
Abstract
Background
Microbiological indicators are commonly used in the assessment of public health risks associated with fecal contamination of freshwater ecosystems. Sediments are a reservoir of microorganisms, and can thus provide information on past pollution events, not obtainable through the testing of surface water. Moreover, pathogens present in sediment may represent future threats to human health. Clostridium perfringens, a typical colonizer of sediments, has been suggested as an alternative indicator of fecal pollution. In order to be suitable for such purpose, the microorganism should be widely distributed in contaminated environments. The objective of this study was thus to determine the composition of the anaerobic community in sediment samples of the lower Tiber basin, in central Italy, through a combined approach involving granulometric analysis of sediment samples, as well as a microbiological and molecular (16S rRNA) analysis of strains.
Results
Granulometry showed a similar, clayey sediment composition, in most sampling sites. The microbiological method, employing, an adaptation of the standard method, proved to be effective in isolating anaerobic bacteria from the environmental matrix for the purpose of genetic analysis. Eighty-three strains of bacteria were isolated and the partial 16S rRNA gene sequenced. While biochemical analysis detected only C. perfringens strains, phylogenetic analysis indicated the presence of three clusters: C. perfringens, C. bifermentans and B. cereus, comprising eight taxa. C. perfringens, the commonest in almost all sediment sampling sites, was present in all sites, and in both seasons (seasonal sampling was carried out only along the Tiber and Aniene rivers). None of the described genetic profiles showed complete similarity with GenBank sequences.
Conclusion
The study underlines the value of C. perfringens as an alternative microbial indicator of fecal contamination in river sediments. This is supported by the bacterium's presence in all sampling sites, and in both seasons, coupled with its detectability using commercial diagnostic kits.
The study also illustrates the presence of an anaerobic community of considerable biodiversity in the lower Tiber basin, with C. perfringens as its main component. The 16S rRNA analysis, while confirming the phylogenetic relationships among isolated species, also showed haplotype patterns different from those present in the NCBI database.
Similar content being viewed by others

Background
Wastewater effluents play an important role as sources of fecal contamination in freshwater environments. These nonpoint sources of fecal pollution are broadly distributed in both urban and agricultural areas. Standard-based water quality assessment is an essential component of monitoring programs for the protection of human health. Microbiological indicators such as total coliforms, fecal coliforms and fecal streptococci are widely used both in monitoring programs of this kind [1, 2] and in human health risk assessment [3, 4]. These indicators may also point to the presence of other non-bacterial pathogens such as enteric viruses and parasitic protozoans [5, 6]. Several studies have highlighted the value of sediments as indicators of pollution. Indeed, sediments – as potential reservoirs for bacteria and viruses in aquatic environments – are able to provide information on past pollution events no longer detectable in water samples, as well as on the presence of pathogens, which may in some cases pose a future threat to human health.
The higher occurrence of microbial indicators of fecal contamination in sediments as compared to other aquatic environments has been attributed to the sorption of microorganisms onto particles suspended in water, which subsequently settle to the bottom [7–10] and accumulate. Various authors observed higher microbial survival rates in sediment as compared to other aquatic environments, possibly due to the absence of abiotic (UV radiation, salinity, toxic agents) and biotic factors (predation, parasitism, low nutrient availability), which, in surface water, act synergistically to inhibit bacterial growth [11–17]. Others showed higher rates of microbial activity in the sediment surface layer, which is commonly rich in organic matter and thus able to provide suitable habitats for different strains of anaerobes [18, 19].
The occurrence of anaerobic microorganisms in freshwater environments is generally linked to the input of sewage outfalls. Some of these organisms are able to survive adverse environmental conditions by forming resting cells (spores) [20, 21]. Clostridium perfringens, a Gram-positive anaerobic spore-forming bacterium of the genus Clostridium, has been suggested [22–28] and successfully used as an alternative indicator of fecal contamination in aquatic environments due to its wide distribution in nature and to its adaptation to a variety of habitats such as soils, sediments and sewages. Moreover, this organism has been found in air, dust, water, and even food [29, 30]. Spores produced by this organism are very resistant to disinfection and their presence in filter units indicate treatment inefficiencies [31].
Studies of C. perfringens in freshwater ecosystems have been fewer than those performed on marine environments. Robles et al. confirmed the value of C. perfringens as a conservative indicator of fecal contamination from sewage disposal in high-mountain lakes [32]. Sorensen et al. were able to detect traces of unfiltered sewage in small streams by comparing the concentrations of C. perfringens spores downstream from wastewater treatment plants [33].
Although the World Health Organization recommends C. perfringens as a useful indicator of fecal pollution in water quality surveys [34], this microorganism has been adopted in Europe exclusively as an additional source of water quality information [35–37].
The sequence of the 16S rRNA gene has been widely used as a phylogenetic marker to study genetic relationships between different strains of bacteria (phylogeny). The analysis of this gene can therefore be considered a standard method for the identification of bacteria at the family, genus and species levels [38–44], and has in fact been included in the latest edition of Bergey's Manual of Systematic Bacteriology [45].
The objective of this study was to determine the composition of the anaerobic community in sediment samples of the lower Tiber catchment area, in central Italy, through a combined approach involving granulometric analysis of sediment samples, followed by microbiological and molecular (16S rRNA) analyses of bacterial strains isolated from these samples.
Results
Sediments and microbiological analysis
With the exception of samples collected from one of the tributaries, the Farfa river, the granulometric composition of all samples was mostly clayey (Table 1).
The microbiological method used in this study has allowed us to isolate 83 strains of anaerobic bacteria from the sediment samples. To ensure successful growth of colonies, each sample was tested in three suspension aliquots. Replicate sets yielded similar bacterial colony counts (CFU per gram of dry weight). The biochemical test was able to detect isolated strains of C. perfringens, but not other species within the genus.
Analysis of 16S rRNA fragments
Eighty three PCR products derived from Gram-positive sulphite-reducing bacteria collected in this study were aligned and matched against NCBI database sequences for the closest phylogenetic neighbors. The resulting phylogenetic tree, based on the neighbor-joining method, is given in Fig. 1. Thirty one haplotypes were observed (Table 2), with a total of 202 variable sites, of which 180 were informative. The transition/transversion (ti/tv) ratio was 1.1. Base frequencies were as follows: A = 0.261, C = 0.216, G = 0.312 and T = 0.211. The higher frequency of G corresponds to what is generally observed among spore-forming bacteria.

Phylogenetic tree constructed with the Hasegawa-Kishino-Yano (1985) method, for a 720 bp fragment of the 16S rRNA coding region. The tree shows the genetic relationships between haplotypes of spore-forming bacterial strains isolated from sediments of the lower Tiber basin. Numbers above branches show bootstrap values expressed as percentages of 100 replications. Database accession numbers are given in bold. Bar, 0.02% sequence divergence.
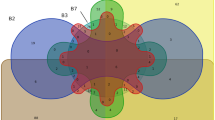
All phylogenetic analyses clearly assigned the bacteria to one of three main clusters of anaerobic bacteria: C. perfringens (I), C. bifermentans (II) and B. cereus (III), grouped into two genuses (Clostridium and Bacillus). Cluster I included the taxa C. perfringens, C. barati, C. thiosulfatireducens and C. butyricum, and cluster II included C. bifermentans, C. glycolicum and C. ghoni. The only member of cluster III was B. cereus, with a haplotype presenting a 26-bp insertion at position 78 in our alignment set. A Multidimensional Scaling (MDS) [46, 47] plot based on the HKY (Hasegawa Kishino Yano) model [48] revealed a marked heterogeneity within the first cluster, along the third dimension (Fig. 2). The haplotype corresponding to C. thiosulfatireducens appeared to be the most distant within its cluster, with a genetic distance value of 0.117. The haplotype corresponding to C. barati appeared to be the closest to C. perfringens, with a distance value of 0.045. Genetic distances between the members of the second cluster ranged from 0.04 to 0.05. As for the third cluster, B. cereus diverged from the other groups, with distance values between 0.39 and 0.29. Mean distance values between the main groups are given in Table 3.

Multidimensional scaling ordination in three dimensions, based on Kruskal's method. The plot shows genetic relationships between anaerobic strains isolated from sediments of the lower Tiber basin. Genetic distances between the three clusters are clearly visible, as well as the incomplete homology between the environmental strains isolated (squares), and those previously published in GenBank (circles).
None of the genetic profiles described in this study showed complete similarity with those previously published in the NCBI database, with similarity scores ranging between 0.95 and 0.99.
The geographical and frequency distribution of the eight taxonomic units identified is given in Fig. 3. C. perfringens was found to be the most prevalent in all but one of the sampling sites, accounting for between 33% and 100% of anaerobic beacteria identified. As expected, more biodiversity was observed downstream than upstream, both along the stem of the Tiber and along its tributaries. The nucleotide sequence data reported in this study have been deposited in the NCBI (GenBank) database [accession numbers EF153846 to EF153928].

Geographical distribution and frequencies of the genetic profiles observed in different seasons (left figure = cold season; right figure = warm season).
Discussion
The rate of pollution of the Tiber river increases considerably downstream, with the growing exploitation of its waters for agricultural, urban and industrial activities. The quality of the river's water decreases remarkably after its confluence with its main tributary, the Aniene river, in the city of Rome. Despite several measures taken in order to reduce the impact of human activities on the Tiber ecosystem, illegal sewage discharge persists in a number of locations, attesting to severe human pressures and water mismanagement. The study of microbial indicators of pollution in sediment, rather than water, could contribute valuable information concerning factors affecting freshwater ecosystems. Sediments may prove useful in this respect, in that they "trap" pollutants and constitute a favourable habitat for various anaerobic bacteria.
Preliminary efforts, involving the application of published protocols [49–51], such as the Most Probable Number (MPN) and the Membrane Filtration (MF), entailed several problems, due to the growth of atypical colonies, which interfered with the growth of spore-forming microorganisms. To avoid this inconvenience, we implemented an adaptation of the standard protocol, described in the Methods section. As noted above, each sample was tested in three suspension aliquots yielding similar bacterial counts, which attests to the validity of the method employed. This adaptation allowed us to successfully isolate 83 strains of anaerobic bacteria from our sediment samples.
Subsequent genetic analysis assigned the bacteria to one of three clusters: C. perfringens, C. bifermentans and B. cereus. The biochemical test, on the other hand, was able to detect isolated strains of C. perfringens, but not other species within the genus. The low efficiency of the biochemical analysis on the samples collected is probably related to the fact that commercial kits are specifically developed on the basis of the metabolic pathways of clinical strains, and therefore cannot be as selective in discriminating environmental species. It is noteworthy, however, that such kits were able to detect the most frequently present and widely spread bacterium – C. perfringens.
The combined application of both microbiological isolation and selection of specific primers for the 16S rRNA gene provided a suitable tool for the detection of the sulphite reducing bacteria present in the layers of sediment of the lower Tiber river basin.
The primers used for PCR amplification are efficient only if, before microbiological analysis, samples are submitted to heat treatment, to inactivate vegetative forms of bacteria. The crucial role of such treatment emerged in a recent, yet unpublished study, in which we found a clear association between the efficiency of the primers and preliminary heat treatment of environmental samples.
Our phylogenetic analysis confirmed the previously described heterogeneity within the genus Clostridium [52–54]. The lack of complete similarity between the genetic profiles found in this study and those present in the NCBI database could be attributed to the former being typical of the geographic region of the lower Tiber basin. Additional comparisons with strains originating from other watersheds remain necessary, however, both in order to confirm the exclusivity of the Tiber haplotypes and to evaluate their geographic ranges.
The greater microbial biodiversity observed in samples collected in early autumn seems to be related to seasonal water regimes and temperature fluctuations. Higher temperatures generally promote eutrophication and thus the establishment of anaerobic conditions. The input of sewage is generally enhanced by a higher frequency of rainfall during this season, which is generally followed by unpredictable increments in pollutants that are partly adsorbed by the sediment matrix. The effects of such events on water macrofauna, notably in the form of high fish mortality, are particularly evident following heavy rainfalls. These events can promote the development of suitable habitats for the growth of different species of anaerobes. Further studies should therefore be conducted in order to detect possible relationships between outfall types and the composition of anaerobic microbial communities in river sediments.
The ability of sediment to record former pollution events and the pathogenic nature of sulphite-reducing spore-forming bacteria such as C. perfringens, underscore the importance of bacteriological analysis of sediment for the purposes of microbiological risk assessment of inland water ecosystems [55].
Conclusion
This study underlines the value of Clostridium perfringens as a microbial indicator of fecal contamination in river sediments. The presence of this bacterium in all sediment sampling sites, as well as in both seasons (along the main stem of the Tiber, where samples were collected seasonally), combined with the fact that it was the only anaerobic bacterium detectable using commercial biochemical diagnostic kits, lend support to its suitability as an alternative indicator of fecal pollution in water quality surveys.
While further studies are still needed to explore possible relationships between the presence of specific microorganisms in sediments and the effects of such pollution on human health, one may safely assume that sulphite-reducing bacteria – being useful sources of information – are destined to play a key role in the management of freshwater environments. Moreover, information derived from the analysis of sulphite-reducing bacteria in river sediments may prove valuable in water reclamation plans. In this context, the information may be used for purposes such as the evaluation of health risks in a given area, or quality control, to ensure that the health of the water ecosystem in question has in fact been restored or improved.
Although quantitative analysis on the densities of sulphite reducing clostridia in the sediments were not performed in this work, however the qualitative molecular tools employed for the identification of bacteria proved extremely useful not only in confirming the value of C. perfringens as a possible bacterial indicator of fecal contamination, but also in allowing the identification of genetic relationships between species. Further quantitative studies will be useful in order to state the suitability of this group of bacteria as indicators. Despite the usefulness of qualitative molecular tools, however, these cannot currently replace classical methods of routine water quality assessment, the latter being quantitatively more informative and easier to execute. An alternative, faster molecular methodology, which may be examined in future studies, is the direct extraction of microbial genome from the matrix and its amplification using selective primers specifically designed for each taxonomic unit.
Methods
Geographical area studied and sediment sampling
The Tiber River belongs to one of the largest river systems in Italy, with a catchment area of 17,375 km2 . The river is 405 km long, and runs from the Tuscan-Emilian Apennines to the Tyrrhenian Sea, through four administrative regions. The water volume ranges from 60 m3s-1 to 3200 m3s-1 with a yearly average of 230 m3s-1 [56].
Thirteen sampling sites were selected, four along the main stem of the Tiber, four along the Farfa, four along the Treja and one along the Aniene (Table 4). The aim was to allow a comparison of the microbial community composition in sediments upstream and downstream of the city of Rome Sediment samples were collected seasonally from February 2002 to June 2003 for all sites located along Tiber River and Aniene tributarie while sites along Treja and Farfa were sampled only during the hot season. Sampling was performed using a bucket attached to a winch, that was immersed at a depth of between 1 and 3 meters below the water surface. Samples were transported and stored in sterile 50 ml tubes, at 4 °C [57]. Granulometric characterization was performed on each sediment sample, based on the different sedimentation speeds of silt, clay and sand particles [58, 59].
Isolation of bacteria
Five grams of sediment from each sample were suspended in 45 ml of sterile phosphate buffered saline (PBS) [K2HPO4 3 g/ml, KH 2PO4 1 g/ml, NaCl 8,5 g/ml; (pH 7,2 ± 0,2)] + 0.1% Tween 80. Three replicates were used for each sample. Suspension samples were then homogenized on a magnetic stirrer (Heidolph, MR 3001) for 30 minutes in order to break up clumps of bacteria. The samples were heat-shocked at 80–85°C for 10 minutes before cultivation, so as to inactivate vegetative bacteria and enhance sporulation. Five ml of the spore-containing suspension was diluted (1:10) with sterile PBS, in 15 ml Falcon vials (BD, USA). For optimal growth, we adopted the following plating technique: 8 ml of SPS (Sulphite Polymixine Sulphadiazine Agar, Oxoid) medium culture was placed in Petri dishes with a 1 ml aliquot of diluted suspension, and incubated in an anaerobic jar equipped with a manometer and a CO2 generator, at 36 ± 1°C for up to 24 hours. Black colonies characteristic of anaerobic bacteria appeared.
Colonies were counted directly, and the results expressed as colony-forming units per gram of dry weight of sediment (CFU/gss). In order to calculate dry weight, about five grams of sediment were weighted and dried, in triplicate, at 120 ± 1°C for 12 hours. The procedure was repeated until a stable weight was reached.
After counting, colonies were transferred into new plates with TSA (Tryptone Soya Agar, Oxoid) medium and incubated in anaerobic conditions as described above. Single colonies were isolated from each of the three replicates and resuspended in sterile water for DNA extraction. Biochemical identification of strains was then performed using API 20A Strips (Biomerieux, France) according to the manufacturer's instructions.
Positive and negative controls were used to validate the microbiological method. A negative control was generated as follows: after sterilization at 121°C for 15 min, five grams of sediment were suspended into 45 ml of sterile PBS, then plated in medium culture as described above. The same procedure, followed by in vitro infection with C. perfringens (ATCC 12918), was used to generate a positive control.
Microbiological analyses and genome extractions were performed within 24 hours from sampling; extracted DNA were redissolved in sterile water, aliquoted in small volumes and stored at -80°C until use. All molecular experiments were completed within six months from extraction.
DNA extraction and 16S ribosomal RNA amplification
One and a half ml of microbial suspension was pelleted by spinning at 6000 rpm for 5 min. Extraction of nucleic acids was carried out following the method described in Ausubel et al. [60]. The pellets were resuspended in 570 μ1 TE buffer [10 mM Tris-HCl, 1 mM EDTA, (pH 8)] and the suspension mixed with 80 μl CTAB/NaCl solution (10% CTAB in 0.7 M NaCl), 30 μl of 10% SDS and 3 μl of Proteinase K (20 mg μ/ml), and incubated at 55°C for 10 min.
An equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) was added, and the mix was centrifuged at 17000 rpm for 5 min. Supernatants were transferred to new tubes, washed with 1 volume of chloroform/isoamyl alcohol (24:1) and spinned at 17000 rpm for 5 min. Supernatants were again transferred to new tubes and DNA precipitated at -20°C with two equivalent volumes of 95% ethanol for 30 min. After precipitation pellets were washed with 70% ethanol, dried in a thermoblock at 40°C, and resuspended in 100 μl of 10 mM TE buffer. A 701 bp fragment of the 16S rRNA coding region was selectively amplified with two primers: 728 forward (5'-GGGGAATATTGCACAATGG-3') and 729 reverse (5'-GGGACTTAACCCAACATCTCA-3'). Primers were designed (using Primer3 software) [61], so as to anneal to conserved positions of different Clostridium species published on the NCBI database (accession numbers: NC_003366, AF371836, X68174, AF479585, AF458779). In particular the forward primer anneal at position 351–371 and the reverse primer at position 1047–1060 of accession number NC_003366 (Clostridium perfringens str. 13, complete genome, GeneID: 988236). Annealing temperature was calculated by the Primer 3 software. The final concentrations of PCR reaction components were: 10 mM PCR Buffer [Tris-HCl (pH 8.8)], 2 mM MgCl2, 0.2 mM of each dNTP, 0,2 mM of the two primers, 1 U Taq polymerase (Roche, Mannheim, Germany).
The PCR reaction was performed in a Personal cycler (Biometra T4, Göttingen, Germany), under the following conditions: an initial denaturation step at 94°C for 2 min, followed by 35 cycles at 94°C for 30 s, annealing at 57°C for 30 s and elongation at 72°C for 60 s. The reaction was terminated with a 10-min final elongation step at 72°C. A negative control (sterile deionized water) and a positive control (C. perfringens genomic DNA) were included in the experiment to confirm the validity of results.
PCR products (5 μl of each reaction mixture) were visualized by electrophoresis on a 1% agarose gel, in TBE (Tris Borate EDTA) buffer (100 mM Tris-HCl, 90 mM boric acid, 1 mM Na2EDTA), with ethidium bromide staining (10 mg/ml), at 90 V for 1 h.
Partial sequencing of the 16S ribosomal RNA gene
Before sequencing, unincorporated nucleotides and primers were removed from PCR products using the Promega DNA Purification System (Madison, WI, USA), following the manufacturer's protocol. The sequencing reaction was prepared using the CEQ 2000 Dye Terminator Cycle Sequencing with the Quick Start Kit (Beckman Coulter, Fullerton, USA), and was performed in a Personal cycler (Biometra T4, Göttingen, Germany) according to the manufacturer's instructions. Sequences were generated with a Beckman 3000 sequencer (Beckman Coulter, Fullerton, USA) and analyzed using Chromas 2.23 software (Technelysium Pty Ltd, Tewantin, Australia). BLAST searches were performed for each of the sequences obtained, to find its closest phylogentic neighbors.
Sequences alignment and analysis
The partial sequences of 16S rRNA genes obtained in this study were aligned to each other using ClustalX software, version 1.81 [62, 63]. Phylogenetic relationships were inferred by the neighbor-joining method [64] using the phylogenetic inference software Paup*, version 4.0 [65]. The evaluation of the most appropriate model of sequence evolution was computed using ModelTest version 3.0 [66]. For the 16S rRNA gene, this resulted in the selection of the HKY85 + G model. Genetic relationships were also plotted by non-metric multidimensional scaling (MDS) using SYSTAT 9.0 software (SPSS-SCIENCE), which provides an ordering of n variables on a similarity/dissimilarity matrix by means of n points in a three dimensional space [Figure 2].
Accession numbers of 16S rRNA sequences
The 83 nucleotide sequences reported in this paper have been deposited in the GSDB, DDBJ; EMBL and NCBI nucleotide sequence databases under accession nos. EF153846 to EF153928. Eighty three environmental isolates of Clostridium: C. bifermentans [GenBank: EF153846 to 153861], C. ghoni [GenBank: 153862 to 153868], C. glycolicum [GenBank: 153869 to 153870], C. perfringens [GenBank: 153871 to 153918], C. barati [GenBank: 153919 to 153921], C. thiosulfatireducens [GenBank: 153922 to 153925] and C. butyricum [GenBank: 153926], and two environmental isolates of Bacillus cereus [GenBank: EF153927, EF153928].
References
Tyagi VK, Chopra AK, Kazmi AA, Kumar A: Alternative microbiol indicators of faecal pollution: Current Perspective. (IAEH)Iran Ass Environ Health. 2006, 3: 205-216.
The European Parliament and the Council of European Union: Directive 2006/7/EC 15 February 2006. Concerning the management of bathing water quality and repealing Directive 76/160/EEC. Official Journal of the European Union. 2006
Berg G: The indicator system. Indicators of Viruses in Water and Food. Edited by: Berg G. 1978, Ann Arbor, MI: Ann Arbor Science, 1-13.
Grabow WOK: Waterborne diseases: Update on water quality assessment and control. Water SA. 1996, 22: 193-202.
Payment P, Franco E: Clostridium perfringens and somatic coliphages as indicators of the efficiency of drinking water treatment for viruses and protozoan cysts. Appl Environ Microbiol. 1993, 59: 2418-2424.
Gleeson C, Gray NF: The coliform index and waterborne disease: problems of microbial drinking water assessment. 1997, London: E. & F.N. Spon, 1
Hendricks CW: Enteric bacterial metabolism of stream sediment eluates. Can J Microbiol. 1971, 17: 551-556.
Grimes JE: The Thread of Discourse. Janua Linguarum Series Minor. 1975, The Hague: Mouton, 207:
Erkenbrecher CW: Sediment bacterial indicators in an urban shellfishing subestuary of the lower Chesapeake Bay. Appl Environ Microbiol. 1981, 42: 481-492.
Pommepuy M, Guillaud JF, Dupray E, Derrien A, Le Guyader F, Cormier M: Enteric bacteria survival factors. Water Sci Technol. 1992, 12: 93-103.
Jones GE: Effect of chelating agents on the growth of Escherichia coli in seawater. J Bacteriol. 1964, 87: 484-499.
Enzinger EM, Cooper RC: Role of bacteria and protozoa in the removal of Escherichia coli from estuarine waters. Appl Environ Microbiol. 1976, 31: 758-763.
Bitton G, Henis Y, Lahav N: Effect of Several Clay Minerals and Humic Acid on the Survival of Klebsiella aerogenes Exposed to Ultraviolet Irradiation. Appl Microbiol. 1972, 23: 870-874.
Roper MM, Marshall KC: Effects of salinity on sedimentation and of particulates on survival of bacteria in estuarine habitats. Geomicrobiol J. 1979, 1: 103-116.
Ghoul M, Bernard T, Cormier M: Evidence that Escherichia coli accumulates glycine betaine from marine sediment. Appl Environ Microbiol. 1986, 56: 551-554.
Gauthier MJ, Munro PM, Breittmayer VA: Influence of prior growth conditions on low nutrient response of Escherichia coli in seawater. Can J Microbiol. 1989, 35: 379-383.
Davies CM, Julian A, Long H, Donald M, Ashbolt NJ: Survival of faecal microorganisms in marine and freshwater sediments. Appl Environ Microbiol. 1995, 61: 1888-1896.
Cavallo RA, Latorre F, Acquaviva MI, Montagna MT, Mele MS: Indicatori microbiologici di inquinamento: corrispondenza acqua-sedimenti del Mar Piccolo di Taranto. Inquinamento. 1996, 3: 54-57.
Li J, Purdy KJ, Takii S, Hayashi H: Seasonal changes in ribosomal RNA of sulphate-reducing bacteria and sulphate reducing activity in a freshwater lake sediment. FEMS Microbiol Ecol. 1999, 28: 31-39. 10.1111/j.1574-6941.1999.tb00558.x.
Bezirtzoglou E, Panagiou A, Savvaidis Y, Maipa V: Distribution of Clostridium perfringens in polluted lake environments. Anaerobe. 1997, 3: 169-172. 10.1006/anae.1997.0101.
Chapelle FH: Microorganisms present in the ground-water environment. Ground-water microbiology and geochemistrynd Edition. Edited by: Chapelle FH. 2000, New York: Wiley J & Sons, 32-57. 2
Bisson JW, Cabelli VJ: Clostridium perfringens as a water pollution indicator. J Water Pollut Control Fed. 1980, 52: 241-248.
Fujioka RS, Shizumura LK: Clostridium perfringens, a reliable indicator of stream water quality. J Water Pollut Control Fed. 1985, 57: 986-992.
Ferguson CM, Coote BG, Ashbolt NJ, Stevenson IM: Relationships between indicators, pathogens and water quality in an estuarine system. Wat Res. 1996, 30: 2045-2054. 10.1016/0043-1354(96)00079-6.
Skanavis C, Yanko WA: Clostridium perfringens as a potential indicator for the Presence of Sewage Solids in Marine Sediments. Mar Pollut Bull. 2001, 42: 31-35. 10.1016/S0025-326X(00)00087-4.
Desmarais TJ, Solo-Gabriele HM, Palmer CJ: Influence of Soil on Faecal Indicator Organisms in a Tidally Influenced Subtropical Environment. Appl Environ Microbiol. 2002, 68: 1165-1172. 10.1128/AEM.68.3.1165-1172.2002.
Solo-Gabriele H, Wolfert M, Desmarais T, Palmer C: Sources of E. coli to a SubTropical Coastal Environment. App Environ Microbiol. 2000, 66 (1): 230-237.
Leeming R, Nichols PD, Ashbolt NJ: Distinguishing sources of fecal pollution in Australian inland and coastal waters using sterol biomarkers and microbial fecal indicators. Research Report No. 204. 1998, Melbourne: Water Services Association of Australia
Niilo L: Clostridium perfringens in animal disease: A review of current knowledge. Can Vet J. 1980, 21: 141-148.
Van Metre P, Mahler B, Furlong E: Urban sprawl leaves its PAH signature. Environ Sci Technol. 2000, 34: 4064-4070. 10.1021/es991007n.
World Health Organization: Guidelines for drinking water quality. Health criteria and other supporting information. 1996, Geneva: World Health Organization, 2: Second
Robles S, Rodrìguez JM, Granados I, Guerrero MC: Sulfite-reducing clostridia in the sediment of high mountain lake (Laguna Grande, Gredos, Spain) as indicators of faecal pollution. Int Microbiol. 2000, 3: 187-91.
Sorensen DL, Eberl SG, Dicksa RA: Clostridium perfringens as a point-source indicator in non-point source polluted streams. Water Res. 1989, 23: 191-197. 10.1016/0043-1354(89)90043-2.
International Hydrological Decade – World Health Organization (IHD-WHO): Water quality surveys; a guide for the collection and interpretation of water quality data. Studies and Reports in Hydrology (UNESCO), no. 23/International Hydrological Decade, 75 – Paris (France). 1978, Geneva: World Health Organization, 57-54.
Cabelli VJ: Obligate anaerobic bacterial indicators. Indicators of Viruses in Water and Food. Edited by: Berg G. 1978, Ann Arbor, MI: Ann Arbor Science, 171-200.
Olivieri VP: Bacterial Indicators of Pollution. Bacterial indicators of pollution. Edited by: Minton NP, Clarke DJ. 1982, Boca Raton, FL: CRC Press, 21-41.
Rhodes MW, Kantor H: Sorbitol-fermenting bifidobacteria as indicators of diffuse human faecal pollution in estuarine watersheds. J Appl Microbiol. 1999, 87: 528-535. 10.1046/j.1365-2672.1999.00845.x.
Woese CR: Bacterial evolution. Microbiol Rev. 1987, 51: 221-271.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ: 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991, 17: 697-703.
Jeng RS, Svircev AM, Myers AL, Beliaeva L, Hunter DM, Hubbes M: The use of 16S and 16S-23S rDNA to easily detect and differentiate common Gram-negative orchard epiphytes. J Microbiol Methods. 2001, 44: 69-77. 10.1016/S0167-7012(00)00230-X.
Harmsen D, Karch H: 16S rDNA for Diagnosing Pathogens: a Living Tree. ASM News. 2004, 19: 19-24.
Lehner A, Tasara T, Stephan R: 16S rRNA gene based analysis of Enterobacter sakazakii strains from different sources and development of a PCR assay for identification. BMC Microbiol. 2004, 4 (1): 43-10.1186/1471-2180-4-43.
Raju D, Waters M, Setlow P, Sarker MR: Investigating the role of small, acid-soluble spore proteins (SASPs) in the resistance of Clostridium perfringens spores to heat. BMC Microbiol. 2006, 6: 50-10.1186/1471-2180-6-50.
Johansson A, Aspan A, Bagge E, Båverud V, Engström Björn, Johansson Karl-Erik: Genetic diversity of Clostridium perfringens type A isolates from animals, food poisoning outbreaks and sludge. BMC Microbiol. 2006, 6: 47-10.1186/1471-2180-6-47.
AAVV: Bergey's Manual of Systematic Bacteriology. Edited by: Garrity G. 2005, Springer, London, UK, 2
Kruskal JB: Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrica. 1964, 29: 1-27. 10.1007/BF02289565.
Kruskal JB: Nonmetric multidimensional scaling: a numerical method. Psychometrica. 1964, 29: 115-129. 10.1007/BF02289694.
Hasegawa M, Kishino H, Yano T: Dating of the human-ape splitting by molecular clock of mitochondrial DNA. J Mol Evol. 1985, 32: 443-445. 10.1007/BF02101285.
APHA-AWWA-WPCF: Standard Methods for the Examination of Water and WasteWater. 1998, Washington DC, USA,: APHA American Public Health Association, 20
Bisson JW, Cabelli VJ: Membrane filter enumeration method for Clostridium perfringens. Appl Environ Microbiol. 1979, 37: 55-66.
Armon R, Payment P: A modified m-CP medium for enumerating C. perfringens from water samples. Can J Microbiol. 1988, 34: 78-79.
Johnson JL, Francis BS: Taxonomy of the clostridia: Ribosomal ribonucleic acid homologie among the species. J Gen Microbiol. 1975, 88: 219-24.
Cato EP, Stackebrandt E: Taxonomy and Phylogeny. Clostridia-Biotechnology handbooks, III series. Edited by: Minton NP, Clarke DJ. 1989, New York and London: Plenum Press, 1-26.
Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hipe H, Frarrow JAE: The phylogeny of the genus Closridium : Proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994, 44: 812-826.
Hill RT, Straube WL, Palmisano AC, Gibson SL, Colwell RR: Distribution of Sewage Indicated by Clostridium perfringens at a Deep-Water Disposal Site after Cassation of Sewage Disposal. Appl Environ Microbiol. 1996, 62: 1741-1746.
ACEA, Azienda Comunale Energia & Ambiente: Attività di controllo della qualità delle acque del bacino del Tevere. Rapporto n. 1 descrizione delle attività ed analisi di variabilità dei dati. Roma. Rapporto tecnico. 1996
Rapporto ICRAM Sedimenti – Scheda 6 – Analisi di spore di clostridi solfito-riduttori. Ministero dell'Ambiente e Tutela del Territorio. 2000
Indorante SJ, Follmer LR, Hammer RD, Koenig PG: Particle-size analysis by a modified pipette procedure. Soil Sci Soc Am J. 1990, 54: 560-563.
Moshrefi N: A new method of sampling soil suspension for particle-size analysis. Soil Sci. 1993, 155: 245-8. 10.1097/00010694-199304000-00002.
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman GJ, Smith JA, Struhl K: Current Protocols in Molecular Biology. 1989, New York: Green Publishing Associates and Wiley-Interscience
Rozen S, Skaletsky HJ: Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Edited by: Krawetz S, Misener S. 2000, Totowa, NJ: Human Press, 365-386.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids. 1997, 24: 4876-4882. 10.1093/nar/25.24.4876.
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ: Multiple sequence alignment with Clustal X. Trends in Bioch Sci. 1998, 23: 403-405. 10.1016/S0968-0004(98)01285-7.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0. 2002, Sunderland, Massachusetts: Sinauer Associates
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14: 817-818. 10.1093/bioinformatics/14.9.817.
Acknowledgements
We wish to thank Dr MariaElena Beltrami for her assistance in data analysis and Dr Francesca Anna Aulicino for her kind comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SM performed most of the experiments, and participated in the interpretation of data and in the discussion of results. MI designed and performed part of the molecular analysis and was involved in the interpretation of data. AD planned the microbiological analysis. MM carried out the phylogenetic analysis. ME participated as a consultant and hosted SM in his laboratory for DNA sequencing. EP carried out the sampling of sediment. GL critically revised the manuscript. LM initiated the study, designed the experiments, and participated in the discussion of results. SM, MI and LM participated in the writing of the manuscript. All authors read and approved the final manuscript.
Stefania Marcheggiani, Marcello Iaconelli and Laura Mancini contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Marcheggiani, S., Iaconelli, M., D'angelo, A. et al. Microbiological and 16S rRNA analysis of sulphite-reducing clostridia from river sediments in central Italy. BMC Microbiol 8, 171 (2008). https://doi.org/10.1186/1471-2180-8-171
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-8-171