
Abstract
Background
An ever growing body of evidences is emerging concerning metabolism hormones, neurotransmitters or stress-related biomarkers as effective modulators of eating behavior and body weight in mammals. The present study sought at examining the density and affinity of two proteins related to neurotransmission and cell metabolism, the serotonin transporter SERT and the cholesterol import-benzodiazepine site TSPO (translocator protein), in a rodent leptin-lacking mutant, the obese ob/ob mouse. Binding studies were thus carried out in brain or peripheral tissues, blood platelets (SERT) and kidneys (TSPO), of ob/ob and WT mice supplied with a standard diet, using the selective radiochemical ligands [3H]-paroxetine and [3H]-PK11195.
Results
We observed comparable SERT number or affinity in brain and platelets of ob/ob and WT mice, whilst a significantly higher [3H]-PK11195 density was reported in the brain of ob/ob animals. TSPO binding parameters were similar in the kidneys of all tested mice. By [3H]-PK11195 autoradiography of coronal hypothalamic-hippocampal sections, an increased TSPO signal was detected in the dentate gyrus (hippocampus) and choroids plexus of ob/ob mice, without appreciable changes in the cortex or hypothalamic-thalamic regions.
Conclusions
These findings show that TSPO expression is up-regulated in cerebral regions of ob/ob leptin-deficient mice, suggesting a role of the translocator protein in leptin-dependent CNS trophism and metabolism. Unchanged SERT in mutant mice is discussed herein in the context of previous literature as the forerunner to a deeper biochemical investigation.
Similar content being viewed by others


Background
The mechanisms of action of appetite hormones and related networks operating on body weight control have not been entirely established in mammals; the study of these pathways in models of mutant rodents, is contributing to elucidate the issue. In particular, the obese ob/ob mouse, mutant for the leptin (OB hormone) gene, is currently used to evaluate the pathogenesis of human obesity and type 2 diabetes: it lacks the functional leptin, develops hyperphagia, insulin resistance, hyperglycemia and hypercholesterolemia together altered immune response, impaired locomotor activity and fertility [1]. Leptin release from adipose cells is stimulated by insulin and glucocorticoids while counter-regulatory hormones inhibit its secretion [2].
In the hypothalamus, leptin exerts a double action: it up-regulates the release of anorexigenic neuropeptides as melanotropic hormone (α-MSH), CART (cocaine/amphetamine-regulated transcript) and CRH (corticotropin-releasing hormone) while inhibiting secretion of orexigenic neuropeptide Y (NPY), MCH (melanin-concentrating hormone), orexins, and AGRP (agouti-related peptide), all signals that, at the opposite, increases appetite and reduces energy consumption. Thus, ob/ob mice present a significant impairment of leptin-dependent hypothalamic pathways, accompanied by adrenal hypertrophy and increased corticosteroid secretion during diurnal rhythms [3, 4]. The use of ob/ob animals has shown the involvement of catecholamines and serotonin (5-HT) in the leptin-deficient syndrome: in mutant animals, dopaminergic agonists reduce food intake and restrain metabolic dysfunctions, while SSRI antidepressant treatment decreases hyperphagia and hyperglycemia [1]. Some authors have observed a reduced expression of 5-HT transporter (SERT) mRNA in dorsal raphe nuclei of ob/ob animals accompanied by an altered locomotor activity [5]. Despite all these evidences, to our knowledge, there are still controversial opinions concerning the existence of interactions between leptin, 5-HT transmission and stress signals.
The aim of this study was therefore to investigate the expression of two pivotal proteins involved in 5-HT transmission and metabolism, SERT and the translocator protein (TSPO), in central and peripheral tissues of ob/ob mice in comparison with wild-type (WT) animals. SERT was evaluated given that it modulates the 5-HT re-uptake inside serotonergic neurons, a key mechanism permitting the activation/desensitization of 5-HT receptors within the synaptic cleft. SERT is also localized in periphery where it regulates platelet aggregation, gut peristalsis and immune response. An altered SERT has been reported in several complex human disorders, such as psychiatric diseases, pain and eating disturbances [6]. Density and affinity values of the TSPO protein were instead investigated as potential markers of stress-response and cholesterol metabolism in ob/ob obese mice. The TSPO protein is in fact the mitochondrial import of cholesterol with binding sites for benzodiazepines [7, 8], also implicated in steroidogenesis [7–9]. The TSPO molecular complex is prevalently located on the mitochondrial membrane and is formed by: the main subunit, the isoquinolinic binding protein (18 KDa, identified as TSPO), the benzodiazepine binding portion VDAC (32 KDa, voltage dependent anionic channel) and the adenine nucleotide translocator ANT, which is also a target for benzodiazepines. Associated components of the complex are StAR (steroidogenesis acute regulatory) and PAP7 (associated protein 7) proteins, implicated in the steroidogenesis process [8]. Nonetheless, the precise function of TSPO is unknown [10]. TSPO has been indeed related to a variety of biological functions and processes, such as protection against reactive oxygen species, regulation of cell apoptosis, immunity and porphyrin transport [9]. Moreover, TSPO expression has been found changed in several diseases and pathological conditions [9], including psychiatric diseases and fibromyalgia where altered densities of both SERT and TSPO have been reported [11–13].
SERT and TSPO proteins were thus appraised by means of binding assays carried out in either neuronal or peripheral districts of ob/ob and WT mice. SERT was measured through the high-affinity SSRI ligand [3H]-paroxetine on brain and platelets, whereas TSPO was assessed by the isoquinilinic compound [3H]-PK11195 on brain and kidney. We also performed autoradiographic studies on limbic-hypothalamic coronal sections of mouse brain. Some previous works have in fact shown changes in SERT and 5-HT receptor subtypes at the level of the hypothalamus and limbic areas of mice fatten by a palatable diet [14, 15]. Plasma chemical analyses were carried out in animals to evaluate their metabolic state.
Results
Analysis of blood chemical parameters
As shown in Table 1, ob/ob mice had higher levels of total cholesterol, glucose and High-Density-Lipoprotein (HDL) in comparison with WT mice. Concerning the other blood parameters examined, no relevant between-genotype difference was reported. γ-GT enzyme activity did not exceed 40 U lt-1 (0-40 U lt-1) in ob/ob and mutant animals, indicating a normal hepatic function in all mice.
[3H]-paroxetine and [3H]-PK11195 binding parameters
[3H]-paroxetine Scatchard analysis revealed a single population of high-affinity binding sites in all membrane preparations. [3H]-paroxetine equilibrium binding experiments (mean ± SD, 4 separate experiments in duplicate), showed no significant difference in either SERT density (Bmax, fmol/mg protein) or affinity (KD, nM) in both brain and platelets of ob/ob vs WT animals (Table 2).
[3H]-PK11195 binding Scatchard analysis also fitted with a single population of high-affinity sites in brain and kidneys. As shown in Table 3, no variation of [3H]-PK11195 binding affinity (KD, nM) was observed in brain and kidneys (mean ± SD, 4 separate experiments performed in duplicate) of different animals, whilst a significant increased Bmax was observed in the ob/ob brain (p < 0.05). Kidney Bmax resulted unchanged from comparison analysis of the two genotypes. Figure 1 depicts a representative linear transformation Scatchard, analysis obtained in brain membranes from ob/ob and WT mice, displaying an increased TSPO number in mutant animals.

[3H]-PK11195 binding assay in WT and ob/ob brain membranes. Scatchard lines are representative of [3H]PK11195 saturation binding of 4 separate experiments carried out in brain membranes of obese ob/ob (□) and WT (■) mice.
Autoradiography of brain coronal sections
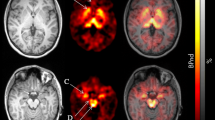
Figure 2 depicts [3H]-PK11195 and [3H]-paroxetine autoradiography on coronal brain sections carried out at the hypothalamic-hippocampal level. TSPO binding sites labeled by [3H]-PK11195 (Figure 2.a,b) resulted unevenly distributed in these brain regions, with the highest expression in the cerebral cortex and hypothalamus. [3H]-PK11195 density signal was found increased in the dentate gyrus (hippocampus) of ob/ob mice (Figure 2b), together a binding raise in correspondence of peri-ventricular areas, at the level of the choroids plexus, especially that surrounding the dorsal third ventricle.

[3H]-PK11195 (a,b) and [3H]-paroxetine (c,d) autoradiography in coronal hypothalamic-hippocampal sections of WT and ob/ob brain. The increased signal corresponding to [3H]-PK11195 binding in sections of ob/ob mice is indicated by arrows (hippocampal region and choroids plexus-third ventricle). cc: cerebral cortex; hi: hippocampal region; hy: hypothalamus; th: thalamus.
Figure 2 (c,d) also shows brain sections labeled by [3H]-paroxetine. These sections differed for about 55 μm from the [3H]-PK11195 ones (Figure 2.a,b), displaying a quite diffuse SERT labeling in cortex, hippocampus, hypothalamus and thalamus. Despite some decreased signal in the ob/ob cortex (Figure 2.d) vs. the WT one (Figure 2.c), these changes were not significant.
Discussion
Since many years, the ob/ob mouse is conceived as a suitable experimental model for studying neuropeptide substrates and metabolic pathways implicated in human obesity and type 2 diabetes [1]: these rodents bear a nonsense mutation in the coding region of the leptin gene causing the lack of secretion of a functional peptide. An important aspect should be considered when using this mutant model: human obesity is rarely due to a single gene mutation [4] and obese humans prevalently present high circulating levels of leptin [16] together desensitized, leptin-resistant pathways; by contrast, ob/ob mice are defective in leptin production. Differences between these two conditions have not been still elucidated at the biochemical level. The ob/ob syndrome can be reversed by exogenous administration of leptin or by leptin gene transfection [1], proving that leptin receptors and networks are functional. On the other side, the study of ob/ob mice permits to dissect leptin-dependent neuroendocrine loops, involved in appetite control and energy balance dysfunctions. Indeed, as already mentioned, leptin pathways, co-modulation or connection with corticotropin releasing factor (CRF), NPY, α-MSH and neurotransmitter systems have not been fully characterized [2]. Specifically, energy expenditure balance and feeding behavior are regulated by redundant pathways: monoamines and leptin are both able to modulate food intake at the hypothalamic levels, but it is not precisely known if and how these signal molecules interact [17]. Some authors also consider leptin a signal evolved to prevent starvation rather than food plenty [18]. Beside alterations of glucose metabolism, another important feature of the ob/ob syndrome is the increased blood cholesterol in mutant animals. Cholesterol is the precursor of steroidogenesis, being a main, high-affinity ligand for the benzodiazepine site-translocator protein TSPO [7, 9]. Reduced cholesterol levels have been evidenced inside macrophages of ob/ob mice, along with a diminished capacity in inflammatory response [19], supporting metabolic and hormonal cross-talks between immune response, inflammation and body weight signals [20, 21]. Interestingly, platelet TSPO and SERT densities have been found altered in fibromyalgia [12, 13], panic disorders [11, 22] and suicide attempters [23, 24]. This prompted us to preliminary assess the equilibrium-binding parameters of SERT and TSPO proteins, either in brain or high expression peripheral tissues, circulating platelets and kidneys, of ob/ob mice: to our knowledge, this is the first study that simultaneously evaluates SERT and TSPO expression in distinct anatomical district of a rodent genetic model of obesity.
Prior to SERT and TSPO analyses, blood chemical parameters were determined in ob/ob and WT animals to monitor cell metabolism: higher total cholesterol and glucose concentrations were observed in mutant vs. WT animals, according to data provided by the commercial source. Blood levels of the γ-GT enzyme were low in ob/ob and WT mice, indicating the absence of hepatic alterations. Moreover, ob/ob mice presented similar circulating triglyceride or calcium levels to those measured in WT animals. Concerning binding results, no difference of SERT density or affinity was reported in brain and platelets of ob/ob and control mice. Also, [3H]-paroxetine autoradiographic sections showed no appreciable binding differences between animals. This finding could signify that leptin-dependent pathways are altered in ob/ob mice without affecting 5-HT transmission. On the other hand, the fact that ob/ob mice show no changes in SERT density or affinity (present results) but a reduced SERT mRNA [5] is intriguing. Since leptin has been found able to decrease SERT binding sites in the rat brain [25], the ob/ob mouse could maintain the capacity at counteracting the decrease of SERT transcripts at the protein level, in the absence of SERT (or SERT-related) gene mutations. In fact, SERT underlies posttranslational regulation, trafficking or protein inactivation causing a differential distribution within cell compartments and/or discrepancies between mRNA and protein expression, as reported during megakaryoblastic differentiation [26]. Leptin-lacking mice could present an altered 5-HT responsiveness together a modified SERT reserve/function (uptake), without significant differences in binding sites. A deeper biochemical analysis of SERT should be therefore carried out, including flow cytofluorimetry, immunoprecipitation, gene expression, proteomic-functional (uptake) studies, in the context of 5-HT (or other monoamine) levels in blood and tissues of these animals. Concerning TSPO binding results, this protein was found increased in the ob/ob mouse brain while being similar in kidneys of mutant and WT animals. The [3H]PK11195 autoradiography of hippocampal-hypothalamic sections has revealed an up-regulation of TSPO density in two brain regions of ob/ob mice, the dentate gyrus of hippocampus and choroids plexus, indicating that TSPO number variations in ob/ob mouse brain are region-dependent. These results also underline that leptin chronic deficiency affects brain protein patterns.
The interpretation of our TSPO finding is difficult. In fact, to date, the precise role of TSPO within CNS is not understood. A brain region-specific regulatory mechanism in response to hypercholesterolemia and hyperglycemia could be active in these animals. Brain TSPO is mainly localized on glial cells and can be modulated by protein kinase C signaling [7] and the cAMP-protein kinase A pathway [9], activated by G-protein coupled receptors, including, therefore, metabotropic 5-HT receptor subtypes. In hippocampus and choroids plexus there could be an unbalance of regulatory signaling cascades, resulting in the enhancement of TSPO number. This could depend upon many factors, such as different receptor sub-type localization and activation: hippocampus and choroids plexus are brain regions at high expression of 5-HT1 and 5-HT2C receptors, respectively, and insulin inhibits choroids plexus 5-HT2C receptors [27]. Additional difficulties in interpreting our result come from the observation that increased TSPO has been associated with either tissue/neuronal damage or repair. Some authors have reported protective effects of TSPO agonists in experimental diabetic neuropathy [28], suggesting, hypothetically, reparative actions of brain TSPO in such a disease.
The same as for SERT, a deeper study of TSPO gene and protein expression together the investigation of its function and drug activities in ob/ob animals by means of different methodologies, including leptin treatment, is essential to confirm present results as well as to understand leptin-dependent neuronal trophism, metabolism and transmission. Another variable to consider is the different age of mutant animals: in our study, 4 month old mice were examined, when all symptoms of the "leptin-lacking syndrome" are present [1], but this does not exclude the diverse SERT and TSPO expression at other development or aging stages.
Conclusion
Present results indicate that leptin-lacking mutant mice have an augmented density of TSPO in the hippocampus and choroids plexus, without changes of SERT protein number. Central TSPO variations are not paralleled by changes in periphery (kidney).
Despite limitations due to the small sample of animals investigated, our study indicates that the obese ob/ob mouse can be a challenging animal model to elucidate the mammalian leptin-dependent neuroendocrine-neurosteroid signaling involved in appetite control.
Methods
Materials
[3H]-paroxetine (specific activity, 24 Ci/mmol) and [3H]-PK11195 (specific activity, 69.9 Ci/mmol) were purchased from Perkin-Elmer Life Science (Milan, Italy). Hyperfilm MP were obtained from Amersham-Pharmacia (UK). All other reagents were obtained from normal commercial sources.
Animals
5 ob/ob (C57BL6V-Lep ob) and 5 wild-type (C57BL/6J) adult (age: 15-17 weeks) male mice were obtained from Harlan (UK) and kept under standard laboratory conditions and feeding: animals were housed two per cage in saw-dust-lined cages at 22°C under a 12 h light/dark cycle, with free access to normal diet and tap water. All experimental procedures were carried out following the guidelines of the International European ethical standards for the care and use of laboratory animals (Community Council Directive 86-609). All protocols were approved by the Ethical Deontological Committee for animal experimentation of the University of Pisa.
Sample preparation
Brain and kidney tissues
For experiments, all mice were sacrificed by cervical dislocation, in the morning, between 8.30-9.30 am, without modifying diet conditions or access to food. Kidneys and brains were rapidly removed and treated for membrane preparation and autoradiography. Brains were taken after removing cerebellum.
Blood collection for plasma chemical analysis
The day of sacrifice, blood was withdrawn by cardiac puncture of anesthetized mice and collected inside plastic tubes containing lithium-heparin to separate plasma. Blood was then centrifuged at 2000 g for 3-5 min at room temperature by means of a Micro-Centrifuge (Menarini, Italy). Glucose, total cholesterol, High Density Lipoprotein (HDL), triglycerides, calcium and γ-glutamil transpeptidase (γGT) were evaluated by an automated procedure. Briefly, plasma was automatically dispensed on a multi-layered reagent strip, specific for each dosage, and the resulting colorimetric reaction was analyzed by the automatic spectrophotometric analyzer SPOTCHEM SP-4410 (Menarini, Italy).
Blood collection for platelet separation
For separation of platelets used in [3H]-paroxetine binding assays, blood, withdrawn as above indicated, was kept in tubes containing 3% ACD (20 mM acid citric,110 mM sodium citrate, 5 mM dextrose) and centrifuged at 200 g for 15 min at room temperature. The supernatant, corresponding to platelet-rich plasma (PRP), was further centrifuged at 10,000 g for 10 min. The resulting pellet was washed by centrifugation in physiological solution at 10,000 g for 10 min and used for membrane preparation in [3H]-paroxetine binding assays (SERT).
Membrane preparation
Preparation of cerebral membranes
To isolate cerebral membrane fractions, after sacrifice, brains (obtained as above indicated) from animals of each genotype (n = 4) were weighted, suspended in ice-cold 1:10 (w/v) 50 mM Tris-HCl, pH 7.4 (Tris-HCl buffer), containing 0.32M sucrose and protease inhibitors (200 μg/ml bacitracine, 160 μg/ml benzamidine, 20 μg/ml soy bean trypsin inhibitor), disrupted by means of an ultra-turrax homogenizer and centrifuged at 1000 g for 5 min at 4°C. Supernatants were then re-centrifuged at 48,000 g for 15 min at 4°C. For [3H]-paroxetine binding assay, resulting pellets were suspended in 1:10 volumes (w/v) of ice-cold Tris-HCl buffer, containing 120 mM NaCl, 5 mM KCl, protease inhibitors (as above indicated) and treated as previously described [29]. For [3H]-PK11195 binding, after the centrifugation step at 48,000 g for 15 min at 4°C, pellets were suspended in 1:10 volumes (w/v) of ice-cold Tris-HCl buffer without Na/K salts and washed twice in the same buffer by centrifugation at 48,000 g for 15 min at 4°C. Final brain membrane pellets were stored at -80°C until [3H]-paroxetine or [3H]-PK11195 binding assay.
Preparation of platelet and kidney membranes
For [3H]-paroxetine binding assay, platelet pellets (obtained as described previously), were suspended 1:10 (w/v) in 5 mM Tris-HCl buffer containing 5 mM EDTA and protease inhibitors, homogenized by ultra-turrax and treated as previously described [30] to obtain membrane-enriched fractions.
Kidney membranes for [3H]-PK11195 binding assays were prepared following a procedure slightly modified from Selleri and co-authors [31]. Briefly, tissue samples were diluted 1:10 (w/v) in ice-cold 50 mM Tris-HCl, pH 7.4, 0.32M sucrose, 1 mM EDTA (buffer A) containing protease inhibitors. After homogenization, tissue suspensions were centrifuged at 600 g for 10 min at 4°C. Supernatants (S1) were collected and kept on ice while pellets were re-suspended in the same buffer and re-centrifuged again as above indicated. The resulting supernatants (S2) were mixed to S1 and centrifuged at 6,500 g for 20 min at 4°C. Pellets were then suspended in 50 mM Tris-HCl, pH 7.4 (buffer B) homogenized and centrifuged again at 48,000 g for 10 min at 4°C. Final pellets, resulting from both procedures, were stored at -80°C until assay.
Protein assay
Total protein concentration was determined in brain, platelet and kidney membrane preparations according to the method of Bradford (Bio-Rad), using γ-globulins as the standard.
[3H]-paroxetine and [3H]-PK11195 saturation binding experiments
[3H]-paroxetine binding (SERT) was carried out as follows [12]: membranes (50-100 μg protein/tube) were incubated with increasing radioligand concentrations (0.025-5 nM) in 50 mM Tris-HCl buffer, pH 7.4, containing 120 mM NaCl, 5 mM KCl (final volume, 2 ml). Non specific binding was assessed in the presence of 10 μM fluoxetine, used as the unlabelled displacer. All samples were assayed in duplicate and incubated 60 min at 22-25°C. TSPO binding was instead appraised as previously indicated [32]: brain and kidney membranes (25-100 μg protein/tube) were incubated with increasing concentrations of [3H]-PK11195 (0.5-8 nM), in a final volume of 0.5 ml assay buffer, 50 mM Tris-HCl, pH 7.4. Non specific binding was assessed in the presence of 1 μM PK11195 as the cold displacer. All samples were assayed in duplicate and incubated 90 min at 0°C.
In all binding experiments, incubation was halted by adding 5 ml of cold assay buffer. Samples were immediately filtered under vacuum through glass fiber GF/C filters and washed 3 times with 5 ml of assay buffer. Filters were then placed in pony vials with 4 ml of scintillation cocktail, and radioactivity measured by means of a β-counter (Packard LS 1600).
[3H]-paroxetine and [3H]-PK11195 autoradiography on brain coronal sections
After animal sacrifice, the brain of ob/ob and WT mice was rapidly removed, placed in an embedding medium (O.C.T.), frozen in liquid nitrogen and stored at -85°C until use. Coronal sections (15 μm) at the hippocampal-hypothalamus levels obtained by cryostat were mounted onto gelatin-coated slides and stored at -30°C until assay. The day of assay, sections were allowed to equilibrate at room temperature for 10 min and pre-incubated in Tris-HCl buffer (50 mM, pH 7.4) for 15 min at 4°C. Sections were then incubated for: 1) 60 min in Tris-HCl buffer, containing 120 mM NaCl, 5 mM KCl, with 0.5 nM [3H]-paroxetine; 2) 90 min at 4°C in Tris-HCl buffer with 5 nM [3H]-PK11195 (TSPO). After incubation, all sections were washed twice with the Tris-HCl incubation assay buffer at 4°C for 30 sec, then with distilled water and rapidly dried under a cold air steam. Non-specific binding was determined by adding to the incubation solution an excess (10 μM) of unlabelled fluoxetine (SERT) or PK11195 (TSPO). Autoradiograms were generated by exposing tissue sections in tritium-sensitive films for 60 days at -85°C. Films were developed in Kodak D-19 for 5 min at 15°C and fixed in Kodak rapid Fix. After film exposure, tissue sections were fixed in 10% formalin and stained with hematoxylin-eosin for anatomical identification.
Data analysis
Equilibrium-saturation binding data, the maximum binding capacity (Bmax, fmol/mg protein) and the dissociation constant (Kd, nM) were analyzed by means of the iterative curve-fitting computer program EBDA and LIGAND [33]. Statistical analysis was performed by means of Student's t test (Graph pad version 3-4 program, San Diego, CA, USA). To improve visualization of [3H]-paroxetine and [3H]-PK11195 autoradiography, the gray scale, acquired by a X-ray scanner of autoradiographic films, has been translated into a color scale using the Image J software (Figure 2).
Disclosure
The authors declare that they have no conflicts of interests.
References
Lindström P: The physiology of obese-hyperglycemic mice [ob/ob mice]. The Sci World J. 2007, 7: 666-685.
Myers MG: Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. 2004, 59: 287-304. 10.1210/rp.59.1.287.
Saito M, Bray GA: Diurnal rhythm for corticosterone in obese (ob/ob) diabetes (db/db) and goldthioglucose-induced obesity in mice. Endocrinology. 1983, 113: 2181-2185. 10.1210/endo-113-6-2181.
Beck B: Neuropetides and obesity. Nutrition. 2000, 16: 916-923. 10.1016/S0899-9007(00)00410-X.
Collin M, Håkansson-Ovesjö ML, Misane I, Ögren SO, Meister B: Decreased 5-HT transporter mRNA in neurons of the dorsal raphe nucleus and behavioral depression in the obese leptin-deficient ob/ob mouse. Mol Brain Res. 2000, 81: 51-61. 10.1016/S0169-328X(00)00167-4.
Murphy DL, Fox MA, Timpano KR, Moya PR, Patterson AM, Holmes A, Lesch KP, Wendland JR: How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene, which functions to influence all cellular serotonin systems. Neuropharmacology. 2008, 55: 932-960. 10.1016/j.neuropharm.2008.08.034.
Batarseh A, Papadopoulos V: Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol Cell Endocrinol. 2010, 327: 1-12. 10.1016/j.mce.2010.06.013.
Falchi AM, Battetta B, Sanna F, Piludu M, Sogos V, Serra M, Melis M, Putzolu M, Diaz G: Intracellular cholesterol changes induced by translocator protein (18 kDa) TSPO/PBR ligands. Neuropharmacology. 2007, 53: 318-329. 10.1016/j.neuropharm.2007.05.016.
Rone MB, Fan J, Papadopoulos V: Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease state. Biochim Biophys Acta. 2009, 1791: 646-658.
Gavish M, Bachman AN, Shoukrun R, Katz Y, Veenman L, Weisinger G, Weizman A: Enigma of the peripehral benzodiazepine receptor. Pharmacol Rev. 1999, 51: 629-650.
Pini S, Martini C, Abelli M, Muti M, Gesi C, Montali M, Chelli B, Lucacchini A, Cassano GB: Peripheral-type benzodiazepine receptor binding sites in platelets of patients with panic disorder associated to separation anxiety symptoms. Psychopharmacology. 2005, 181: 407-411. 10.1007/s00213-005-2247-x.
Bazzichi L, Giannaccini G, Betti L, Mascia G, Fabbrini L, Italiani P, De Feo F, Giuliano T, Giacomelli C, Rossi A, Lucacchini A, Bombardieri S: Alteration of serotonin transporter density and activity in fibromyalgia. Arthritis Res Ther. 2006, 8: R99-10.1186/ar1982.
Bazzichi L, Giannaccini G, Betti L, Italiani P, Fabbrini L, Defeo F, Giacomelli C, Giuliano T, Rossi A, Uccelli A, Giusti L, Mascia G, Lucacchini A, Bombardieri S: Peripheral benzodiazepine receptors on platelets of fibromyalgic patients. Clin Biochem. 2006, 39: 867-872. 10.1016/j.clinbiochem.2006.06.002.
Park S, Harrold JA, Widdowson PS, Williams G: Increased binding at 5-HT1A, 5-HT1B and 5-HT2A receptors and 5-HT transporters in diet-induced obese rats. Brain Res. 1999, 847: 90-97. 10.1016/S0006-8993(99)02055-7.
Huang XF, Huang X, Han M, Chen F, Storlien L, Lawrence AJ: 5-HT2A/2C receptor and 5-HT transporter densities in mice prone or resistant to chronic high-fat diet-induced obesity: a quantitative autoradiography study. Brain Res. 2004, 1018: 227-235. 10.1016/j.brainres.2004.05.093.
Considine RV, Sinha MK, Heiman ML: Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996, 334: 292-295. 10.1056/NEJM199602013340503.
Ramos EJB, Meguid MM, Campos ACL, Coelho JCU: Neuropeptide Y, a-melanocyte-stimulating hormone and monoamines in food intake regulation. Nutrition. 2005, 21: 269-279. 10.1016/j.nut.2004.06.021.
Jequiert E: Leptin signaling, adiposity and energy balance. Ann NY Acad Sci. 2002, 967: 379-388. 10.1111/j.1749-6632.2002.tb04293.x.
Kjerrulf M, Berke Z, Aspegren A, Umaerus M, Nilsson T, Svensson L, Camejo-Hurt E: Reduced cholesterol accumulation by leptin deficient (ob/ob) mouse macrophages. Inflamm Res. 2006, 55: 300-309. 10.1007/s00011-006-0087-8.
Giannaccini G, Giusti L, Santini F, Marsili A, Betti L, Mascia G, Pelosini C, Baroni S, Ciregia F, Fabbrini L, Lucacchini A, Vitti P, Pinchera A: Tubby protein in human lymphocytes from normal weight and obese subjects. Clin Biochemistry. 2007, 40: 806-809. 10.1016/j.clinbiochem.2007.03.020.
Matarese G, La Cava A: The intricate interface between immune system and metabolism. Trends Immunol. 2004, 25: 193-200. 10.1016/j.it.2004.02.009.
Marazziti D, Rossi A, Dell'Osso L, Palego L, Placidi GP, Giannaccini G, Lucacchini A, Cassano GB: Decreased platelet [3H]-paroxetine binding in untreated panic disorder patients. Life Sci. 1999, 65: 2735-2741. 10.1016/S0024-3205(99)00542-1.
Marazziti D, Dell'Osso B, Baroni S, Masala I, Di Nasso E, Giannaccini G, Conti L: Decreased density of peripheral benzodiazepine receptors in psychiatric patients after a suicide attempt. Life Sci. 2005, 77: 3268-3275. 10.1016/j.lfs.2005.04.031.
Marazziti D, Dell'Osso L, Rossi A, Masala I, Baroni S, Armani A, Giannaccini G, Di Nasso E, Lucacchini A, Cassano GB: Decreased platelet [3H]-paroxetine binding sites in suicide attempters. Psychiatry Res. 2001, 103: 125-131. 10.1016/S0165-1781(01)00281-5.
Charnay Y, Cusin I, Vallet PG, Muzzin P, Rohner-Jeanrenaud F, Bouras C: Intracerebroventricular infusion of leptin decreases serotonin transporter binding sites in the frontal cortex of the rat. Neurosci Lett. 2000, 283: 89-92. 10.1016/S0304-3940(00)00951-4.
Giannaccini G, Betti L, Palego L, Schmid L, Fabbrini L, Pelosini C, Gargini C, Da Valle Y, Lanza M, Marsili A, Maffei M, Santini F, Vitti P, Pinchera A, Lucacchini A: Human Serotonin Transporter Expression during Megakaryocytic Differentiation of MEG-01 Cells. Neurochem Res. 2010, 35: 628-635. 10.1007/s11064-009-0112-8.
Hurley JH, Zhang S, Bye LS, Marshall MS, Depaoli-Roach AA, Guan K, Fox AP, Yu L: Insulin signaling inhibits the 5-HT2C receptor in choroid plexus via MAP-kinase. BMC Neurosci. 2003, 9: 4-10.
Giatti S, Pesaresi M, Cavaletti G, Bianchi R, Carozzi V, Lombardi R, Maschi O, Lauria G, Garcia-Segura LM, Caruso D, Melcangi RC: Neuroprotective effects of a ligand of translocator protein-18KDa (Ro5-4864) in experimental diabetic neuropathy. Neuroscience. 2009, 164: 520-529. 10.1016/j.neuroscience.2009.08.005.
Rotondo A, Giannaccini G, Betti L, Chiellini G, Marazziti D, Martini C, Lucacchini A, Cassano GB: The serotonin transporter from human brain: Purification and partial characterization. Neurochem Int. 1996, 28: 299-307. 10.1016/0197-0186(95)00080-1.
Ramacciotti CE, Coli E, Paoli R, Marazziti D, Dell'Osso L: Serotonergic activity measured by platelet [3H]-paroxetine binding in patients with eating disorders. Psychiatry Res. 2003, 118: 33-38. 10.1016/S0165-1781(03)00059-3.
Selleri S, Bruni F, Costagli C, Costanzo A, Guerrini G, Ciciani G, Costa B, Martini C: 2-Arylpyrazolo[1,5-a]pyrimidin-3-yl Acetamides. New Potent and Selective Peripheral Benzodiazepine Receptor Ligands. Bioorg Med Chem. 2001, 9: 2661-2671. 10.1016/S0968-0896(01)00192-4.
Martini C, Chelli B, Betti L, Montali M, Mancuso M, Giannaccini G, Rocchi A, Murri L, Siciliano G: Peripheral benzodiazepine binding sites in platelets of patients affected by mitochondrial diseases and large scale mitochondrial DNA rearrangements. Mol Med. 2002, 8: 841-846.
Mc Pherson GA: KINETIC, EBDA, LIGAND, LOWRY: A collection of radio-ligand binding analysis programs. 1985, Biosoft, Cambridge. U.K
Acknowledgements
The present work is supported by a grant of "Ministero dell'Istruzione dell'Università e della Ricerca" (M.I.U.R.) to Prof. G. Giannaccini.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
GG, FS and AL conceived the study, participated in its design and coordination and helped to draft the manuscript. AlP participated in the study coordination and manuscript revision. GG, LP, LB, AnP and MM drafted the manuscript. LB, LS and ML carried out all radioligand binding studies. AnP carried out autoradiography experiments. LF and CP were responsible of tissue and blood sampling from animals and carried out blood chemical analyses. LB and LP elaborated experimental results.
All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Giannaccini, G., Betti, L., Palego, L. et al. Serotonin transporter (SERT) and translocator protein (TSPO) expression in the obese ob/ob mouse. BMC Neurosci 12, 18 (2011). https://doi.org/10.1186/1471-2202-12-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2202-12-18