
Abstract
Background
Low temperatures play key roles in the development of most plant diseases, mainly because of their influence on the expression of various virulence factors in phytopathogenic bacteria. Thus far, studies regarding this environmental parameter have focused on specific themes and little is known about phytopathogenic bacteria physiology under these conditions. To obtain a global view regarding phytopathogenic bacteria strategies in response to physiologically relevant temperature changes, we used DNA microarray technology to compare the gene expression profile of the model bacterial pathogen P. syringae pv. phaseolicola NPS3121 grown at 18°C and 28°C.
Results
A total of 236 differentially regulated genes were identified, of which 133 were up-regulated and 103 were down-regulated at 18°C compared to 28°C. The majority of these genes are involved in pathogenicity and virulence processes. In general, the results of this study suggest that the expression profile obtained may be related to the fact that low temperatures induce oxidative stress in bacterial cells, which in turn influences the expression of iron metabolism genes. The expression also appears to be correlated with the profile expression obtained in genes related to motility, biofilm production, and the type III secretion system.
Conclusions
From the data obtained in this study, we can begin to understand the strategies used by this phytopathogen during low temperature growth, which can occur in host interactions and disease development.
Similar content being viewed by others

Background
During their life cycles, phytopathogenic bacteria possess an epiphyte growth stage during which they can grow and reproduce on the surface of a plant without causing disease. However, when conditions are favorable, the bacteria enter a pathogenic stage that leads to disease development. It is known that a complex interaction between key factors must exist for the development of the disease in plants, represented as “disease triangle”. This involves the interaction of a susceptible host, a virulent pathogen, and environmental conditions favorable for disease development [1, 2]. Regarding environmental conditions, temperature is a key factor in most plant diseases, which are favored mainly by low temperatures [1, 3, 4]. The influence of low temperature on disease development is predominantly due to the expression of various pathogenicity and/or virulence factors by the pathogens, which influences plant health. Several bacterial phytopathogens, such as Pseudomonas syringae and Erwinia sp., produce disease in their host plants in response to low temperature, which appears to be the cue for these phytopathogens to produce virulence factors, including toxins, cell-wall degrading enzymes, and effector proteins [4]. Thus, low temperatures are an important environmental parameter in the development of most diseases in plants.
One of the most common and economically important diseases is the bean disease (Phaseolus vulgaris L.) known as “halo blight” because it causes major field crop losses. This disease, caused by the bacterial pathogen P. syringae pv. phaseolicola, affects both the leaves and pods [5–7]. Cool temperatures (less than 25°C) favor disease development, a condition that also favors production of the major virulence factor of the pathogen, known as phaseolotoxin [8, 9]. Phaseolotoxin is a non-host specific and chlorosis-inducing toxin that acts as a reversible inhibitor of the enzyme ornithine carbamoyltransferase (OCTase; EC2.1.3.3), which catalyzes the conversion of ornithine to citruline in the arginine biosynthetic pathway [10, 11]. The production of phaseolotoxin by P. syringae pv. phaseolicola is regulated mainly by temperature and is optimally produced at 18°C-20°C, whereas at 28°C (the optimal growth temperature for this bacterium), the toxin is not detected [8, 9]. Genes whose products are involved in phaseolotoxin synthesis are clustered within of a chromosomal region known as the “Pht cluster”, whose expression is also low temperature (18°C) dependent [12]. Thus, like other phytopathogenic bacteria, low temperatures play an important role in P. syringae pv. phaseolicola for the development of halo blight disease. However, research related to this environmental parameter has focused only on phaseolotoxin production and little is known about the bacterium physiology under these conditions. Likewise, studies performed in other phytopathogenic bacteria have focused on specific topics regarding low temperature function [4].
Global knowledge about the strategies used by these phytopathogens, in terms of temperature change which influences virulence stage and disease development, is very scarce and most of these studies have focused on animal pathogens where high temperature caused this effect [13, 14]. Therefore, this study was undertaken with the objective to understand how phytopathogenic bacteria, in particular the bacterial pathogen P. syringae pv. phaseolicola NPS3121, respond to temperature changes related to the development of the most of plant diseases.
Results and discussion
Low temperature (18°C) negatively affects the growth rate of P. syringaepv. phaseolicola NPS3121
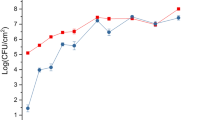
To obtain a global view regarding the strategies used by P. syringae pv. phaseolicola NPS3121 in response to physiologically relevant temperature changes, we used DNA microarray technology. We compared gene expression profiles in the P. syringae pv. phaseolicola NPS3121 wild-type (wt) strain grown at 18°C and 28°C in M9 minimal media. These temperatures represent conditions that either favor the development of the disease (18°C) or do not (28°C) [8]. Initially, to evaluate the effect of temperature and establish the growth stage for this study, we performed bacterial growth curves of the P. syringae pv. phaseolicola NPS3121 strain grown under the conditions mentioned above. The results showed that at low temperature (18°C), the growth rate of the bacteria decreases approximately 0.5-fold relative to 28°C (Figure 1A). This behavior was reproducible in all performed kinetics. The effect of low temperature on the growth rate of several Pseudomonas syringae strains, including pv. phaseolicola, had been previously observed with similar results to this study [15]. Because previous results from our group indicated that during the transition phase, low temperature-induced differential expression in the phaseolotoxin synthesis genes (Pht cluster) occurs [12], we performed this study with cells harvested during this growth stage, which allowed us to use this cluster as a control for the microarray and ensure the virulent stage of the bacterium. Thus, parallel cultures of P. syringae pv. phaseolicola NPS3121 grown at 28°C and 18°C were harvested at the transition phase and RNA was extracted. The results presented in this work reflect the adapted state and significant genes, whose expression is differentially maintained over long-term growth at a given temperature.

Low temperature decreases the bacterial growth rate and favors phaseolotoxin production. Panel A shows the bacterial growth curves of P. syringae pv. phaseolicola NPS3121 grown at 18°C and 28°C. The low temperature decreases the growth rate of the bacterium and favors phaseolotoxin production (panel B). The circles indicate the growth stage in which the RNA extraction was performed.
Differentially expressed genes at 18°C are distributed throughout the chromosome and comprise several functional categories
The differentially expressed genes were identified using a cut-off criteria of ≥1.5 for up-regulated and ≤0.6 for down-regulated genes (p-value ≤ 0.05). A total of 236 differentially regulated genes were identified, of which 133 were up-regulated and 103 were down-regulated at 18°C relative to 28°C. Analyses about the distribution and location of the genes in the P. syringae pv. phaseolicola 1448A sequenced genome, showed that the differentially expressed genes at 18°C are not located in a single chromosomal region of P. syringae pv. phaseolicola, but rather are distributed throughout the genome. Furthermore, only down-regulated genes were distributed in both plasmids of this strain (Figure 2). This pattern of distribution had been observed in preliminary assays, in which a Tn5-derived promoter probe was used to search for genes whose expression was temperature dependent; however, the authors reported the location of only a few genes throughout the genome [16].

Distribution and location of differentially expressed genes at 18°C in the P. syringae pv. phaseolicola genome. Differentially regulated genes were analyzed using the GenoMap software and their distribution and location in the bacterium genome was determined. The red bars depict the distribution of up-regulated genes and the green bars represent the down-regulated genes at 18°C.
For the purposes of this study, the differentially regulated genes were analyzed and manually grouped into categories based on their putative role in biological processes (Tables 1 and 2). In general, data analyses show that the majority of the differentially regulated genes relate to the pathogenicity and/or virulence process of the bacterium.
Control and reliability of the microarray: Pht cluster genes and phaseolotoxin synthesis are induced at 18°C
As expected, the microarray data showed that the Pht cluster genes (Cluster 1), involved in phaseolotoxin synthesis, were up-regulated at 18°C relative 28°C, thus confirming the previously reported data [12]. Thirteen of the 23 genes that comprise the Pht region were highly expressed at 18°C relative 28°C, which was consistent with the conditions of phaseolotoxin synthesis observed in the growth inhibition assays (Figure 1B). Only 13 of the 23 Pht cluster genes were activated because only these genes are printed on our microarray. However, these genes represent the five transcriptional units that comprise the Pht region [12]. To validate the microarray data, one gene from each transcriptional unit was selected for validation of their expression pattern by RT-PCR analyses (Figure 3). The variability in Pht cluster gene expression levels observed could suggest different regulation mechanisms for each of them. Thus far, it is known that there is transcriptional regulation for this group of genes mediated by temperature and only IHF protein has been identified as directly involved in the regulation of some of them [12, 17]. The results regarding the Pht cluster can also be used as control of the microarray, ensuring the reliability of the results obtained in this study.

Microarray validation using RT-PCR analyses. RT-PCR validates the microarray results. a Corresponds to expression levels obtained in the microarray for these genes. The remaining genes do not show expression levels because they were not printed on the microarray.
Genes involved in non-ribosomal peptide synthesis are induced at low temperature
Another group of genes that was up-regulated at 18°C in P. syringae pv. phaseolicola NPS3121 comprise Cluster 2, corresponding to genes involved in non-ribosomal peptide synthesis (NRPS) [18]. NRPS is an alternative pathway that allows production of polypeptides via a different mechanism than the traditional translation pathway. Peptides are created by enzymatic complexes called synthetases. Through NRPS, some bacteria produce several secondary metabolites, such as siderophores, antibiotics, or toxins that contribute to the fitness and/or pathogenicity of the bacterium [19, 20]. In our microarray, six genes that encode four hypothetical proteins (PSPPH_4544, PSPPH_4546, PSPPH_4549, and PSPPH_4555), a facilitator family protein (PSPPH_4553), and an arginine aminomutase (PSPPH_4554) were highly up-regulated at 18°C. These genes are located in a 27 kbp fragment, which also encodes a polyketide synthetase domain protein (PSPPH_4547) and a non-ribosomal peptide synthetase (PSPPH_4550). This region is delimited by genes encoding for transposases (PSPPH_4538 and PSPPH_4559), which were also induced in our microarray (Table 1, Cluster 2). Of all the genes of this region, only six genes were printed on the microarray and all of these were induced in the conditions evaluated. RT-PCR analyses confirmed the profile expression and temperature dependence of PSPPH_4553 and PSPPH_4554, thus validating the microarrays results (Figure 3). The expression of at least one of these genes (PSPPH_4550) in temperature dependence had been previously observed with similar results [21]. In P. syringae pv. phaseolicola NPS3121, it has been suggested that NRPS genes are part of a genomic island (IG) acquired by horizontal transfer and is postulated to be involved in phaseolotoxin synthesis during peptide assembly. However, only the PSPPH_4550 gene has been demonstrated to have a role in this process [21]. Based on this hypothesis, the profile expression obtained for this group of genes at 18°C could be congruent with the differential expression of the Pht cluster genes and the conditions for phaseolotoxin synthesis. However, the RT-PCR results for the PSPPH_4547 gene showed that the expression of this gene is independent of temperature, presenting constitutive behavior at both temperatures (Figure 3). Knowledge regarding the role of this P. syringae pv. phaseolicola gene group is limited and experimental work is still necessary. Likewise, is necessary to evaluate whether there is a relationship between these genes and phaseolotoxin synthesis genes, as has been previously proposed, or whether these genes participate in different biological processes that contribute to the fitness of the bacterium in low temperatures.
In P. syringaepv. phaseolicola NPS3121, the Type VI secretion system (T6SS) is regulated by temperature
Recently, a new secretion system has been recognized, called the Type VI secretion system (T6SS). This system is encoded within the genomes of most Gram negative bacteria, including plant, animal, and human pathogens, as well as environmental strains. The T6SS components are usually encoded by a gene cluster that is thought to form a genomic island whose composition and number varies among species [22–24]. The in silico analyses have revealed that the genome of P. syringe pv. phaseolicola 1448A carries only one putative T6SS gene cluster (HSI) that comprises the region from PSPPH_0119 to PSPPH_0135. Furthermore, several genes putatively encoding some proteins of this system are scattered in the genome of this bacterium [24]. The microarrays results showed the induction of eight genes encoding proteins putatively involved in the T6SS in P. syringe pv. phaseolicola NPS3121 (Cluster 3). The PSPPH_0122 gene encodes a hemolysin-coregulated (Hcp) protein homolog, in addition to be an essential component of the secretion machinery, acts as an effector protein that is secreted through this system. The PSPPH_0124 gene encodes a hypothetical protein and the PSPPH_0125 gene encodes the IcmF protein, which in conjunction with the DotU protein (PSPPH_0126), act as associated structural proteins that anchor the secretion system in the cell membrane [25]. Within this cluster is also the PSPPH_0131 gene encoding the hsiG protein and the PSPPH_0135 gene that encodes a hypothetical protein. The expression of these genes at low temperature was confirmed by RT-PCR (Figure 3). Although PSPPH_ 4978, PSPPH_ 4979, and PSPPH_ 4984, which encode prophage PSPPH06 proteins, are not involved in T6SS, these genes were include within this group because their adjacent genes (PSPPH_ 4980 and PSPPH_ 4985) putatively encode Hcp proteins [24], which may be responsible for the induction levels obtained. This finding is being evaluated in our laboratory. The T6SS has been shown to play a key role in the virulence and pathogenesis of diverse bacterial pathogens, in some cases, by the secretion of effector proteins or toxins. However, its complete mechanism of action is poorly understood. The function of this system is not restricted to pathogenic processes because the T6SS also participates in other processes such as biofilm formation, stress sensing, symbiosis, root colonization, and nodule formation [26, 27]. The role of the putative T6SS gene cluster in P. syringae pv. phaseolicola NPS3121 has not been evaluated so more experimental work is required. However, it has been demonstrated that T6SS in P. syringae pv. syringae B728a, which is phylogenetically identical to P. syringae pv. phaseolicola T6SS, it is not essential for leaf colonization and development of the disease [28]. Several reports have demonstrated that expression of the T6SS gene cluster is tightly regulated in different environmental conditions and low temperatures contribute to the expression of these genes in some pathogens [29]. This phenomenon is similar to our observation that low temperature (18°C) regulates T6SS genes expression. To our knowledge, this is the first report about expression of these genes of P. syringae pv. phaseolicola NPS3121 and the influence of low temperature on their expression.
Cell envelope-associated changes are induced by low temperature
A universal response to low temperature includes changes in the lipid composition of membranes to help cope with the decrease in membrane fluidity caused by the cold. Microorganisms respond by increasing the unsaturated fatty acids level in membrane phospholipids, which helps to maintain membrane homeoviscosity so that its function is not affected. There are a variety of mechanisms that can alter membrane phospholipid composition in response to temperature change [30]. The conversion of saturated fatty acids into unsaturated fatty acids by desaturases enzymes is one of these pathways [30, 31]. In our microarray and RT-PCR analyses (Figure 3, Cluster 1), the desI gene encoding a fatty acid desaturase was induced at 18°C, which might be involved in the unsaturation process, in a similar manner to the reported desA and des genes from Synechosysteis sp. PCC6803 and Bacillus subtilis, respectively. It has been observed that deletion of the des gene in B. subtilis produces a cold-sensitive phenotype and slower growth, thus demonstrating its role during adaptation to low temperatures [32]. In P. syringae pv. phaseolicola NPS3121, the desI gene is part of the phtD transcriptional unit of the Pht cluster (Cluster 1). Mutation of this gene produces a non-toxigenic phenotype relative to the wt strain. However, the relationship of desI with phaseolotoxin synthesis is still unknown [12]. Additionally, it has been observed that mutation in the desI gene decreases the growth rate at 18°C relative to the wt strain, suggesting a cold-sensitivity in the mutant strain (unpublished data).
Another of the mechanisms reported to be involved in membrane lipid composition changes correspond to de novo synthesis. The fabF and lpxP genes induced by low temperature participate in this process [33]. β-ketoacyl-ACP synthase II, the fabF gene product, converts palmitoleic acid to cis-vaccenic acid, which is in turn transferred by an acyltransferase (LpxP) into lipid A, a component of polysaccharides [33, 34]. Although these two genes were not found in our microarray, several genes involved in cell wall biogenesis and membrane synthesis were identified (Cluster 4). These include the murA gene (PSPPH_4139) that is involved in peptidoglycan synthesis (a major component of cell wall), the PSPPH_4682 gene involved in polysaccharide synthesis, as well as three genes PSPPH_4669, PSPPH_3226, and galU (PSPPH_2260) that encode an acetyl-, glycosyl- and uridyl- transferase, respectively, which are likely associated with the transfer of these groups during polysaccharides synthesis. Additionally, it has been demonstrated that during cell envelope biogenesis, there is an increase in outer membrane lipoproteins, which increase connections with the cell wall [34, 35]. In our analyses four genes (PSPPH_ 1464, PSPPH_2654, PSPPH_2842, and PSPPH_3810) encoding lipoproteins were induced, which may be related to outer membrane synthesis. The microarray results suggest that membrane component synthesis is activated in the conditions of our study and these changes are likely related to cell envelope remodeling to adapt to low temperatures.
Low temperature induces expression of motility genes in P. syringaepv. phaseolicola NPS3121
Cluster 5 comprises genes induced at 18°C that are involved in bacterium motility. The data suggest that chemotaxis and rotation of flagella processes function in low temperatures on P. syringae pv. phaseolicola NPS3121. Two genes, PSPPH_3880 that encodes the membrane-bound methyl accepting chemotaxis protein (MCP)-like receptor WspA, and PSPPH_3881, that encodes the CheW-like scaffolding protein WspB, showed high transcripts levels at 18°C relative 28°C (Table 1). WspA and WspB are related to the chemotaxis process. Chemotaxis, as well as other types of taxis (e.g., thermotaxis), enables bacteria to approach beneficial environments and escape from hostile ones. Depending on the parameter monitored, bacteria will respond by either swimming toward attractants or retreating from repellants. Thus, the signal sensed by chemotaxis causes changes in flagellum motility [36]. Flagella gene expression is activated by the FleQ protein, which appears to be the highest regulator in the hierarchical flagellar biogenesis regulatory cascade in P. aeruginosa[37]. FleQ (PSPPH_3387) was induced in our study at 18°C and its expression was validated by RT-PCR (Figure 3), suggesting that the motility of P. syringae pv. phaseolicola NPS3121 is favored under this condition. Furthermore, four genes related to pili formation, which is also involved in bacterial movement, were induced at low temperature: PSPPH_0730 that encodes type IV pilus-associated protein, PSPPH_1200 that encodes a pili assembly chaperone, PSPPH_0818 that encodes PilD protein, and PSPPH_0820 that encodes PilB protein. Each of these genes has been associated with P. syringae pv. phaseolicola virulence because of their role in adhesion to the surface of host plants to initiate infection [38]. It has been reported that RpoN sigma factor regulates the expression of genes required for pili and flagella biosynthesis in P. aeruginosa[37, 39]. Our microarray data and RT-PCR assays showed that the PSPPH_4151 gene (Cluster 8), which encodes the RpoN protein, was induced at 18°C, suggesting a similar regulation may occur in our strain (Figure 3). The results obtained suggest that P. syringae pv. phaseolicola NPS3121 motility is regulated by temperature, similar to those observed in the pathogens Helicobacter pylori and E. coli, whose motility patterns are altered by temperature changes [33, 40].
To assess whether these changes in the gene expression generate a motility phenotype in P. syringae pv. phaseolicola related to temperature, we evaluated the motility pattern of this bacterium at 18°C and 28°C. To ensure that the bacteria were in the same physiological condition as when the microarray analysis was performed, P. syringae pv. phaseolicola NPS3121 cells grown at 18°C and 28°C (OD 600: 1.1 and 1.2) were inoculated in semisolid M9 media containing 0.3%, 0.4%, and 0.5% agar and incubated at 18°C and 28°C. The results showed that under these conditions the bacterium was not motile despite gene induction at 18°C (Figure 4A). Additionally, motility assays in KB media were performed, using the conditions that have demonstrated motility in related pathovars [41, 42]. Plates and glass tubes with semisolid KB media were used to evaluate motility at the mentioned temperatures. Again, the P. syringae pv. phaseolicola NPS3121 strain was not motile under these conditions compared to P. syringae pv. tomato DC3000 and P. syringae pv. tabaci, which showed motility at both temperatures and where it was observed that low temperatures appear to affect their motility (Figures 4B and 4C). This non-motile phenotype of P. syringae pv. phaseolicola NPS3121 had been previously reported [41, 43], and further experiments are required to determine the conditions in which this bacterium can be motile and to evaluate the effect of low temperature in this process.

Motility Tests of the P. syringae pv. phaseolicola NPS3121 strain. (A) Motility assays performed in semisolid M9 media containing 0.3%, 0.4%, and 0.5% agar at 18°C and 28°C. (B) Motility assays in semisolid KB media containing 0.3% (left) and 0.5% (right) agar. (C) The results obtained using the stab technique in M9 and KB media.
Low temperature induces oxidative stress and iron metabolism
Another group of genes differentially expressed at 18°C correspond to genes related to iron metabolism (Cluster 6). Iron fulfills a vital role in virtually all organisms because of its participation in several cellular processes. Because iron is in short supply in many habitats, bacteria secrete siderophores, compounds that are specific iron chelators, to mobilize inside the cell through membrane receptor molecules [44]. Two genes, PSPPH_3753 that encodes a protein related to siderophore synthesis and PSPPH_1923 that is involved in pyoverdine synthesis (a major siderophore of the fluorescent Pseudomonas sp.), were induced at 18°C relative to 28°C [45]. Likewise, the gene encoding sigma factor protein PvdS, which is required for expression of pyoverdine synthesis genes, was induced under these conditions [46]. The induction of this PvdS protein was validated by RT-PCR analysis (Figure 3). One gene encoding the regulatory protein FecR (PSPPH_2117) and proteins involved in iron transport were also included in this group. It is known that in P. aeruginosa, the Fur protein is the master regulator of iron homeostasis. It represses pyoverdine synthesis via negative regulation of the pvdS gene under high iron concentrations. However, in iron-limiting conditions, Fur repression is released and transcription can occur [47]. It has been reported that PvdS sigmulon is conserved among the fluorescent pseudomonads, including the P. syringae group [46]. Although the fur gene was not printed on our microarray, the functional status of Fur protein can be inferred as inactive because the genes regulated by this protein are induced in the conditions evaluated. This expression profile simulates conditions of iron deficiency.
To phenotypically evaluate changes in the expression of siderophores synthesis genes in function of temperature, we performed quantitative analyses of siderophores at 18°C and 28°C. The results of these assays showed that at 18°C, the amount of siderophores in the culture supernatant was higher (58.6 ± 0.39 μM) compared to when the bacterium is grown at 28°C (20.53 ± 0.844 μM). Thus, the results demonstrate that low temperature induces siderophores production by the bacterium.
Additionally, it has been reported that in several bacteria, the Fur protein positively regulates the expression of genes involved in various pathways in response to large iron amounts, such as oxidative stress genes (e.g. catalases) [47]. In our microarray, the PSPPH_3274 gene (encoding the catalase KatB) was induced at 18°C, which would be inconsistent with our hypothesis about an inactive status for the Fur protein at low temperatures. However, several reports have indicated an association between decreased environmental temperature and increased intracellular oxidative stress in bacteria. Thus, there is evidence that free radical production (superoxide O2-, hydrogen peroxide H2O2, or hydroxyl radical HO-) in bacterial cells is stimulated at low temperatures, apparently in an iron-independent manner. Therefore, the expression of oxidative stress adaptation genes, such as catalases, increase considerably [48, 49]. A similar response may occur in our strain, justifying the observed induction in the catalase gene, as low temperature induces free radical production in cells, in turn increasing catalase production. The expression of the gene encoding catalase (KatB) was evaluated by RT-PCR analysis (Figure 3). Furthermore, it has been reported that iron-starvation inducible genes are also induced in response to oxidative stress in P. aeruginosa. This response appears to be due to a transient loss of Fur repressor function [50]. These observations are consistent with our data and support our hypothesis about the inactive status of the Fur protein at low temperatures.
Additionally, within Cluster 6, we also found PSPPH_1309, which encodes the cysteine desulfurase IscS, and PSPPH_1311, which encodes iron-sulfur cluster assembly protein IscA, both components of ISC (iron-sulfur cluster) system essential in the biogenesis of iron-sulfur (Fe-S) proteins in bacteria. It has been observed that some pathways involved in Fe-S cluster assembly operate under iron starvation and oxidative stress conditions [51, 52], which agrees with the results obtained.
On the other hand, several reports have indicated a correlation exists between the uptake-transport iron system and motility process and biofilm formation. Thus, iron deficiency stimulates twitching motility, a form of surface motility that is inconsistent with microcolonies and biofilm formation [53]. This is consistent with the results obtained in our experiments, because iron metabolism genes and siderophores production are induced, simulating iron deficiency conditions, and motility processes appear to be favored, whereas biofilm or extracellular polysaccharide formation is decreased (see data below).
Hypothetical proteins and proteins with unknown function are induced at 18°C
Among the differentially regulated genes induced at 18°C, we found 15 genes that hypothetically encode conserved proteins (Cluster 7). Additionally, Cluster 8 has genes that could not be grouped into any specific biological process but showed high transcript levels at 18°C relative to 28°C. Within this cluster are genes encoding various transcriptional regulators, a gene that encodes an ATP-dependent helicase, DinG family (PSPPH_1406), and the PSPPH_4151 gene that encodes RNA polymerase sigma-54 factor RpoN whose expression was validated by RT-PCR assays (Figure 3).
Low temperature represses alginate synthesis in P. syringaepv. phaseolicola NPS3121
With regard to repressed genes identified at 18°C, we found a group of genes involved in alginate synthesis (Cluster 9). The genes enconding AlgX (PSPPH_1112), AlgG (PSPPH_1113), AlgE (PSPPH_1114), AlgK (PSPPH_1115), and AlgD (PSPPH_1118), as well as the PSPPH_1119 gene that encodes a hypothetical protein, were included in this cluster. Alginate is an extracellular polysaccharide (EPS) produced by bacteria that is secreted into growth media and involved mainly in biofilm formation. Production of this co-polymer by P. syringae and P. aeruginosa has been previously reported [54, 55]. Alginate production by P. syringae has been associated with increased epiphytic fitness, resistance to desiccation and toxic molecules, and the induction of water-soaked lesions on infected leaves. Studies have shown that alginate functions in the virulence of some P. syringae strains and facilities the colonization and/or dissemination in plants [55]. Although P. syringae pv. phaseolicola virulence is favored by low temperature, alginate production by this strain appears to be repressed under these conditions. RT-PCR analyses confirmed the repression mediated by low temperatures of algD, the first gene of the alginate biosynthetic operon (Figure 3). The repression of alginate genes mediated by low temperature also has been observed in P. syringae pv. syringae, where the expression of algD, was induced at 28°C and significantly lower at 18°C [56]. To validate the microarrays results in P. syringae pv. phaseolicola NPS3121, the effect of temperature on EPS production (including alginate) was evaluated. Quantitative analyses showed that at 18°C the production of EPS is lower (76.65 ± 4.09 μg) compared to when the bacterium is grown at 28°C (192.43 ± 14.11 μg). Thus, the results demonstrate that the low temperatures decrease EPS production by the bacterium. Alginate gene regulation is complex and varies between species. In P. aeruginosa, it has been reported that sigma factor-54 (RpoN) represses algD expression by sigma factor antagonism [57]. A similar phenomenon could be occurring in our strain, because the expression levels of the rpoN gene (PSPPH_4151) are consistent with the low expression of alginate genes. Furthermore, it has been reported that a coordinated expression exists between flagellum synthesis and EPS production. In P. aeruginosa, the FleQ protein, a master regulator of flagella genes, represses the expression of genes involved in EPS synthesis, leading to planktonic cells. When this repression is released, the flagellum genes are repressed and EPS production is favored [58]. The alginate gene repression observed in our microarray, could also be due to repression exerted by FleQ protein, which is induced in our experiment, in a similar manner to what occurs in P. aeruginosa. Thus, the results of the microarray are consistent with the fact that EPS production (e.g., alginate) is decreased at low temperatures whereas expression of motility genes is favored.
Genes involved in plant-pathogen interactions are affected by low temperature
Another group of genes repressed at 18°C comprise Cluster 10 and correspond to genes related to plant-pathogen interactions (Table 2). Four of these genes encode Type III effector proteins (T3EFs): HopAB1, HopW1, HopD1, and AvrB2, but the other genes are involved in cell wall degrading enzyme synthesis, such as pectin lyase (PSPPH_3992) and polygalacturonase (PSPPH_A0072). The repression of some T3EFs genes and cell wall degrading enzyme genes was validated by RT-PCR (Figure 3). The classification of these genes as pathogenicity and/or virulence factors in P. syringae pv. phaseolicola has been previously reported [18]. It known that phytopathogenic bacteria suppress plant innate immunity and promote pathogenesis by injecting directly into host cells effector proteins (T3EFs) by a type III protein secretion system (T3SS). However, in the majority of cases, neither the mode of action nor the targets of these effector proteins within the plant are known [59]. In addition, phytopathogens synthesize and secrete various cell wall degrading enzymes, which facilitate pathogen entry and nutrient release for its growth [1]. Thermoregulation of these genes has been observed in other bacterial phytopathogens, where their expression is favored at low temperatures, a phenomenon opposite to data obtained in our experiments [4]. However, it has been reported that in P. syringae pv. tomato DC3000, iron bioavailability regulates the expression of T3SS component genes. Thus, high iron concentrations induced expression of genes such as hrpRS and hrpL, which in turn regulates the expression of T3SS genes, by an as yet unknown mechanism [60]. Based on this, our microarray results might be explained by the fact that the uptake-transport iron genes were induced, mimicking iron limiting conditions, which could lead to the observed repression of T3SS genes.
Genes related to the Type IV secretion system (T4SS) are repressed at 18°C
Another group of genes differentially repressed at 18°C comprise Cluster 11. They include genes related to the type IV secretion system (T4SS), which is closely related to systems involved in the conjugal transfer of DNA (Table 2). Nine of these genes encode conjugal transfer proteins and two encode transcriptional regulators, all within plasmid B (pPh1448B) of P. syringae pv. phaseolicola (Figure 2) [18]. The pPh1448B plasmid belongs to the well-described pPT23A plasmid family, whose members have been demonstrated to play an important role in the interaction of the P. syringae pathogen with host plants [61]. Many of the pPT23A family plasmids are known to be conjugative plasmids. The putative T4SS encoded in the pPh1448B plasmid has been classified as a type IVA system, due to its high similarity with the type IV secretion genes of Agrobacterium tumefaciens (the virB operon and virD4) [61]. In addition to functioning in conjugation, some bacterial T4SSs are capable of delivering effector proteins or toxins to host cells, thus acting as virulence factors [62]. However, in the case of P. syringae pv. phaseolicola 1448A T4SS, it has been suggested to have a role in conjugal transfer of DNA rather than virulence-related protein translocation [18]. Thermoregulation of some T4SS genes in various bacteria has already been reported, similar to our results in this study [4]. More experimental work is necessary to elucidate the role of these genes in P. syringae pv. phaseolicola NPS3121 and their relationship to temperature.
Low temperature represses the heat shock response
Another group of genes repressed at 18°C correspond to those encoding heat-shock proteins (Cluster 12). Genes that encode the HslVU and GrpE heat-shock proteins, as well as the genes encoding DnaK, GroEL, and ClpB chaperones were included in this cluster. Heat shock proteins (HSPs) are a class of functionally related proteins that are responsible for monitoring the state of protein folding in cells. They function as molecular chaperones, facilitating the folding of partially or fully unfolded proteins. Their expression is increased when cells are exposed to elevated temperatures or other stresses, to cope with protein damage. If however, the temperature decreases, a reverse response is observed and heat-shock gene transcription decreases [63]. This latter behavior is similar to the results obtained in our experiments, where the low temperature decreased the transcript levels of heat-shock genes. In E. coli, HSP synthesis is repressed during growth at low temperatures [64]. A similar response has been observed in P. putida, where low temperatures also decrease the expression of these genes [65].
Transcription and replication are repressed by low temperature
Cluster 13 includes genes involved in nucleic acid synthesis. Two of these genes (PSPPH_4598 and PSPPH_4599) encode RNA polymerase beta subunits involved in mRNA synthesis. Three of these genes (PSPPH_2495, PSPPH_B0043, and PSPPH_A0002) are related to the replicative process of DNA synthesis. This result suggests that both processes are affected by low temperature in P. syringae pv. phaseolicola NPS3121, which is consistent with the decreased growth rate observed. This behavior is similar what was previously observed in P. putida where low temperature also reduces proteins involved in the transcription and replication processes [65].
Finally, similar to the analysis and clustering of activated genes, repressed genes at 18°C that hypothetically encode conserved proteins were grouped into Cluster 14. Likewise, those genes whose products could not be grouped into any specific biological process were included in Cluster 15. The relationship of these genes to the physiology of the bacterium to low temperatures remains unknown and more experimental work is required.
Conclusions
In general, the results of the microarray provided us with a global view regarding the physiology of P. syringae pv. phaseolicola NPS3121 at 18°C, a temperature that favors disease development and produces the major virulence factor of the bacterium. The results showed that common processes in response to low temperature, such as cell-envelope remodeling, transcription, translation, and the heat-shock response, are also affected in this bacterial phytopathogen. In addition, low temperatures influence phaseolotoxin synthesis as well as the expression of various virulence factors involved in disease development. Furthermore, our data show low temperature-dependent expression of T6SS, thus being the first report about the expression of this cluster of genes in P. syringae pv. phaseolicola. In general, the expression profile obtained in this study suggest that low temperatures generate an oxidative stress in the bacterium, which leads to expression of uptake-transport iron genes (simulating iron starvation conditions) that in turn are related to the expression of various processes such as motility, biofilm production, and T3SS. From the data obtained in this study, we can begin to understand the temperature dependent strategies used by this phytopathogen during host interactions and disease development.
Methods
Bacterial growth conditions and RNA isolation
The P. syringae pv. phaseolicola NPS3121 strain was grown at 18°C and 28°C in M9 minimal media supplemented with 0.8% glucose as the carbon source. The growth conditions were as follows: pre-inoculums (25 mL) of P. syringae pv. phaseolicola were grown in M9 minimal media overnight at 28°C. The cells were washed once with M9 media and inoculated into 200 mL of M9 minimal media at optical density (OD 600nm) 0.1. To evaluate the effect of temperature, the cultures were incubated at 18°C or 28°C and grown until they reached the transition phase (OD600nm 1.1 at 18°C and 1.2 at 28°C) and RNA was extracted. For RNA isolation, the cells were recovered by centrifugation at 10,000 rpm for 10 min at 4°C, washed with sterile deionized water, and stored at −80°C. The supernatants from each culture were removed for phaseolotoxin production assays. Total RNA was extracted using the TRIzol Reagent following the manufacturer´s instructions (Invitrogen, CA, USA). A second purification step was performed using RNeasy MinElute spin columns (Qiagen, CA, USA) to remove any contaminating DNA. RNA was eluted in 50 μL of diethylpirocarbonate (DEPC)-treated water. RNA concentration was determined using a ND-1000 spectrophotometer (NanoDrop). RNA integrity was verified by analytical agarose gel electrophoresis.
Phaseolotoxin assays
Phaseolotoxin production by P. syringae pv. phaseolicola was assayed using the E. coli JM103 strain growth inhibition assay as previously described [66]. In each case, plates containing arginine (10 mM) were used as controls to confirm that growth inhibition was due to phaseolotoxin effects.
Microarray processing, data acquisition, and statistical analyses
Previously, our group constructed a DNA microarray of P. syringae pv. phaseolicola NPS3121 (GEO accession number GPL7115) with 1911 probes of an average length of 2.4 kbp, which represents approximately 1X of the P. syringae pv. phaseolicola NPS3121 genome. This microarray contains also several PCR products corresponding to various genes with known functions that were printed as controls [67]. To perform this study, we used this P. syringae pv. phaseolicola NPS3121 DNA microarray. Each microarray experiment was repeated six times: two technical replicates with the same RNA samples and three biological replicates using RNA isolated from a different culture. cDNA synthesis, labeling, hybridization, washing, and chip scanning were performed at the Microarray Core Facility at CINVESTAV-LANGEBIO. Hybridized microarray slides were scanned (GenePix 4000, Axon Instruments, Inc) at a 10-μm resolution, adjusting the laser and gain parameters to obtain similar levels of fluorescence intensity in both channels. The spot intensities were quantified using Axon GenePrix Pro 6.0 image analysis software. First, an automatic spot finding and quantification option of the software was used. Subsequently, all spots were inspected individually and in some cases, the spot diameters were corrected or the spots were removed from the analyses. The mean of the signals and the median of backgrounds were used for further analyses. Raw data were imported into the R.2.2.1 software. Background signals were subtracted using Robust Multichip Analysis (RMA) whereas the normalization of the signal intensities within slides was carried out using “print-tip loess” method and the LIMMA package. Normalized data were log2 transformed and fitted into mixed model ANOVAs using the mixed procedure. The p-values of the low temperature (18°C) effect were adjusted using the False Discovery Rate method (FDR). Differentially expressed genes were identified using cut-off criteria of ≥1.5 for up-regulated and ≤0.6 for down-regulated genes (FDR, p-value ≤ 0.05). Analyses of distribution and the location of differentially expressed genes in the P. syringae pv. phaseolicola 1448A sequenced genome were performed using the GenoMap software [68].
Microarray validation by reverse transcription-PCR analyses
To validate the microarray data, we performed reverse transcription (RT)-PCR analyses. The expression levels of several genes with different biological functions were evaluated by this technique. These experiments involved independent biological experiments from those used for microarray analyses. DNA-free RNA was obtained as described above and the integrity of the RNA was evaluated by agarose gel electrophoresis. Total RNA (200 ng) was used for RT-PCR using the Superscript one-step kit (Invitrogen). Controls used for each set of primers were 1) PCR without the reverse transcription step to verify the absence of DNA, 2) RT-PCR performed without RNA templates to detect any contaminating DNA/RNA, 3) PCR performed using genomic DNA as a template to ensure primer fidelity, and 4) amplification of a portion of the 16S rRNA operon as an internal control of the reaction. The RT reaction was performed at 50°C for 30 min. PCR amplification was performed at 94°C for 2 min for 1 cycle; 94°C for 30 s, 55–58°C for 30 s, and 72°C for 1.0 min for 20–28 cycles; and 72°C for 10 min for 1 cycle .
Molecular biology techniques
Routine techniques were performed using standard protocols [69]. Genomic DNA of P. syringae pv. phaseolicola NPS3121 was isolated as described previously [70]. PCR products were amplified with Platinum supermix (Invitrogen). Primers were designed using Vector NTI Software (Invitrogen), with reference to the previously reported sequence of the 1448A strain (Gene Bank accession no. CP000058) [18]. The oligonucleotide primers used in this study are listed in Additional file 1.
Motility assays
To evaluate the motility of P. syringae pv. phaseolicola NPS3121 and the influence of temperature on this process, three strategies were used. The swimming and swarming motility of P. syringae pv. phaseolicola NPS3121 were assessed on semisolid KB plates containing 0.3% and 0.5% agar, respectively, as described in previous studies [41, 42]. The cells were grown in KB broth overnight at 28°C, and harvested and resuspended in KB to OD600 = 1. 50 μL of bacterial suspensions were inoculated on filter disks (6 mm in diameter) and placed in the center of the plate. Plates were incubated for 24 h at 28°C and 18°C before photography. A second strategy was performed to evaluate the swimming and swarming motility of P. syringae pv. phaseolicola NPS3121. To ensure that the bacteria were in the same physiological condition as when the transcriptome analysis was performed, the P. syringae pv. phaseolicola NPS3121 strain was grown in M9 media at 28°C and 18°C until they reached the transition phase. Bacterial suspensions (50 μL) were inoculated on filter disks (6 mm in diameter) and placed in the center of semisolid M9 plates containing 0.3%, 0.4%, and 0.5% agar. Plates were incubated for 48 h at 28°C and 18°C. Finally, motility was also evaluated using the stab technique in semisolid KB and M9 media (0.3% and 0.5% agar) in glass tubes. The tubes were incubated at 28°C and 18°C for 48 h. As controls, we used the P. syringae strains pv. tomato DC3000 and pv. tabaci PTBR2004. Experiments were performed three times with three replicates per treatment.
Quantification of siderophores
Siderophore production into the culture supernatant by bacterial strains was determined using chrome azurol S (CAS) liquid assays as previously described [71]. Briefly, the P. syringae pv. phaseolicola NPS3121 strain was grown in M9 media at 28°C and 18°C until they reached the transition phase. The supernatant was recovered by centrifugation at 8,000 rpm for 15 min at 4°C and filtered through a 0.45-μm-pore-size filter (Millipore). For siderophore quantification, a standard curve was prepared with desferoxamine mesylate. Experiments were performed three times with four replicates per treatment.
Quantitative analyses of EPS
The quantification of EPS was performed as previously described with some modifications [72, 73]. The P. syringae pv. phaseolicola NPS3121 strain was grown in M9 media at 28°C and 18°C until they reached the transition phase [the growth stage in which the microarrays analysis was performed and the repression of EPS synthesis genes (alginate) was observed]. The bacterial cells were harvested by centrifugation at 8,000 rpm for 15 min at 4°C. After centrifugation, the supernatant was mixed with three volumes of ice-cold 95% ethanol (with stirring) for 24 h at −20°C to precipitate the extracellular polysaccharide (EPS). EPS was recovered by centrifugation at 10,000 rpm for 20 min at 4°C. The pellet was washed twice with 95% ethanol and once with absolute ethanol. Quantification of the EPS was performed using the phenol-sulfate method. Total EPS was measured using a glucose standard curve. Experiments were performed three times with four replicates per treatment.
Microarray data accession
The microarray data from this study is available on the GEO database at http://ncbi.nlm.nih.gov/geo with the accession number GSE38423.
References
Agrios GN: Plant Pathology. 1997, California: Academic Press, 4
Hirano SS, Upper CD: Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev. 2000, 64: 624-653. 10.1128/MMBR.64.3.624-653.2000.
Colhoun J: Effects of environmental factors on plant disease. Ann Rev Phytopatol. 1973, 11: 343-364. 10.1146/annurev.py.11.090173.002015.
Smirnova A, Li H, Weingart H, Aufhammer S, Burse A, Finis K, Schenk A, Ullrich MS: Thermoregulated expression of virulence factors in plant associated bacteria. Arch Microbiol. 2001, 176: 393-399. 10.1007/s002030100344.
Mitchell RE: Bean halo-blight toxin. Nature. 1976, 260: 75-76. 10.1038/260075a0.
Mitchell RE: Isolation and structure of a chlorosis inducing toxin of Pseudomonas phaseolicola. Phytochemistry. 1976, 15: 1941-1947. 10.1016/S0031-9422(00)88851-2.
Mitchell RE, Bieleski RL: Involvement of phaseolotoxin in Halo blight of beans. Plant Physiol. 1977, 60: 723-729. 10.1104/pp.60.5.723.
Goss RW: The relation of temperature to common and halo blight of beans. Phytopathology. 1970, 30: 258-264.
Nüske J, Fritsche W: Phaseolotoxin production by Pseudomonas syringae pv. phaseolicola: the influence of temperature. J Basic Microbiol. 1989, 29: 441-447. 10.1002/jobm.3620290713.
Ferguson AR, Johnston JS: Phaseolotoxin: chlorosis, ornithine accumulation and inhibition of ornithine carbamoyltransferase in different plants. Physiol Plant Pathol. 1980, 16: 269-275. 10.1016/0048-4059(80)90041-7.
Templeton MD, Sullivan PA, Shepherd MG: The inhibition of ornithine transcarbamoylase from Escherichia coli W by phaseolotoxin. Biochem J. 1984, 224: 379-388.
Aguilera S, López-López K, Nieto Y, Garcidueñas-Piña R, Hernández-Guzmán G, Hernández-Flores JL, Murillo J, Álvarez-Morales A: Functional characterization of the gene cluster from Pseudomonas syringae pv. phaseolicola NPS3121 involved in synthesis of phaseolotoxin. J Bacteriol. 2007, 189: 2834-2843. 10.1128/JB.01845-06.
Smoot LM, Smoot JC, Graham MR, Somerville GE, Sturdevant DE, LUx Migliaccio CA, Sylva GL, Musser JM: Global differential gene expression in response to growth temperature alteration in group A Streptococcus. Proc Natl Acad Sci. 2001, 98: 10416-10421. 10.1073/pnas.191267598.
White-Zielger CA, Malhowski AJ, Young S: Human body temperature (37°C) increases the expression of iron, carbohydrate and amino acid utilization genes in Escherichia coli K12. J Bacteriol. 2007, 189: 5429-5440. 10.1128/JB.01929-06.
Young JM, Luketina RC: The effects on temperature on growth in vitro of Pseudomonas syringae and Xanthomonas pruni. J Appl Bacteriol. 1977, 42: 345-354. 10.1111/j.1365-2672.1977.tb00702.x.
De Ita ME, Marsch-Moreno R, Guzman P, Álvarez-morales A: Physical map of the chromosome of the phytopathogenic bacterium Pseudomonas syringae pv. phaseolicola. Microbiol. 1998, 144: 493-501. 10.1099/00221287-144-2-493.
Arvizu-Gómez J, Hernández-Morales A, Pastor-Palacios G, Brieba L, Álvarez-Morales A: Integration Host Factor (IHF) binds to the promoter región of the phtD operon involved in phaseolotoxin synthesis in P. syringae pv. phaseolicola NPS3121. BMC Microbiol. 2011, 11: 90-10.1186/1471-2180-11-90.
Joardar V, Lindeberg M, Jackson RW, Selengut J, Dodson R, Brinkac LM, Daugherty SC, DeBoy R, Durkin AS, Giglio MG, Madupu R, Nelson WC, Rasovitz MJ, Sullivan S, Crabtree J, Creasy T, Davidsen T, Haft DH, Zafar N, Zhou L, Halpin R, Holley T, Khouri H, Feldblyum T, White O, Fraser CM, Chatterjee AK, Cartinhour S, Schneider DJ, Mansfield J, Collmer A, Buell R: Whole genome sequence analysis of Pseudomonas syringae pv phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J Bacteriol. 2005, 187: 6488-6498. 10.1128/JB.187.18.6488-6498.2005.
Bender CL, Alarcón-Chaidez F, Gross DC: Pseudomonas syringae Phytotoxins: Mode of action, regulation and biosynthesis by peptide and polyketide synthetases. Microbiol Mol Biol Rev. 1999, 63: 266-292.
Finking R, Marahiel MA: Biosynthesis of nonribosomal peptides. Annu Rev Microbiol. 2004, 58: 453-488. 10.1146/annurev.micro.58.030603.123615.
De la Torre-Zavala S, Aguilera S, Ibarra-Laclette E, Hernández-Flores JL, Hernández-Morales A, Murillo J, Álvarez-Morales A: Gene expression of Pht cluster genes and a putative non-ribosomal peptide synthetase required for phaseolotoxin production is regulated by GacS/GacA in Pseudomonas syringae pv. phaseolicola. Res Microbiol. 2011, 20: 1-11.
Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I: Dissecting the bacterial type VI secretion system by a genome wide in silico analysis what can be learned from available microbial genomic resources?. BMC Genomics. 2009, 10: 104-10.1186/1471-2164-10-104.
Cascales E: The type VI secretion toolkit. EMBO Rep. 2008, 9: 735-741. 10.1038/embor.2008.131.
Sarris PF, Skandalis N, Kokkinidis M, Panopoulos NJ: In silico analysis reveals multiple putative type VI secretion systems and effector proteins in Pseudomonas syringae pathovars. Mol Plant Pathol. 2010, 11: 795-804.
Bönemann G, Pietrosiuk A, Mogk A: Tubules and donuts:a type VI secretion story. Mol Microbiol. 2010, 76: 815-821. 10.1111/j.1365-2958.2010.07171.x.
Bingle LEH, Bailey CM, Pallen MJ: Type VI secretion: a beginner´s guide. Curr Opinion Microbiol. 2008, 11: 3-8. 10.1016/j.mib.2008.01.006.
Filloux A, Hachain A, Bleves S: The bacterial type VI secretion machine: yet another player for protein transport across membranes. Microbiol. 2008, 154: 1570-1583. 10.1099/mic.0.2008/016840-0.
Records AR, Gross D: Sensor kinases RetS and LadS regulate Pseudomonas syringae type VI secretion and virulence factors. J Bacteriol. 2010, 192: 3584-3596. 10.1128/JB.00114-10.
Bernard CS, Brunet YR, Gueguen E, Cascales E: Nooks and crannies in type VI secretion regulation. J Bacteriol. 2010, 192: 3850-3860. 10.1128/JB.00370-10.
Thieringer HA, Jones PG, Inouye M: Cold Shock and adaptation. Bioessays. 1998, 20: 49-57. 10.1002/(SICI)1521-1878(199801)20:1<49::AID-BIES8>3.0.CO;2-N.
Ray MK: Cold stress response of low temperature adapted bacteria. 2006, In: Amere S. Sreedhar and Usha K. Srinivas editors. Stress response A molecular biology approach, 1-23.
Weber MHW, Klein W, Muller L, Niess UM, Marahiel MA: Role of the Bacillus subtilis fatty acid desaturase in membrane adaptation during cold shock. Mol Microbiol. 2001, 39: 1321-1329. 10.1111/j.1365-2958.2001.02322.x.
Weber MHW, Marahiel MA: Bacterial cold shock responses. Sci Progress. 2003, 86: 9-75. 10.3184/003685003783238707.
Wick LM, Egli T: Molecular components of physiological stress responses in Escherichia coli. Adv Biochem Engin/Biotechnol. 2004, 89: 1-45. 10.1007/b93957.
Yang L, Zhou D, Liu X, Han H, Zhan L, Gou Z, Zhang I, Qin C, Wong HC, Yang R: Cold-induced gene expression profiles of Vibrio parahemolyticus: a time-course analysis. FEMS Microbiol Lett. 2008, 291: 50-58.
Eisenbach M: Bacterial Chemotaxis. Embryonic encyclopedia of life sciences. 2001, London England: Ltd, Macmillan publisher
Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R: A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol. 2003, 50: 809-824. 10.1046/j.1365-2958.2003.03740.x.
Romantschuk M, Bamford DH: The causal agent of halo blight in bean Pseudomonas syringae pv. phaseolicola attaches to stomata via its pili. Microb Pathog. 1986, 1: 139-148. 10.1016/0882-4010(86)90016-1.
Ishimoto KS, Lory S: Formation of pilin in Pseudomonas aeruginosa requires the alternative σ factor (RpoN) of RNA polymerase. Proc Natl Acad Sci USA. 1989, 86: 1954-1957. 10.1073/pnas.86.6.1954.
Hang Y, Zhou D, Pang X, Song Y, Zhang L, Bao J, Tong Z, Wang J, Guo Z, Zhai J, Du Z, Wang X, Zhang X, Wang J, Huang O, Yang R: Microarray analysis of temperature induced transcriptome of Yersinia pestis. Microbiol Inmmunol. 2004, 48: 791-805.
Deng X, Xiao Y, Lan L, Zhou JM, Tang X: Pseudomonas syringae pv. Phaseolicola mutants compromised for type III secretion system gene induction. Mol Plant Microbe Int. 2009, 22: 964-976. 10.1094/MPMI-22-8-0964.
Burch AY, Shimada BK, Mullin SWA, Dunlap CA, Bowman MJ, Lindow SE: Pseudomonas syringae coordinates production of a motility-enabling surfactant with flagellar assembly. J Bacteriol. 2012, 194: 1287-1298. 10.1128/JB.06058-11.
Kong HS, Roberts DP, Patterson CD, Kuehne SA, Heeb S, Lakshman DK, Lydon J: Effect of overexpressing rsmA from Pseudomonas aeruginosa on virulence of select phytotozin-producing strains of P. syringae. Phytopatol. 2012, 102: 575-587. 10.1094/PHYTO-09-11-0267.
Braun V, Hantke K, Koster W: Bacterial iron transport:mechanisms, genetics and regulation. Metal Ions Biol Syst. 1998, 35: 67-145.
Visca P, Imperi F, Lamont LL: Pyoverdine siderophores:from biogenesis to biosignificance. Trends Microbiol. 2006, 10.1016/j.tim.2006.11.004.
Swingle B, Thete D, Moll M, Myers CR, Schneider DJ, Cartinhour S: Characterization of the PvdS-regulated promoter motif in Pseudomonas syringae pv. tomato DC3000 reveals regulon members and insights regarding PvdS function in other pseudomonads. Mol Microbiol. 2008, 68: 871-889. 10.1111/j.1365-2958.2008.06209.x.
Vasil ML: How we learnt about iron acquisition in Pseudomonas aeruginosa: a series of very fortunate events. Biometals. 2007, 20: 587-601. 10.1007/s10534-006-9067-2.
Chattopadhyay MK, Raghu G, Sharma YVRK, Rajasehharan MV, Shivaji S: Increase in oxidative stress at low temperature in an Antarctic bacterium. Curr Microbiol. 2011, 62: 544-546. 10.1007/s00284-010-9742-y.
Smirnova GV, Zakirova ON, Oktyabrskii ON: The role of antioxidant systems in the cold stress response of Escherichia coli. Microbiol. 2001, 70: 45-50. 10.1023/A:1004840720600.
Palma M, DeLuca D, Worgall S, Quadri LEN: Transcriptome analysis of the response of Pseudomonas aeruginosa to hydrogen peroxide. J Bacteriol. 2004, 186: 248-252. 10.1128/JB.186.1.248-252.2004.
Outten FW, Djaman O, Storz G: A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol Microbiol. 2004, 52: 861-872. 10.1111/j.1365-2958.2004.04025.x.
Zheng M, Wang X, Templeton LJ, Smulski DR, LaRosa RA, Storz G: DNA microarray mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol. 2001, 183: 4562-4570. 10.1128/JB.183.15.4562-4570.2001.
Patriquin GM, Banin E, Gilmour C, Tuchman R, Greenberg EP, Poole K: Influence of quorum sensing and iron on twitching motility and biofilm formation in Pseudomonas aeruginosa. J Bacteriol. 2008, 190: 662-671. 10.1128/JB.01473-07.
May TB, Shinabarger D, Maharaj R, Kato J, Chu L, Devault JD, Roychoudhury S, Zielinski NA, Berry A, Rothmel RK, Misra TK, Chakrabarty AM: Alginate synthesis by Pseudomonas aeruginosa: a key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clinical Microbiol Rev. 1991, 4: 191-206.
Yu J, Peñaloza-Vazquez A, Chakrabarty AM, Bender CL: Involvement of the exopolysaccharide alginate in the virulence and epiphytic fitness of Pseudomonas syringae pv. syringae. Mol Microbiol. 1999, 33: 712-720. 10.1046/j.1365-2958.1999.01516.x.
Peñaloza-Vázquez A, Kidambi SP, Chakrabarty AM, Bender CL: Characterization of the alginate biosynthetic gene cluster in Pseudomonas syringae pv. syringae. J Bacteriol. 1997, 179: 4464-4472.
Boucher JC, Schurr MJ, Deretic V: Dual regulation of mucoidy in Pseudomonas aeruginosa and sigma factor antagonism. Mol Microbiol. 2000, 36: 341-351. 10.1046/j.1365-2958.2000.01846.x.
Hickman JW, Harwood CS: Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol. 2008, 69: 376-389. 10.1111/j.1365-2958.2008.06281.x.
Block A, Li G, Qing Fu Z, Alfano JR: Phytopathogen type III effector weaponry and their plant targets. Curr Opin Plant Biol. 2008, 11: 396-403. 10.1016/j.pbi.2008.06.007.
Bronstein PA, Filiatrault MJ, Myers CR, Rutzke M, Schneider DJ, Cartinhour SW: Global transcriptional response of Pseudomonas syringae DC3000 to changes in iron bioavailability in vitro. BMC Microbiol. 2008, 8: 209-10.1186/1471-2180-8-209.
Zhao Y, Ma X, Sundin GW: Comparative genomic analysis of the pPT23A plasmid family of Pseudomonas syringae. J Bacteriol. 2005, 187: 2113-2126. 10.1128/JB.187.6.2113-2126.2005.
Wallden K, Rivera-Calzada A, Waksman G: Type IV secretion systems:versatility and diversity in function. Cell Microbiol. 2010, 12: 1203-1212. 10.1111/j.1462-5822.2010.01499.x.
Wagner R: Regulation networks. Transcription regulation in prokaryotes. 2000, USA: Oxford University Press, 264-335.
Kandror O, Golgberg AL: Trigger factor is induced upon cold shock and enhances viability of Escherichia coli at low temperatures. Proc Natl Acad Sci USA. 1997, 94: 4978-4981. 10.1073/pnas.94.10.4978.
Fonseca P, Moreno R, Rojo F: Growth of Pseudomonas putida at low temperature global transcriptomic and proteomic analyses. Env. Microbiol Rep. 2011, 10.1111/j.1758-2229.2010.00229.x.
Staskawicz BB, Panopoulos NJ: Rapid and sensitive microbiological assay for phaseolotoxin. Phytopatol. 1979, 69: 663-666. 10.1094/Phyto-69-663.
Hernández-Morales A, De La Torre-Zavala S, Ibarra-Laclette E, Hernández- Flores JL, Jofre-Garfias AE, Martínez-Antonio A, Álvarez Morales A: Transcriptional profile of Pseudomonas syringae pv phaseolicola NPS3121 in response to tissue extracts from a susceptible Phaseolus vulgaris L. cultivar. BMC Microbiol. 2009, 9: 257-10.1186/1471-2180-9-257.
Sato N, Ehira S: GenoMap a circular genome data viewer. Bioinformatics. 2003, 19: 1583-1584. 10.1093/bioinformatics/btg195.
Sambrook J, Fritsch EF, Maniatis T: Molecular cloning: a laboratory manual. 1989, New York: Cold Spring Harbor, 2
Chen WP, Kuo TT: A simple and rapid method for the preparation of gram negative bacterial genomic DNA. Nucleic Acids Res. 1993, 21: 2260-10.1093/nar/21.9.2260.
Alexander DB, Zuberer DA: Use of chrome azurol S reagents to evaluate siderophore production by rhizosphere bacteria. Boil fertile soils. 1991, 12: 39-45. 10.1007/BF00369386.
May TB, Chakghbarty AM: Isolation and assays of Pseudomonas aeruginosa alginate. 1994, Methods in enzymology: Academis Press Inc
Taguchi F, Ogawa Y, Takeuchi K, Suzuki T, Toyoda K, Shiraishi T, Ichinose Y: A homologue of the 3 oxoacyl- (acyl carrier protein) synthase III gene located in the glycosylation island of Pseudomonas syringae pv. tabaci regulates virulence factors via N-acyl homoserine lactona and fatty acid synthesis. J Bacteriol. 2006, 188: 8376-8384. 10.1128/JB.00763-06.
Acknowledgements
We are grateful to Biol. Ismael Hernández-González for analyzing the distribution of differentially regulated genes. This work was funded by grants from CONACYT to A A-M (research grant).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have declared that no competing interests exist.
Authors’ contributions
JLA-G and AH-M contributed to experimental design, performed experiments, analyzed data, and drafted the manuscript. JRP-A performed experiments and analyzed data. AA-M conceived the study, contributed to experimental design, and edited the manuscript. All authors read and approved the final manuscript.
Jackeline Lizzeta Arvizu-Gómez, Alejandro Hernández-Morales contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Arvizu-Gómez, J.L., Hernández-Morales, A., Aguilar, J.R.P. et al. Transcriptional profile of P. syringaepv. phaseolicola NPS3121 at low temperature: Physiology of phytopathogenic bacteria. BMC Microbiol 13, 81 (2013). https://doi.org/10.1186/1471-2180-13-81
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-81