induced up-regulation of proinflammatory cytokines in the hippocampus, and that brain p38α MAPK should be considered as a credible molecular target in future drug development campaigns for AD and related neurodegenerative disorders.

Strategy for development of a p38α mitogen-activated protein kinase (MAPK) inhibitor. (A) The p38α MAPK inhibitor MW01-2-069A-SRM was developed using a structure-assisted and computational modeling design strategy, along with consideration of compound molecular properties. The inhibitor is based on a 3-amino-6-phenyl pyridazine scaffold (MW01-3-183WH) found in other central nervous system (CNS)-active compounds [46, 68]. A pyridinyl pharmacophore characteristic of many p38 MAPK inhibitors was added to the scaffold. Modeling forecasted that MW01-2-069A-SRM could be accommodated by the p38α MAPK structure. The smaller Thr106 gatekeeper residue allowed the phenyl ring of the compound to occupy a hydrophobic pocket while the nitrogen of the pyridine ring could make a critical H-bond interaction with the amide bond between Met109 and Gly110. These interactions are important for selectivity and affinity for the p38α MAPK isoform. MW01-2-069A-SRM was a p38α MAPK inhibitor, with an IC50 of approximately 0.8 μM, and was relatively selective for p38α MAPK; at 20 μM, the compound showed complete inhibition of p38α MAPK, partial inhibition of p38β MAPK, and no inhibition of p38δ MAPK, p38γ MAPK, or 40 other protein kinases [41]. (B) Validation of the design approach and modeling of predicted interactions was done by testing analogs that did not show the predicted interactions with the kinase. For example, the scaffold compound MW01-3-183WH, which lacks the pyridine ring, is inactive. The inactive analog MW01-4-199SRM has a pyridine nitrogen in a different structural orientation, which should compromise activity due to distance constraints and altered electronegativity. Another inactive analog, MW01-6-189WH, has an identical composition to MW01-2-069A-SRM, but has its pyridine ring in a different position on the pyridazine scaffold. These data show that the pyridine pharmacophore must be introduced adjacent to the phenyl group in the molecular context of the scaffold, as found in MW01-2-069A-SRM, to produce a p38α MAPK inhibitor with good affinity and selectivity.
Conclusion
p38α MAPK is emerging as an attractive target for CNS disorders where increases in proinflammatory cytokines appear to play a role in disease progression. The development of orally bioavailable, brain-penetrant, CNS-active p38α MAPK inhibitors provides not only a valuable research tool for testing hypotheses about the role of p38α MAPK in CNS disorders, but also represents a foundation for future drug discovery efforts to develop potential neurodegenerative disease-modifying therapeutics that target this critical gene-regulating protein kinase.
Abbreviations
- Aβ:
-
amyloid-beta
- CNS:
-
central nervous system
- IL:
-
interleukin
- MAPK:
-
mitogen-activated protein kinase
- MSK1:
-
mitogen and stress activated protein kinase 1
- TNF:
-
tumor necrosis factor.
References
Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Watterson DM: Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target based discovery approaches. Int Rev Neurobiol. 2007, 82: 277-296.
Kyriakis JM, Avruch J: Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001, 81: 808-869.
O'Neill LA: Targeting signal transduction as a strategy to treat inflammatory diseases. Nat Rev Drug Discov. 2006, 5: 549-563.
Saklatvala J: The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004, 4: 372-377.
Han J, Lee JD, Bibbs L, Ulevitch RJ: A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994, 265: 808-811.
Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR: A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994, 372: 739-746.
Mielke K, Herdegen T: JNK and p38 stress kinases-degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000, 61: 45-60.
Schieven GL: The biology of p38 kinase: a central role in inflammation. Curr Topics Med Chem. 2005, 5: 921-928.
Roux PP, Blenis J: ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004, 68: 320-344.
Guo X, Gerl RE, Schrader JW: Defining the involvement of p38alpha MAPK in the production of anti- and proinflammatory cytokines using an SB 203580-resistant form of the kinase. J Biol Chem. 2003, 278: 22237-22242.
Cuenda A, Rousseau S: p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007, 1773: 1358-1375.
O'Keefe SJ, Mudgett JS, Cupo S, Parsons JN, Chartrain NA, Fitzgerald C, Chen SL, Lowitz K, Rasa C, Visco D, Luell S, Carballo-Jane E, Owens K, Zaller DM: Chemical genetics define the roles of p38α and p38β in acute and chronic inflammation. J Biol Chem. 2007, 282: 34663-34671.
Shi Y, Gaestel M: In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol Chem. 2002, 383: 1519-1536.
Zarubin T, Han J: Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15: 11-18.
Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J: Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994, 78: 1039-1049.
Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M: MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999, 1: 94-97.
Culbert AA, Skaper SD, Howlett DR, Evans NA, Facci L, Soden PE, Seymour ZM, Guillot F, Gaestel M, Richardson JC: MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. J Biol Chem. 2006, 281: 23658-23667.
Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Müller M, Gaestel M, Resch K, Holtmann H: The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999, 18: 4969-4980.
Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M: MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002, 277: 3065-3068.
Deak M, Clifton AD, Lucocq LM, Alessi DR: Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17: 4426-4441.
Saccani S, Pantano S, Natoli G: p38-dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002, 3: 69-75.
Vermeulen L, De Wilde G, Van Damme P, Berghe Vanden W, Haegeman G: Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003, 22: 1313-1324.
Olson CM, Hedrick MN, Izadi H, Bates TC, Olivera ER, Anguita J: p38 mitogen-activated protein kinase controls NF-kappaB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen- and stress-activated protein kinase 1 in response to Borrelia burgdorferi antigens. Infect Immun. 2007, 75: 270-277.
Funding AT, Johansen C, Kragballe K, Otkjaer K, Jensen UB, Madsen MW, Fjording MS, Finnemann J, Skak-Nielsen T, Paludan SR, Iversen L: Mitogen- and stress-activated protein kinase 1 is activated in lesional psoriatic epidermis and regulates the expression of pro-inflammatory cytokines. J Invest Dermatol. 2006, 126: 1784-1791.
Jia Q, Zhou HR, Shi Y, Pestka JJ: Docosahexaenoic acid consumption inhibits deoxynivalenol-induced CREB/ATF1 activation and IL-6 gene transcription in mouse macrophages. J Nutr. 2006, 136: 366-372.
Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ: Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995, 270: 7420-7426.
Andreakos E: Targeting cytokines in autoimmunity: new approaches, new promise. Expert Opin Biol Ther. 2003, 3: 435-447.
Morel J, Berenbaum F: Signal transduction pathways: new targets for treating rheumatoid arthritis. Joint Bone Spine. 2004, 71: 503-510.
Bolos J: Structure-activity relationships of p38 mitogen-activated protein kinase inhibitors. Mini Rev Med Chem. 2005, 5: 857-868.
Kaminska B: MAPK signaling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005, 1754: 253-263.
Miwatashi S, Arikawa Y, Kotani E, Miyamoto M, Naruo K, Kimura H, Tanaka T, Asahi S, Ohkawa S: Novel inhibitor of p38 MAP kinase as an anti-TNF-alpha drug: discovery of N-[4-[2-ethyl-4-(3-methylphenyl)-1,3-thiazol-5-yl]-2-pyridyl]benzamide (TAK-715) as a potent and orally active anti-rheumatoid arthritis agent. J Med Chem. 2005, 48: 5966-5979.
Mbalaviele G, Anderson G, Jones A, De Ciechi P, Settle S, Mnich S, Thiede M, Abu-Amer Y, Portanova J, Monahan J: Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. J Pharmacol Exp Ther. 2006, 317: 1044-1053.
Medicherla S, Ma JY, Mangadu R, Jiang Y, Zhao JJ, Almirez R, Kerr I, Stebbins EG, O'Young G, Kapoun AM, Luedtke G, Chakravarty S, Dugar S, Genant HK, Protter AA: A selective p38 alpha mitogen-activated protein kinase inhibitor reverses cartilage and bone destruction in mice with collagen-induced arthritis. J Pharmacol Exp Ther. 2006, 318: 132-141.
Dambach DM: Potential adverse effects associated with inhibition of p38alpha/beta MAP kinases. Curr Top Med Chem. 2005, 5: 929-939.
Goldstein DM, Gabriel T: Pathway to the clinic: inhibition of p38 MAP kinase. A review of ten chemotypes selected for development. Curr Top Med Chem. 2005, 5: 1017-1029.
Dominguez C, Powers DA, Tamayo N: p38 MAP kinase inhibitors: many are made, but few are chosen. Curr Opin Drug Discov Devel. 2005, 8: 421-430.
Lee MR, Dominguez C: MAP kinase p38 inhibitors: clinical results and an intimate look at their interactions with p38α protein. Curr Med Chem. 2005, 12: 2979-2994.
Peifer C, Wagner G, Laufer S: New approaches to the treatment of inflammatory disorders with small molecule inhibitors of p38 MAP kinase. Curr Top Med Chem. 2006, 6: 113-149.
Stein B, Yang MX, Young DB, Janknecht R, Hunter T, Murray BW, Barbosa MS: p38-2, a novel mitogen-activated protein kinase with distinct properties. J Biol Chem. 1997, 272: 19509-19517.
O'Dwyer ME, Druker BJ: The role of the tyrosine kinase inhibitor STI571 in the treatment of cancer. Curr Cancer Drug Targets. 2001, 1: 49-57.
Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM: A novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007, 4: 21.
Wrobleski ST, Doweyko AM: Structural comparison of p38 inhibitor-protein complexes: a review of recent p38 inhibitors having unique binding interactions. Curr Top Med Chem. 2005, 5: 1005-1016.
Craft JM, Watterson DM, Marks A, Van Eldik LJ: Enhanced susceptibility of S100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human β-amyloid. Glia. 2005, 51: 209-216.
Mrak RE, Griffin WS: Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005, 26: 349-354.
Tuppo EE, Arias HR: The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005, 37: 289-305.
Hu W, Ralay Ranaivo H, Roy SM, Behanna HA, Wing LK, Munoz L, Guo L, Van Eldik LJ, Watterson DM: Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. Bioorg Med Chem Lett. 2007, 17: 414-418.
Bhat NR, Zhang P, Lee JC, Hogan EL: Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998, 18: 1633-1641.
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE: Glial-neuronal interactions in Alzheimer's disease: the potential role of a 'cytokine cycle' in disease progression. Brain Pathol. 1998, 8: 65-72.
Li Y, Liu L, Barger SW, Mrak RE, Griffin WST: Vitamin E suppression of microglial activation is neuroprotective. J Neurosci Res. 2001, 66: 163-170.
Jeohn GH, Cooper CL, Wilson B, Chang RC, Jang KJ, Kim HC, Liu B, Hong JS: p38 MAP kinase is involved in lipopolysaccharide-induced dopaminergic neuronal cell death in rat mesencephalic neuron-glia cultures. Ann N Y Acad Sci. 2002, 962: 332-346.
Kim SH, Smith CJ, Van Eldik LJ: Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1β production. Neurobiol Aging. 2004, 25: 431-439.
Xie Z, Smith CJ, Van Eldik LJ: Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004, 45: 170-179.
Griffin WS, Liu L, Li Y, Mrak RE, Barger SW: Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006, 3: 5.
Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS: Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001, 39: 341-348.
Ferrer I: Stress kinases involved in tau phosphorylation in Alzheimer's disease, tauopathies and APP transgenic mice. Neurotox Res. 2004, 6: 469-475.
Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH: Reactivating kinase/p38 phosphorylates τ protein in vitro. J Neurochem. 1997, 69: 191-198.
Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A: Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol. 2001, 60: 1190-1197.
Ferrer I, Blanco R, Carmona M, Puig B: Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J Neural Transm. 2001, 108: 1397-1415.
Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J: Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2005, 2: 3-18.
Li Y, Liu L, Barger SW, Griffin WS: Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003, 23: 1605-1611.
Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F: Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38 MAPK pathway. Neurobiol Dis. 2002, 11: 257-274.
Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW: Activation of c-Jun N terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002, 22: 3376-3385.
Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye ZN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G: p38 kinase is activated in the Alzheimer's disease brain. J Neurochem. 1999, 72: 2053-2058.
Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF: Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration. J Alzheimers Dis. 2001, 3: 41-48.
Sun A, Liu M, Nguyen XV, Bing G: p38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp Neurol. 2003, 183: 394-405.
Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA: SB 23 a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharm Exp Ther. 9063, 296: 312-321.
Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC: SB 23 a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 9063, 892: 70-77.
Contreras JM, Rival YM, Chayer S, Bourguignon JJ, Wermuth CG: Aminopyridazines as acetylcholinesterase inhibitors. J Med Chem. 1999, 42: 730-741.
Acknowledgements
This research was supported in part by a grant from the Alzheimer's Drug Discovery Foundation, and NIH grants AG031311 (DMW) and AG013939 (LVE). ASB is a postdoctoral trainee previously supported by NIH T32 AG000260 and currently supported by NIH F32 AG032842.
This article has been published as part of BMC Neuroscience Volume 9 Supplement 2: 2008 Proceedings of the 8th International Conference on Alzheimer's Disease Drug Discovery The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2202/9?issue=S2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ASB coordinated literature searches and data analysis. LdA assisted with literature searches and figure preparation. DMW and LVE conceived of the project, and drafted the manuscript with the assistance of the other authors. All authors read and approved the final manuscript.
Aaron S Borders, Lucia de Almeida contributed equally to this work.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Borders, A.S., de Almeida, L., Van Eldik, L.J. et al. The p38α mitogen-activated protein kinase as a central nervous system drug discovery target. BMC Neurosci 9 (Suppl 2), S12 (2008). https://doi.org/10.1186/1471-2202-9-S2-S12
Published:
DOI: https://doi.org/10.1186/1471-2202-9-S2-S12