
Abstract
Background
The Ferric uptake regulator (Fur) is a transcriptional regulator that controls iron homeostasis in bacteria. Although the regulatory role of Fur in Escherichia coli is well characterized, most of the studies were conducted under routine culture conditions, i.e., in ambient oxygen concentration. To reveal potentially novel aspects of the Fur regulon in Salmonella enterica serovar Typhimurium under oxygen conditions similar to that encountered in the host, we compared the transcriptional profiles of the virulent wild-type strain (ATCC 14028s) and its isogenic Δfur strain under anaerobic conditions.
Results
Microarray analysis of anaerobically grown Δfur S. Typhimurium identified 298 differentially expressed genes. Expression of several genes controlled by Fnr and NsrR appeared to be also dependent on Fur. Furthermore, Fur was required for the activity of the cytoplasmic superoxide disumutases (MnSOD and FeSOD). The regulation of FeSOD gene, sodB, occurred via small RNAs (i.e., the ryhB homologs, rfrA and rfrB) with the aid of the RNA chaperone Hfq. The transcription of sodA was increased in Δfur; however, the enzyme was inactive due to the incorporation of iron instead of manganese in SodA. Additionally, in Δfur, the expression of the gene coding for the ferritin-like protein (ftnB) was down-regulated, while the transcription of the gene coding for the nitric oxide (NO·) detoxifying flavohemoglobin (hmpA) was up-regulated. The promoters of ftnB and hmpA do not contain recognized Fur binding motifs, which indicated their probable indirect regulation by Fur. However, Fur activation of ftnB was independent of Fnr. In addition, the expression of the gene coding for the histone-like protein, H-NS (hns) was increased in Δfur. This may explain the observed down-regulation of the tdc operon, responsible for the anaerobic degradation of threonine, and ftnB in Δfur.
Conclusions
This study determined that Fur is a positive factor in ftnB regulation, while serving to repress the expression of hmpA. Furthermore, Fur is required for the proper expression and activation of the antioxidant enzymes, FeSOD and MnSOD. Finally, this work identified twenty-six new targets of Fur regulation, and demonstrates that H-NS repressed genes are down-regulated in Δfur.
Similar content being viewed by others


Background
The Ferric uptake regulator (Fur) is a metal-dependent regulator of transcription and post-transcription in bacteria, which senses metal concentration and/or the redox state of the cells (reviewed in [1]). The classical model of the regulatory role of Fur depicts transcriptional repression through ferrous iron that results in Fur-Fe2+ binding to the operator site of a target gene [2, 3]. Fur-Fe2+ binding to DNA are presumed to be homodimeric; however, multimeric complexes have been reported [4, 5]. In addition, the metal cofactor present in vivo is controversial, due to the ability of the Fur protein to bind different divalent cations, in vitro [6]. For example, Fur represses aerobactin biosynthesis using ferrous iron, cobalt, or manganese [2]. Moreover, most researchers studying Fur binding to promoter sequences, in vitro, employ manganese instead of ferrous iron due to the reactivity of ferrous iron with oxygen. However, evidence exists that Fur regulates specific genes differently in the presence of ferrous iron or manganese [7]. Fur also contains zinc for protein stability [8, 9]. This indicates that the availability of the metal cofactor to pathogens residing in the host dictates the activity of Fur.
The regulatory role of Fur has been demonstrated in numerous pathogenic and non-pathogenic organisms growing in the presence of ambient oxygen [10–19]. However, research has shown that the oxygen concentration in the host is low. For example, the oxygen sensitive [20], Fnr (Fumarate nitrate reduction) was shown to be essential for virulence in Salmonella enterica serovar Typhimurium (S. Typhimurium) [21], Shigella flexnari [22], Neisseria meningitidis [23], and Pseudomonas aeruginosa [24]. In addition, the expression of the dimeric Cu-Zn superoxide dismutase (SodCI), one of the virulence determinants in S. Typhimurium, within the J774.1 cell line was shown to be Fnr-dependent [25]. Fnr is a transcriptional regulator that is active as a homodimer and contains an oxygen labile iron sulfur cluster (4Fe-4S) [26]. Fnr can serve either as an activator or as a repressor of transcription, depending on the target gene. For instance, under anaerobic conditions, Fnr represses the cytochrome c oxidase (cyoABCDE) and the cytochrome bd complex (cydAB), while activating genes important for utilizing alternative electron acceptors such as fumarate [21]. Therefore, it is reasonable to conclude that O2 concentration within the host is low enough to activate Fnr in S. Typhimurium residing within cells of the innate immune system. This in vivo low oxygen concentration appears to be sufficient to cause a shift in the redox state of iron from ferric to ferrous. Indeed, when S. Typhimurium is within macrophages, repression of the Fur regulated iroBCDE promoter occurs regardless of the presence of the host metal transporter Nramp1 [27, 28]. This demonstrates that during intracellular growth of S. Typhimurium, the state of oxygen tension and iron valence are adequate for the activation of both Fnr and Fur, respectively. Recently, we demonstrated the role of Fur in HilA expression and virulence in S. Typhimurium, which is mediated by the negative regulation of H-NS by Fur under anaerobic conditions [29].
H-NS is a DNA binding protein that is associated with the nucleoid of Gram-negative enteric bacteria (reviewed in [30]). Deletion of hns is considered lethal unless an additional mutation occurs in either the alternative sigma factor, rpoS, or the transcription factor, phoP [31]. H-NS binding can alter the topology of DNA and influence gene regulation [32]. Typically, H-NS exhibits a repressive role in gene regulation, especially of genetic loci associated with virulence [31, 33–35]. H-NS preferentially binds to AT rich segments of DNA, which are characteristic of horizontally acquired Salmonella pathogenicity islands (SPIs) [36]. Interestingly, H-NS also represses genes associated with anaerobic metabolism including those responsible for the degradation of L-threonine, encoded by the tdc operon, and are induced under anaerobic conditions [37]. H-NS binds the tdc locus and represses its transcription [31], thereby linking amino acid catabolism with H-NS regulation. In addition, Fur is known to activate SPI1 via the activation of the positive regulators of SPI1 (i.e., HilA and HilD) [38, 39]. This activation is, in part, indirect where Fur represses the expression of hns, which represses the expression of hilA and hilD [29]. Thus, Fur indirectly activates SPI1 via its repression of hns, demonstrating that iron metabolism can influence genes regulated by H-NS.
Our goal here was to compare the transcriptome of wild-type (WT) S. Typhimurium to an isogenic strain lacking the fur gene (Δfur) in cells growing under anaerobic conditions (i.e., conditions resembling that encountered by the pathogen during infection [40]). To accomplish that goal, we used DNA microarray analysis and operon reporter fusions. We found that Fur directly or indirectly regulates 298 genes (~6.5% of the genome); of these, 49 contained a putative Fur binding site. Interestingly, Fnr controls 15 of these 49 genes [21] and 12 of the 15 genes contain putative binding sites for both Fur and Fnr. This suggests a regulatory link between oxygen and iron availability through the action of these two global regulators, Fur and Fnr. Furthermore, Fur was required for the activity of both cytoplasmic superoxide dismutases (MnSOD and FeSOD). We also found that the anaerobic expression of ftnB (encoding a ferritin-like protein) and hmpA (encoding the NO· detoxifying flavohemoglobin) was dependent on both Fur and Fnr. However, the promoters of ftnB and hmpA do not contain recognizable Fur binding motifs indicating their indirect regulation by Fur. Increased expression of H-NS, a known repressor of ftnB, tdc operon, and other genes, in Δfur may account for their activation by Fur. Finally, we have also identified twenty-six genes as new targets of Fur regulation in S. Typhimurium.
Methods
Bacterial strains, plasmids, growth conditions, and reagents
S. Typhimurium (ATCC 14028s) was used throughout this study, and for the constructing gene knockouts. Bacterial strains and plasmids used are listed in Table 1. Primers used were purchased from Integrated DNA Technologies (Coralville, IA) and are listed (Additional file 1: Table S1).
All knockouts were constructed using λ Red mediated methodologies in the host strain carrying pKD46. The cells were grown in Luria-Bertani (LB) medium to an optical density (OD600) of 0.3 at which point 50 mM arabinose was added for 90 min [41]. The culture was centrifuged, electroporated with 1 μg of purified PCR product of the gene of interest, recovered in SOC media (20 g tryptone, 5 g yeast extract, 0.5 g NaCl, per liter plus 20 mM glucose) for 3 h, plated on LB agar with the appropriate antibiotic, and incubated at 37°C. Transformants were verified by PCR followed by DNA sequencing. P22 phage transduction was used to move the mutations into the specified genetic backgrounds of S. Typhimurium 14028s. Colony PCR was used to confirm the genotype(s). Transductants were purified on Evans-Blue-Uranine (EBU) agar plates.
The medium used throughout this study was a buffered (pH = 7.4) LB containing 100 mM MOPS and 20 mM xylose (LB-MOPS-X) [21, 29, 42, 43]; where indicated, kanamycin and ampicillin were used at 55 μg ml-1 and 100 μg ml-1, respectively. Anaerobic conditions were maintained in a Coy anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) filled with anaerobic gas mixture (10% H2, 5% CO2, and 85% N2). Media were equilibrated in the anaerobic chamber for at least 48 h prior to use. Aerobic conditions were maintained by shaking at 200 RPM at 37°C in a New Brunswick gyratory water bath. Growth was determined by measuring changes in OD600 over time. The ferrous iron chelator, 2, 2' dipyridyl (dip), was purchased from Sigma-Aldrich (St. Louis, MO) and used at 200 μM. PCR reagents were from Promega (Madison, WI).
RNA isolation
For the microarray experiments, independent anaerobic cultures of 14028s and Δfur (KLM001) were used to inoculate three independent flasks (150 ml of anoxic LB-MOPS-X) for each strain. The three independent cultures of 14028s and Δfur were grown to an OD600 of 0.30 to 0.35 (~ four generations) and treated with RNAlater (Qiagen) to fix the cells and preserve the quality of the RNA as described previously [21, 43]. Total RNA was extracted and its quality was assured before aliquots of the RNA samples were stored at -80°C for use in the microarray as previously described [21, 43].
Microarray studies
Serovar Typhimurium microarray slides were prepared and used as previously described [21, 43, 44]. The SuperScript Indirect cDNA labeling system (Invitrogen, Carlsbad, CA) was used to synthesize the cDNA for the hybridizations. Each experiment consisted of two hybridizations, on two slides carried-out at 42°C overnight. Dye swapping was performed to avoid dye-associated effects on cDNA synthesis. The slides were washed at increasing stringencies and the microarrays were scanned for the Cy3 and Cy5 fluorescent signals with a ScanArray 4000 microarray scanner from GSI Lumonics (Watertown, MA). The intensity of each spot was expressed as the sum of the intensities of the pixels included in a circle positioned over the spot. The background was the sum of the intensities of an identical number of pixels surrounding the circled spot.
Data analysis
Values of Cy3 and Cy5 for each spot were normalized over the total intensity for each dye to account for differences in total intensity between the scanned images. The data from the microarray analysis were evaluated by two methods as previously described [21, 43]. Briefly, the data were evaluated by a pair-wise comparison, calculated with a two-tailed Student's t test and analyzed by the MEAN and TTEST procedures of SAS-STAT statistical software (SAS Institute, Cary, NC) the degrees of freedom for the t test were calculated as described previously [21, 43]. The t statistic was performed using the, two-tailed, heteroscedastic TTEST function of Excel software (Microsoft Corporation, Redmond, WA). The signal intensity at each spot from Δfur and the WT was analyzed and used to calculate median expression ratios and standard deviations for ORFs showing at least 2.5-fold change and p < 0.05 [21, 43].
Microarray data
The microarray data are accessible via GEO accession number GSE18441 at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18441.
Logo graph and promoter analysis
The information matrix for the generation of the Fur logo was produced using the alignment of the Escherichia coli Fur binding sequences, available at http://arep.med.harvard.edu/ecoli_matrices/. To account for slight variation in nucleotide usage between E. coli and Salmonella, a second alignment for S. Typhimurium was built using the 5' regions of the homologous genes used to build the E. coli information matrix. The new alignment was used to generate an information matrix specific for S. Typhimurium. A graphical representation of the matrix through a logo graph was obtained with Weblogo software (version 2.8.1, 18 October 2004), available at http://weblogo.berkeley.edu. The information matrix was used to scan the 5' region (from the position -400 to +50) of the genes with significant variations of transcripts using the Patser software (version 3d), available at http://rsat.ulb.ac.be/rsat/.
If a sequence corresponding to a Fur binding motif was identified, then this sequence was given a weighted score [45].
Construction of transcriptional lacZfusions
Single-copy genomic transcriptional lacZ fusions were constructed as described previously [46]. Briefly, 300 ng of pCP20 was transformed into mutant strains; cultures were transferred twice at 30°C, and checked for loss of the antibiotic marker. Plasmids with a single FRT site upstream of promoterless lacZY were transformed into mutant strains carrying pCP20 and incubated at 37°C on an LB-agar plate with kanamycin. Transformants were transferred three times at 40°C, verified by PCR, and transduced into appropriate background(s).
β-galactosidase assay and "Differential Plot" presentation of the data
The β-galactosidase assay was used to assess expression of transcriptional fusions in cultures growing at steady state. This was accomplished by 50-fold dilution of anaerobically grown overnight (~17 hr) cultures into fresh medium and once a steady state of growth was established, the cells were re-inoculated into fresh LB-MOPS-X medium to an OD600 ~0.02. β-galactosidase assays were conducted during growth and the activity (U/ml) [47] was plotted against changes in OD600 in the form of a differential plot [48, 49]; which are usually recommended for determining the rate of synthesis of an mRNA or a protein relative to the total rate of synthesis in the cell. The slope of the linear regression of this type of plot represents the differential rate of synthesis (i.e., Specific Activity, Units/OD600) during the steady state of growth. The intrinsic advantages of using this method (i.e., differential rate) over the commonly used method (i.e., one-time point assays) are well documented [50–53]. Data shown were from three independent cultures with standard deviation.
Preparation of cell-free extracts and SOD activity gels
Cultures were grown anaerobically overnight, diluted to ~0.02 OD600 in LB-MOPS-X, and cells were harvested at OD600 ~0.25. Further cell growth and de novo protein synthesis were minimized by adding chloramphenicol (50 μg ml-1) and ice to the cultures. In addition, 50 μg ml-1 chloramphenicol was included at each step of sample preparation and handling. The cultures were sealed anaerobically and the cells collected by centrifugation at 5,000 × g at 4°C. Cells were washed with phosphate buffer (pH 7.8, 50 mM potassium phosphate containing 0.1 mM EDTA, KPi), centrifuged again, and resuspended in the same buffer. Cells were sonicated on ice for 15 sec on and 30 sec off for 15 min of total sonication time. Cell debris was cleared by centrifugation at 19,000 × g for 30 min at 4°C, and the supernatant was dialyzed against KPi in dialysis membranes with an 8,000 molecular weight cut-off. Dialyzed cell-free extracts were centrifuged at 20,000 × g for 30 min at 4°C, and the supernatant was stored at -80°C until use. Protein concentration was determined by the Lowry method [54]. Superoxide dismutase activity gels were performed using native 10% acrylamide gels as described previously [55].
Fumarate reductase activity
Fumarate reductase activity (FRD) was assayed from cell-free extracts as described previously [56]. Briefly, cells were grown, cell-free extracts were prepared as described above, and the fumarate dependent oxidation of reduced benzyl viologen was determined. Specific activity of FRD is expressed as μmole of reduced benzyl viologen oxidized per minute per milligram of total protein.
Measurements of total [Mn]
Independent anaerobic cultures were diluted to OD600 ~0.02 and grown until OD600 0.35 in a Coy anaerobic chamber. Chloramphenicol was added at 50 μg ml-1, samples were sealed anaerobically, and centrifuged at 12,000 × g for 20 min at 4°C. Samples were washed with KPi as above, centrifuged, and resuspended in 2 ml of buffer. Samples were dried and treated with 3 M nitric acid overnight at room temperature then quickly boiled. Total manganese content was determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) at North Carolina State University Analytical Service Laboratory. Total manganese and iron was measured in LB medium as above using a 5X concentration of medium.
Results
Growth of Δfurunder anaerobic and aerobic conditions
Iron is an essential element for redox reactions in biology. However, it is an important factor in oxygen toxicity due to its involvement in hydroxyl radicals (HO·) formation via Fenton chemistry [57]. Therefore, we compared the effects of a deletion of fur on growth kinetics under both anaerobic and aerobic conditions. Data in Figure 1 demonstrate that Δfur was not compromised in its growth kinetics under either anaerobic or aerobic conditions.

Growth kinetics of Δ fur (black square) compared to 14028s (white square). Cells were grown in LB-MOPS-X medium as described in Methods; (A) Anaerobic growth; (B) Aerobic growth.
Effect of Fur on the anaerobic transcriptome of S. Typhimurium
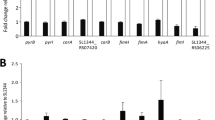
Under anaerobic conditions, the absence of fur resulted in the differential expression of 298 genes (Additional File 2: Table S2). These genes were organized by Cluster of Orthologous Groups (COGs) and the numbers of genes within each COG are shown in Table 2. The absence of fur resulted in increased expression (i.e., Fur acted as a repressor) of 226 genes. However, the absence of Fur resulted in decreased expression (i.e., Fur acted as an activator) of 72 genes, most likely via an indirect mechanism.
A Fur information matrix, specific for S. Typhimurium, was generated (Figure 2), and used to scan the upstream regions of the 298 genes identified as differentially expressed in response to deletion of fur. We identified 49 genes that contain a putative Fur binding site (Table 3 - columns 1 & 2 and Additional file 2: Table S2).

Logo graph of the information matrix from the alignment of Fur-regulated genes in S. Typhimurium. The height of each column of characters represents information, measured in bits, for that specific position and the height of each individual character represents the frequency of each nucleotide.
a. Fur as a repressor
Genes associated with metal homeostasis were up-regulated in Δfur. These included the well characterized genes/operons involved in iron homeostasis (i.e., entABEC, iroBCDE, iroN, fes, tonB, fepA, bfr, bfd), Mn2+ transport genes (i.e., sitABC), and copper resistance (i.e., cutC) [58–65] (Additional file 2: Table S2).
Expressions of genes involved in xylose metabolism (xylBR) were increased 3.7 and 2.9-fold, respectively, in Δfur relative to the WT (Additional file 2: Table S2). In addition, the glycolytic genes pfkA and gpmA were 3.3-and 5.6-fold higher in Δfur, respectively (Additional file 2: Table S2). Two genes, STM1586 (coding for a putative periplasmic protein) and sitA were up-regulated 76.1 and 53.8-fold, respectively, in Δfur (Additional file 2: Table S2). These two genes exhibited the highest differential expression in Δfur. Intriguingly, the microarray data showed that the gene for adenloysuccinate synthetase (purA), which is required for adenosine 5' monophosphate synthesis, was up-regulated 3.5-fold in Δfur. Incidentally, purA mutants are known to be highly attenuated and have been used in developing in vivo expression technology (IVET) to detect promoters activated during S. Typhimurium infection [66, 67].
Transcription of the cytochrome-o ubiquinol oxidase operon (cyoABCDE) and the high affinity cytochrome-d terminal oxidase genes (cydAB) was repressed by Fur (Additional file 2: Table S2). Interestingly, aerobic expression of cydAB is repressed by H-NS, which is relieved by the response regulator ArcA [68]. In addition, we detected increased expression of hns in Δfur (Additional file 2: Table S2), and earlier work detected in vivo binding of Fur to the upstream region of hns [29]; this strongly indicates that Fur directly represses hns under anaerobic conditions. How or if H-NS may interact in the anaerobic regulation of cydAB under our conditions is unknown, since the repression of cydAB by H-NS does not appear to occur under anaerobic conditions [68].
Genes associated with DNA repair and purine metabolism (nrdAB, nth, recA, and nei) were repressed by Fur under anaerobic conditions (Additional file 2: Table S2), thus implicating Fur as a regulator of DNA repair and de novo synthesis. Fur was found to repress ydiE (STM1346) and a putative Fur binding site was found upstream of the start codon, where the expression of the gene was 7.4-fold higher in the mutant than in the wild-type (Additional file 2: Table S2). In Yersinia enterocolitica, YdiE has a conserved HemP (COG4256) domain, and is encoded within the hemin uptake operon [69]. Although S. Typhimurium is not known to utilize host's heme, previous work has established a Fur binding site upstream of ydiE and hemP in S. Typhimurium and Y. enterocolitica, respectively [16, 69]. This indicates that our bioinformatic analyses indeed agree with experimentally identified Fur binding sites.
b. Fur as an activator
Anaerobic transcription of the fumarate reductase (frdABD) operon and the aspartase gene (aspA) was significantly lower in Δfur (i.e., Fur is serving as an activator); however, the genes coding for the alpha and beta subunits of succinyl-CoA synthetase (sucCD) were up-regulated 4.1 and 2.7-fold, respectively (Additional file 2: Table S2). These genes (i.e., frdABD, aspA, sucCD) and fumAB (fumarate hydratase) are members of the reductive branch of the TCA cycle. We assayed for fumarate reductase (FRD) in cell-free extracts from anaerobic cultures and found that Fur is required for the anaerobic transcription and activity of FRD in S. Typhimurium (Additional file 3: Table S3). In E. coli, the transport of C4-dicarboxylates occurs via two seemingly redundant genes encoded by dcuA and dcuB [70]. In the present study, the dcuB-fumB operon was unaffected by Fur, while the aspA-dcuA operon was significantly down regulated in Δfur and both genes contained a putative Fur box 5' of the start codon (Additional file 2: Table S2).
Genes involved in anaerobic respiration (dmsABC) and ethanolamine utilization (eutSPQTDMEJGHABCLK) were activated by Fur (Additional file 2: Table S2). The mechanism for reduced expression of dmsABC is unclear. Ethanolamine is a significant source of carbon and nitrogen during Salmonella infection [71].
One metabolic pathway that appears impacted by Fur is that required for glycerol metabolism. The genes for glycerol metabolism are located throughout the genome. For instance, glpQT and glpABC are divergently transcribed in two predicted operons. All of these genes were significantly down regulated in Δfur (Additional file 2: Table S2). Furthermore, glpD, and glpKF were all down regulated in Δfur (Additional file 2: Table S2). The down-regulation of these genes suggests that the Δfur strain may be unable to utilize glycerol or transport glycerol- 3 phosphate. The mechanism of this regulation is unclear, but the absence of Fur binding sites in the promoters of any of these genes suggests an indirect mode of regulation. The contribution of glycerol metabolism to infection is unknown.
Another metabolic pathway, the tdc operon (required for the anaerobic transport and metabolism of L-threonine and L-serine [72, 73]) was activated by Fur. The genes in this operon (tdcBCDEG) are activated by tdcA [74]. TdcA is a member of the LysR family of transcriptional activators [75]. Our data showed that the expression of all genes in this operon, tdcABCDEG, was significantly down-regulated in Δfur (Additional file 2: Table S2). However, a Fur binding site was not identified in the promoters of any of the genes in the tdc operon, suggesting its indirect regulation by Fur. Importantly, H-NS is known to directly bind and repress this operon [31, 76]. Therefore, the increased expression of hns in Δfur (Additional file 2: Table S2), may account for the observed effect of Fur on the tdc operon. Mutations in the tdc operon have been shown to reduce invasion and virulence in S. Typhimurium [77, 78]. In addition to the reduced expression of the eut operon, the reduced expression of the tdc operon and hilA may contribute to the observed attenuation of the Δfur strain of S. Typhimurium [29, 79].
Role of Fur in regulation of antioxidant genes
Reactive oxygen and nitrogen species (ROS and RNS, respectively) are important host defense responses during bacterial infection. Our array data (Additional file 2: Table S2) revealed differential regulation of some important antioxidant genes whose products are essential for protecting the cells against ROS and RNS (i.e., superoxide dismutases, ferritin-like protein, and flavohemoglobin). Therefore, we decided to study the expression of these genes in greater detail.
a. Regulation of sodA and sodB
There is plethora of information about the regulation of sodA and sodB in E. coli [80–85], but there is little knowledge about the regulation of these genes in S. Typhimurium [86]. In the present study, the microarray data showed that the anaerobic expression of sodA and sodB in Δfur was > 9-fold higher and > 3-fold lower, respectively, than in the parent WT strain (Additional file 2: Table S2). SodA (MnSOD) and SodB (FeSOD) are the cytosolic superoxide dismutases of S. Typhimurium and they require the cofactors manganese and iron, respectively. These SODs are homodimers, and are fully functional when metalated with the appropriate metals (i.e., manganese for SodA and iron for SodB). However, a heterodimer consisting of SodA(Mn)/SodB(Fe) can still exhibit SOD activity, albeit at a reduced level compared to the homodimer [87]. Thus, in order to see an active hybrid SOD, both SodA and SodB must be expressed. Data in Figure 3A demonstrated that, as in anaerobic E. coli, the WT strain (Lane 1) lacked the activity of both Mn- and Hybrid-SODs, but possessed an active FeSOD. However, Δfur (Figure 3A - Lane 2) was devoid of all three SOD-isozymes. The lack of FeSOD in Δfur was of no surprise, as previous studies in E. coli [83, 84] have established that Fur is indirectly required for the translation of sodB via its repression of the small RNA, ryhB, which works in conjunction with the RNA chaperon protein, Hfq [88, 89]. Indeed, a strain harboring deletions in both Fur and Hfq (ΔfurΔhfq) resulted in restoration of SodB activity (Figure 3A - Lane 4). Furthermore, the high degree of sequence identity in the promoter and the gene sequence of ryhB of E. coli with the two ryhB-like small RNAs, rfrA and rfr of S. Typhimurium [39], suggested that the regulation of sodB in S. Typhimurium is similar to that reported in E. coli [88, 89]. Interestingly, expression of the hybrid SOD appears up-regulated in Δhfq and ΔfurΔhfq (Figure 3A - Lane 3 and 4). The reason for this is unclear, but may be due to the activation of the Hfq-binding small RNA (fnrS) by Fnr, which subsequently represses the expression of sodA [90, 91].

Effects of Fur, Hfq, and manganese on the activity of superoxide dismutases. (A) Effects of Fur and Hfq - Cell-free extracts from anaerobically grown cultures (14028s, Δfur, Δhfq, and ΔfurΔhfq) were prepared as described in the Methods. Equal protein (125 μg/ml) was loaded and following electrophoresis the gel was stained for SOD activity. Lane 1 - 14028s; lane 2 - Δfur; lane 3 - Δhfq; lane 4 - ΔfurΔhfq. (B) Effects of Fur and MnCl2 - Cell-free extracts were prepared from anaerobically grown cultures as in (A) except that 1 mM MnCl2 was added to the media. Equal protein (125 μg/lane) was loaded, elecrophoresed, and stained for SOD as in (A). Lane 1, 14028s + MnCl2; lane 2, 14028s + pfur-ha; lane 3, 14028s + MnCl2 + pfur-ha; lane 4, Δfur + MnCl2; lane 5, Δfur + pfur-ha; lane 6, Δfur + MnCl2 + pfur-ha.
The WT strain of S. Typhimurium possessed neither an active SodA (MnSOD) nor the hybrid enzyme (SodA/SodB), which is not surprising since this is normally the case in WT E. coli [92]. What was surprising is the lack of MnSOD activity in the anaerobic cell-free extracts from Δfur (Figure 3A - Lane 2) in spite of the > 9-fold increase in the transcription of sodA (Additional file 2: Table S2). Therefore, we reasoned that the increased intracellular concentration of free iron in Δfur [93] could result in competition of iron with manganese for the active site of SodA. This would lead to the formation of a non-active form of the enzyme, i.e., SodA-Fe instead of the active SodA-Mn (MnSOD). Analysis of total iron and manganese concentrations in our media showed that it contained ~40-fold more iron than manganese (i.e., ~7.5 μM iron vs. ~0.2 μM manganese). Additionally, the manganese content of anaerobic cultures of the parent strain and of the Δfur strain were low, 0.09 ± 0.01 and 0.08 ± 0.04 μmoles manganese per gram of dry weight, respectively. Therefore, we supplemented the growth media with 1 mM MnCl2 and determined the SOD activities (Figure 3B). If our reasoning was correct, we expected that excess Mn2+ added to the growth media would reveal increased MnSOD activity in Δfur. Indeed, this was the case, as a dramatic increase in MnSOD was observed in Δfur, but not in the parent strain (Figure 3B - lanes 1 vs.4). Also, cultures grown in presence of 1 mM MnCl2 contained 47.2 ± 2.7 and 48.8 ± 2.0 μmoles of manganese per gram of dry weight for the parent strain and for Δfur, respectively. Altered MnSOD activity in Δfur was due entirely to the lack of a functional fur gene since the introduction of a plasmid carrying the fur gene (i.e., pfur-ha) diminished MnSOD activity to that of the parent strain (Figure 3B - Lane 1 and 6). In addition, the plasmid pfur-ha restored FeSOD activity (Figure 3A - lane 5) as well as the phenotypic appearance of the WT strain observed on a Tris buffered chrome azurol agar plates (CAS plates) [94] containing 0.3% xylose [29]. These results indicated that increased transcription of sodA in Δfur did not result in a corresponding increased MnSOD activity due to the excess intracellular free iron and that the addition of Mn2+ negated this effect. On the other hand, the inclusion of excess Mn2+ in the growth medium of the parent strain did not increase MnSOD activity, which indicated that Mn2+ was not a signal for sodA induction. Furthermore, these findings demonstrated an important aspect of metalloenzyme regulation, i.e., the availability of the correct cofactor has a profound impact on enzyme activity.
b. Regulation of ftnB
Microarray data (Additional file 2: Table S2) revealed a 7-fold reduction in the expression of ftnB in Δfur as compared to the parent strain. The expression of ftnB was shown to be activated by Fnr [21]. Therefore, we used a chromosomal ftnB-lacZ transcriptional fusion in Δfur and in Δfnr genetic backgrounds to determine the contribution of each regulator in the expression of ftnB. The deletion of fur reduced the aerobic rate of synthesis of the reporter gene by > 2-fold compared to the parent strain (Figure 4A). 2, 2' dipyridyl (dip) reduced the rate of synthesis of the reporter gene in aerobic conditions (Figure 4A). Although induction of the reporter fusion occurred earlier in the growth phase with dip treated cultures, the rate of synthesis was reduced compared to untreated parent strain. This indicates inhibition by dip (Figure 4A). As expected, the oxygen sensitive regulator Fnr did not impact regulation of ftnB in aerobic conditions (Figure 4A). This indicated that Fur is required for ftnB expression, independent of Fnr. Data in Figure 4B show that the absence of fur resulted in a 2-fold reduction in the rate of synthesis (U/OD600) of ftnB-lacZ under anaerobic conditions. Furthermore, the ferrous iron chelator, dip, reduced the rate of anaerobic synthesis of ftnB-lacZ in the WT strain by > 2-fold (Figure 4B). In Δfur, the rate of synthesis was further reduced (> 10-fold) when compared to the WT parent strain treated with dip (Figure 4B). In addition, the rate of synthesis in the parent strain was greatest under anaerobic conditions due to the active roles of both Fnr and Fur (Figure 4). Collectively, full expression of ftnB is dependent on Fur in aerobic and anaerobic conditions, whereas Fnr is a strong activator in the absence of O2.

Effects of Fur, Fnr and iron chelation on transcription of ftnB. Transcriptional ftnB-lacZ activity was determined in 14028s (squares), Δfur (circles), and Δfnr (triangles) under (A) anaerobic, and (B) aerobic conditions in LB-MOPS-X media without (open symbols) and with (closed symbols) 200 μM of 2, 2' dipyridyl. β-galactosidase assay was conducted throughout the growth of the culture and activity is presented in the form of differential plots with representative data shown in (A) and (B). Best-fit lines, calculated as described in the Methods, are shown in (A) and (B). For (A) and (B), representative data are shown with the differential rate of synthesis (U/OD600) ± standard deviations from three independent experiments listed.
c. Regulation of hmpA
The gene coding for the flavohemoglobin (hmpA), a NO· detoxifying protein [95–98], was differentially expressed in Δfur (Additional file 2: Table S2). Expression of hmpA is repressed by Fnr and another DNA binding protein that contains an iron sulfur cluster, NsrR [21, 95–97, 99]. Repression of hmpA by two regulators that are sensitive to RNS allows derepression of this gene under conditions of increased RNS. Indeed, regulation of hmpA-lacZ was induced ~80-fold by the nitrosating agent sodium nitroprusside in aerobic conditions (B. Troxell and H.M. Hassan, unpublished data). Under anaerobic conditions, hmpA was up-regulated 4-fold in Δfur. Thus, we examined its anaerobic regulation with a chromosomal hmpA-lacZ transcriptional fusion. Figure 5 shows that the WT exhibited very little expression of hmpA-lacZ under anaerobic conditions (Figure 5A); suggesting regulation may be oxygen dependent. Indeed, expression was ~14-fold higher under aerobic conditions than anaerobic conditions (B. Troxell and H.M. Hassan, unpublished data). However, the addition of the iron chelator, dip, resulted in an increased rate of synthesis ~81-fold (Figure 5A). The increased expression of hmpA-lacZ by the addition of dip could have been due to inactivation of Fnr, Fur, and/or NsrR. We narrowed our focus to the roles of Fur and Fnr in regulation of this gene. In Δfur, the reporter activity was up-regulated > 9-fold (Figure 5A), which confirmed the microarray data. The addition of dip increased the rate of synthesis by 25-fold in Δfur. One known repressor of hmpA is Fnr [21, 95–97]. Therefore, we combined the fur and the fnr deletions (ΔfurΔfnr) in the hmpA-lacZ background to determine the role of Fur and Fnr in the regulation of hmpA. Deletion of fnr increased the rate of hmpA-lacZ synthesis by 216-fold as compared to the parent strain (Figure 5B). The synthesis of hmpA-lacZ in the Δfnr mutant background was similar to that seen in the Δfur treated with dip (i.e., 1253 ± 107 and 1403 ± 280 - U/OD600). The lack of an obvious Fur binding motif upstream of hmpA indicates that reporter activity seen in Δfur was likely indirect. The combined deletion of fur and fnr in the hmpA-lacZ strain increased the rate of synthesis 746-fold as compared to the WT strain (i.e., 4328 ± 90 vs. 5.8 ± 2.4 - U/OD600) (Figure 5). Thus, the rate of synthesis of hmpA-lacZ in ΔfurΔfnr was ~3.5-fold higher than the rate of synthesis in Δfnr (i.e., 4328 ± 90 vs. 1253 ± 107 - U/OD600). Since we did not identify a discernable Fur binding site in hmpA, the fact that there is no published report showing Fur binding to the regulatory region of hmpA, and that the expression of hmpA-lacZ in ΔfurΔfnr was ~3.5-fold higher than in Δfnr demonstrates that under anaerobic conditions, Fur is indirectly regulating hmpA-lacZ independent of Fnr.

Fur and Fnr control transcription of hmpA. (A) The transcriptional hmpA-lacZ activity was determined in 14028s and Δfur under anaerobic conditions. The iron chelator 2, 2' dipyridyl (dip) was used at 200 μM; and (B) β-galactosidase activity was measured in Δfnr and ΔfurΔfnr backgrounds under anaerobic conditions - the best-fit lines are shown. For (A) and (B) representative data are shown with the differential rate of synthesis (U/OD600) ± standard deviations from three independent experiments listed.
Identification of new Fur targets
Table 3 shows genes differentially regulated in Δfur that contain a putative Fur binding site located within -400 to +50 nucleotides relative to the translational start site. The putative translocase subunit, yajC, was up-regulated 3.2-fold in Δfur. This gene is predicted to be in the Sec-dependent pathway of protein export. At least one other gene of the Sec-dependent pathway of protein export was up-regulated in Δfur, secY. This gene, secY, is a direct target of Fur regulation in Neisseria meningitides [100, 101]. Indeed, we detected a putative Fur binding site upstream of secY (Additional file 2: Table S2). The role of yajC during infection is unknown, but our results suggest Fur controls Sec-dependent protein secretion.
NrdR is a global transcriptional regulator that controls expression of oxygen-dependent and independent ribonucleotide reductases [102–104]. Expression of nrdR was up-regulated in Δfur and a putative Fur binding site was identified. Although, deletion of fur results in up-regulation of nrdHIEF [105], a class Ib ribonucleotide reductase, we did not detect increased expression of this operon in our conditions. However, we did detect up-regulation of the class Ia ribonucleotide reductase, nrdAB, in Δfur (Additional file 2: Table S2). The class III oxygen sensitive ribonucleotide reductase, encoded by nrdDG, is encoded in an operon. Expression of nrdD, the first gene of this operon, was down-regulated in Δfur 2.5-fold. (Additional file 2: Table S2). Our data indicate that Fur controls the class Ib and III ribonucleotide reductases, either directly or indirectly, under anaerobic conditions.
A putative dehydrogenase (STM1133) was down-regulated 4.2-fold in the Δfur (Table 3). This gene contains a putative Fur binding site on the reverse DNA strand. STM1133 is the final gene in an apparent four gene operon of unknown function (STM1130-1133). The first gene of this operon, STM1130, was also down-regulated 7.9-fold in Δfur (Additional file 2: Table S2); however, a Fur binding site was not identified upstream of STM1130. Interestingly, this operon is composed of the putative N-acetylneuraminic acid mutarotase (STM1130), a putative outer membrane protein (STM1131), a putative sialic acid transporter (STM1132), and a putative NAD (P) binding dehydrogenase (STM1133). Thus, our results suggest Fur controls at least a portion of this operon that may be localized to the bacterial membrane. The importance of these genes during infection is unknown.
Several putative genes appear to be under direct control of Fur. Genes that exhibited reduced expression in Δfur were the putative universal stress protein encoded by ynaF, the putative glutamate synthase (STM2186), the putative sugar kinase (STM3600), and the putative lipoprotein (STM3690). The putative Fur binding site for ynaF and STM3600 is located on the reverse strand for these genes. The mechanism of Fur activation of these putative genes is unknown. In addition, several putative genes exhibited up-regulation in Δfur. A putative glutamic dehydrogenase (STM1795), a putative glutaredoxin (yffB), and a putative protein (yggU), were all up-regulated in Δfur. Interestingly, yffB is predicted to be a glutathione-dependent thiol reductase. The contribution of these genes to infection is unknown.
The TonB siderophore receptor gene, iroN, was up-regulated 9.1-fold in Δfur. Despite the widespread study of siderophores (salmochelins) in Salmonella virulence, we were unable to find any published report that Fur represses iroN. Although Fur repression of the iroBCDE loci is known [59], iroN is encoded downstream of this operon and is transcribed in the opposite orientation. Our results confirm the prediction by Baumler et al that iroN is regulated by Fur [58].
Discussion
Iron is essential in most pathogenic bacteria, which compete rigorously with the host for this element. S. Typhimurium is no exception. The 17-kDa transcriptional regulator, Fur, plays an important role in bacterial iron homeostasis. Although publications of Fur regulation in E. coli and other bacteria are numerous, this is the first report on the global role of Fur in anaerobically grown S. Typhimurium. Indeed, anaerobic metabolism has been shown to be important for pathogens and pathogenesis [21–24, 29].
In this study, we found that, under anaerobic conditions, Fur directly or indirectly affected the expression of 298 genes (Additional file 2: Table S2). A putative Fur binding motif was identified in 49 genes (Table 4. column #1). Also, Table 4 shows evidence of published data demonstrating the role of Fur in their regulation (column #3) and published experimental evidence for Fur binding to the regulatory region of these genes (column #4). The role of other co-regulators is also shown (Table 4, column #5). Interestingly, twelve of the 49 genes contained the binding motifs for both Fnr and Fur (Additional file 4: Table S4).
The appropriate metal cofactor was shown to be essential for detection of MnSOD activity, in spite of the 9-fold increase in sodA transcript for Δfur. Therefore, genetic backgrounds that alter the steady-state [Mn2+] or its competitor [Fe2+] may have dramatic effects on MnSOD activity. Indeed, we were only able to discern the role of Fur in sodA and MnSOD expression with the addition of excess MnCl2 to the growth media. These data are summarized in Figure 6, which depicts the transcriptional, translational, and post-translational role of Fur in sodA and sodB. This implies that disruption of iron homeostasis is likely to have a two-pronged effect, increase in Fenton chemistry and a decrease in MnSOD activity due to iron overload. It appears that the inhibition of MnSOD by iron is evolutionarily conserved. Thus, the mitochondrial Mn2+-cofactored SOD2 has been shown to be inactivated in a similar manner when iron homeostasis was disrupted in yeast [106]. In addition, supplementation of the medium with Mn2+ reduced oxidative stress in a murine model of hemochromatosis [107]. It is unknown if this is due to enhanced MnSOD or if Mn2+ supplementation reduces oxidative stress in other pathological states of altered iron homeostasis.

Role of Fur in the transcriptional, translational and post-translational regulation of sodA and sodB. (A) Repression of sodA by Fur is depicted in addition to the role of Fur in iron homeostasis. Iron is known to bind to the active site of MnSODs that leads to inactivation of the enzyme [106, 124]. Increased expression of MnSOD was detected only when excess Mn2+ was added to the media in order to out compete the Fe2+. Deletion of fur under iron replete conditions results in increase transcription of sodA, but incorportation of Fe2+ into the active site of SodA resulting in SodA-Fe and an inactive enzyme. Addition of excess Mn2+ to the culture media can out compete Fe2+ for the active site of SodA resulting in SodA-Mn and an active enzyme. (B) Indirect regulation of SodB by Fur in S. Typhimurium. The small RNAs rfrA and rfrB of S. Typhimurium are likely to function as their homolog ryhB in E. coli in regards to SodB regulation [88]. Our data confirms the Hfq-dependent function of reduced FeSOD activity in Δfur. Previous work confirmed the role of Hfq and Fur in SodB expression [39]. Deletion of fur results in increased transcription of the sRNAs (rfrA and rfrB) that can pair with mRNA of sodB in an Hfq-dependent fashion and result in the degradation of sodB mRNA. However, a combined deletion of hfq in Δfur results in loss of rfrAB-mediated degradation of sodB, and results in the synthesis of SodB protein that gets activated to FeSOD in the presence of Fe2+.
Our decision to further study ftnB and hmpA was due to our previous findings, where we found that ftnB and hmpA were activated and repressed by Fnr, respectively [21]. The Fnr-dependent expression of ftnB was apparent from the reduced activity in Δfnr under anaerobic conditions, and the reduced activity in the WT strain in presence of oxygen. In addition, iron chelation and the deletion of fur reduced ftnB expression regardless of the oxygen tension. These results indicated that Fur controlled regulation of ftnB is independent of Fnr. Our results are in agreement with earlier work that demonstrated dependence of ftnB expression on Fur [15]. However, they are contrary to a previous report, which determined that Fur exhibited a repressive role on ftnB expression [79]. The reason for this discrepancy is unclear. It is evident from work reported herein and in a previous study in E. coli that ftnB exhibits a strong dependence on low O2 conditions [108]. Furthermore, the earlier study [108] determined that Fnr bound the promoter of ftnB in E. coli and that the Fnr binding site was further upstream than in known Fnr regulated genes. The same investigators [108], postulated that Fnr was unable to induce ftnB and that other regulators were required. However, we have determined that Fnr alone contributes to the activation of ftnB and that Fur is required for full induction of the gene, with Fnr exhibiting a more pronounced role. The lack of a predicted Fur binding site in ftnB indicated that Fur regulation was indirect. The following scenario is proposed to explain these findings and to suggest that the observed regulation of ftnB by Fur is mediated by the histone-like protein H-NS. First, the microarray data showed that Fur negatively regulates the expression of hns and has a predicted Fur binding site (Table 3). Second, we recently demonstrated that Fur binds upstream of hns in a metal dependent fashion [29]. Third, whole genome ChIP analysis demonstrated that H-NS binds to ftnB and the expression of ftnB is up-regulated in the absence of hns [31]. Fourth, the tdc operon is a known target for H-NS repression [31, 76] and was significantly reduced in the absence of fur. Therefore, we propose that the positive regulation ftnB by Fur is mediated by the negative regulation of hns by Fur. Thus removal of Fur (i.e., as in Δfur) results in repression of ftnB by H-NS (see Figure 7). A second possibility is reduced Fnr function (or an additional activator) in Δfur since several Fnr regulated genes were differentially expressed in Δfur. However, our data rule-out this possibility in ftnB regulation by showing the involvement of Fur in the regulation of ftnB under aerobic conditions, where Fnr is inactive.

Representation depicting the role of Fur and H-NS in the regulation of ftnB and the tdc operon. H-NS confirmed binding sites and transcriptional repression [31] were compared with our microarray data and Fur repression of hns [29]. Collectively, the data indicate that Fur-dependent activation of ftnB and the tdc operon may be due to the increased expression of H-NS in Δfur, which represses ftnB and the tdc operon. Thus, under Fur active conditions (left panel), hns is repressed by Fur thereby blocking H-NS repression of ftnB and the tdc operon (signified by the circle with an "X"). While under Fur inactive conditions (right panel), the overexpression of H-NS results in the repression of ftnB and the tdc operon under anaerobic conditions.
H-NS controls diverse functions within the cell and forms complex structures when binding DNA that indicates a central role in DNA topology [109–113]. Similar to Fur, H-NS is a repressor of transcription [31, 34, 35, 114]. This implies that genes controlled by H-NS are regulated by iron through Fur. This interaction also demonstrates interaction between two regulators (Fur and H-NS) functioning in highly conserved physiological events, regulating a potentially toxic, but needed metal and regulating foreign DNA in a concerted manner. Thus, our results provided additional insight into iron-dependent regulation of H-NS.
Another gene regulated by Fnr or Fur was the NO· detoxifying flavohemoglobin protein encoded by the hmpA. This gene (hmpA) is repressed by Fnr and contained a putative Fnr binding site, but did not contain a predicted Fur binding site [21, 95, 96]. Previous work determined that Fur was a repressor of hmpA [115]. However, it was later revealed that the reporter fusion was to the Fur repressed iroC and not to the hmpA [116]. Additionally, a previous report did not reveal a role for Fur in regulation of hmpA [97], while two other studies found a modest effect of Fur on hmpA expression [98, 117]. NsrR is another repressor of hmpA [97]. Thus, hmpA is repressed by two regulators that contain an iron-sulfur cluster. Despite contradictory reports, increased hmpA expression was detected in Δfur. Our initial hypothesis was that this was due to reduced Fnr function in Δfur. To support this hypothesis, we expected reporter activity to be similar in Δfnr and ΔfurΔfnr backgrounds. However, our results did not support this initial hypothesis since ΔfurΔfnr exhibited ~3.5-fold increased expression compared to Δfnr; indicating that Fur regulation was Fnr-independent.
A striking finding was the shared regulation of several genes by Fur and Fnr. Microarray and bioinformatic studies indicated that 12 of the 298 Fur-dependent genes contained a predicted binding site for both Fur and Fnr. Thus, these two global regulators may be directly involved in regulation of these 12 genes (Additional file 4: Table S4). The expression data indicated that Fur and Fnr cooperate in the regulation of these 12 genes. For instance, each gene was regulated in the same manner in Δfur or Δfnr; a gene activated by Fur was also activated by Fnr. Lastly, our investigations indicate that Fur indirectly regulates genes that are under control of Fnr or additional regulators with an iron sulfur cluster (i.e., ftnB and hmpA). Furthermore, the observed reduced expression of the ethanolamine operon, frdABD, and dmsABC in Δfur, suggest altered regulation of operons induced under anaerobiosis (Additional file 2: Table S2). Thus, Fur is an activator of genes that are typically induced under anaerobic conditions. Ethanolamine utilization within the host is important for S. Typhimurium and the Gram-positive pathogen Listeria monocytogenes [118, 119]. In addition, Fnr is an activator of the frd and dms operons, which are responsible for anaerobic utilization of fumarate and dimethyl sulfide as alternative electron acceptors, respectively [120–123]. Our study of the anaerobic expression of hmpA suggests that it is regulated by Fur, independent of Fnr. Clearly, these results suggest Fnr is functional in Δfur and that Fur is regulating genes of anaerobic metabolism (eut, frd, and dms operons) through an unknown mechanism.
Conclusions
We demonstrated that Fur is an activator of ftnB in S. Typhimurium, which is likely due to the de-repression of hns in Δfur. The strong dependence of ftnB expression on O2 indicates that Fnr is crucial in its regulation. Additionally, we presented evidence that Fur indirectly controls hmpA, independent of Fnr. We determined that Fur represses sodA transcription, but is required for the maturation of SodA into an active enzyme, MnSOD. Finally, we identified new target genes regulated by Fur in S. Typhimurium, and our data support the increasing evidence of enhanced H-NS expression in Δfur.
References
Lee JW, Helmann JD: Functional specialization within the Fur family of metalloregulators. Biometals. 2007, 20 (3-4): 485-499. 10.1007/s10534-006-9070-7.
Bagg A, Neilands JB: Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry. 1987, 26 (17): 5471-5477. 10.1021/bi00391a039.
Neilands JB: Siderophores. Arch Biochem Biophys. 1993, 302 (1): 1-3. 10.1006/abbi.1993.1172.
Baichoo N, Helmann JD: Recognition of DNA by Fur: a reinterpretation of the Fur box consensus sequence. J Bacteriol. 2002, 184 (21): 5826-5832. 10.1128/JB.184.21.5826-5832.2002.
Lavrrar JL, Christoffersen CA, McIntosh MA: Fur-DNA interactions at the bidirectional fepDGC-entS promoter region in Escherichia coli. J Mol Biol. 2002, 322 (5): 983-995. 10.1016/S0022-2836(02)00849-5.
Mills SA, Marletta MA: Metal binding characteristics and role of iron oxidation in the ferric uptake regulator from Escherichia coli. Biochemistry. 2005, 44 (41): 13553-13559. 10.1021/bi0507579.
Privalle CT, Fridovich I: Iron specificity of the Fur-dependent regulation of the biosynthesis of the manganese-containing superoxide dismutase in Escherichia coli. J Biol Chem. 1993, 268 (7): 5178-5181.
Jacquamet L, Aberdam D, Adrait A, Hazemann JL, Latour JM, Michaud-Soret I: X-ray absorption spectroscopy of a new zinc site in the fur protein from Escherichia coli. Biochemistry. 1998, 37 (8): 2564-2571. 10.1021/bi9721344.
Althaus EW, Outten CE, Olson KE, Cao H, O'Halloran TV: The ferric uptake regulation (Fur) repressor is a zinc metalloprotein. Biochemistry. 1999, 38 (20): 6559-6569. 10.1021/bi982788s.
Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD: The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci USA. 2008, 105 (33): 11927-11932. 10.1073/pnas.0711752105.
Stojiljkovic I, Baumler AJ, Hantke K: Fur regulon in Gram-negative bacteria. Identification and characterization of new iron-regulated Escherichia coli genes by a fur titration assay. J Mol Biol. 1994, 236 (2): 531-545. 10.1006/jmbi.1994.1163.
McHugh JP, Rodriguez-Quinones F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC: Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J Biol Chem. 2003, 278 (32): 29478-29486. 10.1074/jbc.M303381200.
Mukhopadhyay P, Zheng M, Bedzyk LA, LaRossa RA, Storz G: Prominent roles of the NorR and Fur regulators in the Escherichia coli transcriptional response to reactive nitrogen species. Proc Natl Acad Sci USA. 2004, 101 (3): 745-750. 10.1073/pnas.0307741100.
Mey AR, Wyckoff EE, Kanukurthy V, Fisher CR, Payne SM: Iron and fur regulation in Vibrio cholerae and the role of fur in virulence. Infect Immun. 2005, 73 (12): 8167-8178. 10.1128/IAI.73.12.8167-8178.2005.
Bjarnason J, Southward CM, Surette MG: Genomic profiling of iron-responsive genes in Salmonella enterica serovar Typhimurium by high-throughput screening of a random promoter library. J Bacteriol. 2003, 185 (16): 4973-4982. 10.1128/JB.185.16.4973-4982.2003.
Tsolis RM, Baumler AJ, Stojiljkovic I, Heffron F: Fur regulon of Salmonella typhimurium: identification of new iron-regulated genes. J Bacteriol. 1995, 177 (16): 4628-4637.
Foster JW, Hall HK: Effect of Salmonella typhimurium ferric uptake regulator (fur) mutations on iron- and pH-regulated protein synthesis. J Bacteriol. 1992, 174 (13): 4317-4323.
Ollinger J, Song KB, Antelmann H, Hecker M, Helmann JD: Role of the Fur regulon in iron transport in Bacillus subtilis. J Bacteriol. 2006, 188 (10): 3664-3673. 10.1128/JB.188.10.3664-3673.2006.
Baichoo N, Wang T, Ye R, Helmann JD: Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol Microbiol. 2002, 45 (6): 1613-1629. 10.1046/j.1365-2958.2002.03113.x.
Sutton VR, Mettert EL, Beinert H, Kiley PJ: Kinetic analysis of the oxidative conversion of the [4Fe-4S]2+ cluster of FNR to a [2Fe-2S]2+ Cluster. J Bacteriol. 2004, 186 (23): 8018-8025. 10.1128/JB.186.23.8018-8025.2004.
Fink RC, Evans MR, Porwollik S, Vazquez-Torres A, Jones-Carson J, Troxell B, Libby SJ, McClelland M, Hassan HM: FNR is a global regulator of virulence and anaerobic metabolism in Salmonella enterica serovar Typhimurium (ATCC 14028s). J Bacteriol. 2007, 189 (6): 2262-2273. 10.1128/JB.00726-06.
Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prevost MC, Sansonetti P, Tang CM: Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010, 465 (7296): 355-358. 10.1038/nature08970.
Bartolini E, Frigimelica E, Giovinazzi S, Galli G, Shaik Y, Genco C, Welsch JA, Granoff DM, Grandi G, Grifantini R: Role of FNR and FNR-regulated, sugar fermentation genes in Neisseria meningitidis infection. Mol Microbiol. 2006, 60 (4): 963-972. 10.1111/j.1365-2958.2006.05163.x.
Filiatrault MJ, Picardo KF, Ngai H, Passador L, Iglewski BH: Identification of Pseudomonas aeruginosa genes involved in virulence and anaerobic growth. Infect Immun. 2006, 74 (7): 4237-4245. 10.1128/IAI.02014-05.
Ammendola S, Pasquali P, Pacello F, Rotilio G, Castor M, Libby SJ, Figueroa-Bossi N, Bossi L, Fang FC, Battistoni A: Regulatory and structural differences in the Cu, Zn-superoxide dismutases of Salmonella enterica and their significance for virulence. J Biol Chem. 2008, 283 (20): 13688-13699. 10.1074/jbc.M710499200.
Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ: DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J Biol Chem. 1996, 271 (5): 2762-2768. 10.1074/jbc.271.5.2762.
Taylor CM, Osman D, Cavet JS: Differential expression from two iron-responsive promoters in Salmonella enterica serovar Typhimurium reveals the presence of iron in macrophage-phagosomes. Microb Pathog. 2009, 46 (2): 114-118. 10.1016/j.micpath.2008.11.001.
Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC: Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol. 2003, 47 (1): 103-118.
Troxell B, Sikes ML, Fink RC, Vazquez-Torres A, Jones-Carson J, Hassan HM: Fur negatively regulates hns and is required for the expression of HilA and virulence in Salmonella enterica serovar Typhimurium. J Bacteriol. 2011, 193 (2): 497-505. 10.1128/JB.00942-10.
Fang FC, Rimsky S: New insights into transcriptional regulation by H-NS. Curr Opin Microbiol. 2008, 11 (2): 113-120. 10.1016/j.mib.2008.02.011.
Navarre WW, Porwollik S, Wang YP, McClelland M, Rosen H, Libby SJ, Fang FC: Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006, 313 (5784): 236-238. 10.1126/science.1128794.
Hinton JC, Santos DS, Seirafi A, Hulton CS, Pavitt GD, Higgins CF: Expression and mutational analysis of the nucleoid-associated protein H-NS of Salmonella typhimurium. Mol Microbiol. 1992, 6 (16): 2327-2337. 10.1111/j.1365-2958.1992.tb01408.x.
Main-Hester KL, Colpitts KM, Thomas GA, Fang FC, Libby SJ: Coordinate regulation of Salmonella pathogenicity island 1 (SPI1) and SPI4 in Salmonella enterica serovar Typhimurium. Infect Immun. 2008, 76 (3): 1024-1035. 10.1128/IAI.01224-07.
Olekhnovich IN, Kadner RJ: Role of nucleoid-associated proteins Hha and H-NS in expression of Salmonella enterica activators HilD, HilC, and RtsA required for cell invasion. J Bacteriol. 2007, 189 (19): 6882-6890. 10.1128/JB.00905-07.
Olekhnovich IN, Kadner RJ: Crucial roles of both flanking sequences in silencing of the hilA promoter in Salmonella enterica. J Mol Biol. 2006, 357 (2): 373-386. 10.1016/j.jmb.2006.01.007.
Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC: H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2006, 2 (8): e81-10.1371/journal.ppat.0020081.
Sawers G: A novel mechanism controls anaerobic and catabolite regulation of the Escherichia coli tdc operon. Mol Microbiol. 2001, 39 (5): 1285-1298. 10.1111/j.1365-2958.2001.02316.x.
Teixido L, Carrasco B, Alonso JC, Barbe J, Campoy S: Fur activates the expression of Salmonella enterica pathogenicity island 1 by directly interacting with the hilD operator in vivo and in vitro. PLoS One. 2011, 6 (5): e19711-10.1371/journal.pone.0019711.
Ellermeier JR, Slauch JM: Fur regulates expression of the Salmonella pathogenicity island 1 type III secretion system through HilD. J Bacteriol. 2008, 190 (2): 476-486. 10.1128/JB.00926-07.
Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al: Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010, 467 (7314): 426-429. 10.1038/nature09415.
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000, 97 (12): 6640-6645. 10.1073/pnas.120163297.
Liu X, De Wulf P: Probing the ArcA-P modulon of Escherichia coli by whole genome transcriptional analysis and sequence recognition profiling. J Biol Chem. 2004, 279 (13): 12588-12597.
Evans MR, Fink RC, Vazquez-Torres A, Porwollik S, Jones-Carson J, McClelland M, Hassan HM: Analysis of the ArcA regulon in anaerobically grown Salmonella enterica sv. Typhimurium. BMC Microbiol. 2011, 11: 58-10.1186/1471-2180-11-58.
Porwollik S, Wong RM, Sims SH, Schaaper RM, DeMarini DM, McClelland M: The Delta uvrB mutations in the Ames strains of Salmonella span 15 to 119 genes. Mutat Res. 2001, 483 (1-2): 1-11. 10.1016/S0027-5107(01)00239-1.
Hertz GZ, Stormo GD: Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999, 15 (7-8): 563-577.
Ellermeier CD, Janakiraman A, Slauch JM: Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene. 2002, 290 (1-2): 153-161. 10.1016/S0378-1119(02)00551-6.
Miller JH: Experiments in molecular genetics. 1972, Cold Spring Harbor Laboratory
Monod J: AN OUTLINE OF ENZYME INDUCTION. Recueil Des Travaux Chimiques Des Pays-Bas-Journal of the Royal Netherlands Chemical Society. 1958, 77 (7): 569-585.
Neidhardt FC, Ingraham JL, Schaechter M: Physiology of the bacterial cell: a molecular approach. 1990, Sunderland, Mass.: Sinauer Associates, 331:
Mutalik VK, Nonaka G, Ades SE, Rhodius VA, Gross CA: Promoter strength properties of the complete sigma E regulon of Escherichia coli and Salmonella enterica. J Bacteriol. 2009, 191 (23): 7279-7287. 10.1128/JB.01047-09.
Costanzo A, Nicoloff H, Barchinger SE, Banta AB, Gourse RL, Ades SE: ppGpp and DksA likely regulate the activity of the extracytoplasmic stress factor sigmaE in Escherichia coli by both direct and indirect mechanisms. Mol Microbiol. 2008, 67 (3): 619-632. 10.1111/j.1365-2958.2007.06072.x.
Costanzo A, Ades SE: Growth phase-dependent regulation of the extracytoplasmic stress factor, sigmaE, by guanosine 3',5'-bispyrophosphate (ppGpp). J Bacteriol. 2006, 188 (13): 4627-4634. 10.1128/JB.01981-05.
Hassan HM, Sun HC: Regulatory roles of Fnr, Fur, and Arc in expression of manganese-containing superoxide dismutase in Escherichia coli. Proc Natl Acad Sci USA. 1992, 89 (8): 3217-3221. 10.1073/pnas.89.8.3217.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem. 1951, 193 (1): 265-275.
Beauchamp C, Fridovich I: Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971, 44 (1): 276-287. 10.1016/0003-2697(71)90370-8.
Lemire BD, Weiner JH: Fumarate reductase of Escherichia coli. Methods Enzymol. 1986, 126: 377-386.
Fenton H: Oxidation of tartaric acid in presence of iron. J Chem Soc, Trans. 1894, 65 (65): 899-911.
Baumler AJ, Norris TL, Lasco T, Voigt W, Reissbrodt R, Rabsch W, Heffron F: IroN, a novel outer membrane siderophore receptor characteristic of Salmonella enterica. J Bacteriol. 1998, 180 (6): 1446-1453.
Baumler AJ, Tsolis RM, vanderVelden AWM, Stojiljkovic I, Anic S, Heffron F: Identification of a new iron regulated locus of Salmonella typhi. Gene. 1996, 183 (1-2): 207-213. 10.1016/S0378-1119(96)00560-4.
Gupta SD, Lee BT, Camakaris J, Wu HC: Identification of cutC and cutF (nlpE) genes involved in copper tolerance in Escherichia coli. J Bacteriol. 1995, 177 (15): 4207-4215.
Ikeda JS, Janakiraman A, Kehres DG, Maguire ME, Slauch JM: Transcriptional regulation of sitABCD of Salmonella enterica serovar typhimurium by MntR and Fur. J Bacteriol. 2005, 187 (3): 912-922. 10.1128/JB.187.3.912-922.2005.
Janakiraman A, Slauch JM: The putative iron transport system SitABCD encoded on SPI1 is required for full virulence of Salmonella typhimurium. Mol Microbiol. 2000, 35 (5): 1146-1155. 10.1046/j.1365-2958.2000.01783.x.
Jeon J, Kim H, Yun J, Ryu S, Groisman EA, Shin D: RstA-promoted expression of the ferrous iron transporter FeoB under iron-replete conditions enhances Fur activity in Salmonella enterica. J Bacteriol. 2008, 190 (22): 7326-7334. 10.1128/JB.00903-08.
Tsolis RM, Baumler AJ, Heffron F, Stojiljkovic I: Contribution of TonB- and Feo-mediated iron uptake to growth of Salmonella typhimurium in the mouse. Infect Immun. 1996, 64 (11): 4549-4556.
Kehres DG, Janakiraman A, Slauch JM, Maguire ME: SitABCD is the alkaline Mn(2+) transporter of Salmonella enterica serovar Typhimurium. J Bacteriol. 2002, 184 (12): 3159-3166. 10.1128/JB.184.12.3159-3166.2002.
Mahan MJ, Slauch JM, Mekalanos JJ: Selection of Bacterial Virulence Genes That Are Specifically Induced in Host Tissues. Science. 1993, 259 (5095): 686-688. 10.1126/science.8430319.
Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ: Antibiotic-Based Selection for Bacterial Genes That Are Specifically Induced during Infection of a Host. Proc Natl Acad Sci USA. 1995, 92 (3): 669-673. 10.1073/pnas.92.3.669.
Govantes F, Orjalo AV, Gunsalus RP: Interplay between three global regulatory proteins mediates oxygen regulation of the Escherichia coli cytochrome-d oxidase (cydAB) operon. Mol Microbiol. 2000, 38 (5): 1061-1073.
Stojiljkovic I, Hantke K: Hemin uptake system of Yersinia enterocolitica: similarities with other TonB-dependent systems in gram-negative bacteria. EMBO J. 1992, 11 (12): 4359-4367.
Six S, Andrews SC, Unden G, Guest JR: Escherichia coli possesses two homologous anaerobic C4-dicarboxylate membrane transporters (DcuA and DcuB) distinct from the aerobic dicarboxylate transport system (Dct). J Bacteriol. 1994, 176 (21): 6470-6478.
Roof DM, Roth JR: Ethanolamine utilization in Salmonella typhimurium. J Bacteriol. 1988, 170 (9): 3855-3863.
Goss TJ, Datta P: Escherichia coli K-12 mutation that inactivates biodegradative threonine dehydratase by transposon Tn5 insertion. J Bacteriol. 1984, 158 (3): 826-831.
Goss TJ, Schweizer HP, Datta P: Molecular characterization of the tdc operon of Escherichia coli K-12. J Bacteriol. 1988, 170 (11): 5352-5359.
Hagewood BT, Ganduri YL, Datta P: Functional analysis of the tdcABC promoter of Escherichia coli: roles of TdcA and TdcR. J Bacteriol. 1994, 176 (20): 6214-6220.
Ganduri YL, Sadda SR, Datta MW, Jambukeswaran RK, Datta P: TdcA, a transcriptional activator of the tdcABC operon of Escherichia coli, is a member of the LysR family of proteins. Mol Gen Genet. 1993, 240 (3): 395-402.
Kim MJ, Lim S, Ryu S: Molecular analysis of the Salmonella typhimurium tdc operon regulation. J Microbiol Biotechnol. 2008, 18 (6): 1024-1032.
Lim S, Kim M, Choi J, Ryu S: A mutation in tdcA attenuates the virulence of Salmonella enterica serovar Typhimurium. Mol Cells. 2010, 29 (5): 509-517. 10.1007/s10059-010-0063-6.
Kim M, Lim S, Kim D, Choy HE, Ryu S: A tdcA mutation reduces the invasive ability of Salmonella enterica serovar typhimurium. Mol Cells. 2009, 28 (4): 389-395. 10.1007/s10059-009-0133-9.
Velayudhan J, Castor M, Richardson A, Main-Hester KL, Fang FC: The role of ferritins in the physiology of Salmonella enterica sv. Typhimurium: a unique role for ferritin B in iron-sulphur cluster repair and virulence. Mol Microbiol. 2007, 63 (5): 1495-1507. 10.1111/j.1365-2958.2007.05600.x.
Tardat B, Touati D: Two global regulators repress the anaerobic expression of MnSOD in Escherichia coli:Fur (ferric uptake regulation) and Arc (aerobic respiration control). Mol Microbiol. 1991, 5 (2): 455-465. 10.1111/j.1365-2958.1991.tb02129.x.
Compan I, Touati D: Interaction of six global transcription regulators in expression of manganese superoxide dismutase in Escherichia coli K-12. J Bacteriol. 1993, 175 (6): 1687-1696.
Tsaneva IR, Weiss B: soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J Bacteriol. 1990, 172 (8): 4197-4205.
Dubrac S, Touati D: Fur-mediated transcriptional and post-transcriptional regulation of FeSOD expression in Escherichia coli. Microbiology. 2002, 148 (Pt 1): 147-156.
Dubrac S, Touati D: Fur positive regulation of iron superoxide dismutase in Escherichia coli: functional analysis of the sodB promoter. J Bacteriol. 2000, 182 (13): 3802-3808. 10.1128/JB.182.13.3802-3808.2000.
Niederhoffer EC, Naranjo CM, Bradley KL, Fee JA: Control of Escherichia coli superoxide dismutase (sodA and sodB) genes by the ferric uptake regulation (fur) locus. J Bacteriol. 1990, 172 (4): 1930-1938.
Pomposiello PJ, Demple B: Identification of SoxS-regulated genes in Salmonella enterica serovar typhimurium. J Bacteriol. 2000, 182 (1): 23-29. 10.1128/JB.182.1.23-29.2000.
Clare DA, Blum J, Fridovich I: A hybrid superoxide dismutase containing both functional iron and manganese. J Biol Chem. 1984, 259 (9): 5932-5936.
Masse E, Gottesman S: A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci USA. 2002, 99 (7): 4620-4625. 10.1073/pnas.032066599.
Vecerek B, Moll I, Afonyushkin T, Kaberdin V, Blasi U: Interaction of the RNA chaperone Hfq with mRNAs: direct and indirect roles of Hfq in iron metabolism of Escherichia coli. Mol Microbiol. 2003, 50 (3): 897-909. 10.1046/j.1365-2958.2003.03727.x.
Durand S, Storz G: Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol Microbiol. 2010, 75 (5): 1215-1231. 10.1111/j.1365-2958.2010.07044.x.
Boysen A, Moller-Jensen J, Kallipolitis B, Valentin-Hansen P, Overgaard M: Translational regulation of gene expression by an anaerobically induced small non-coding RNA in Escherichia coli. J Biol Chem. 2010, 285 (14): 10690-10702. 10.1074/jbc.M109.089755.
Hassan HM, Fridovich I: Enzymatic defenses against the toxicity of oxygen and of streptonigrin in Escherichia coli. J Bacteriol. 1977, 129 (3): 1574-1583.
Touati D, Jacques M, Tardat B, Bouchard L, Despied S: Lethal oxidative damage and mutagenesis are generated by iron in delta fur mutants of Escherichia coli: protective role of superoxide dismutase. J Bacteriol. 1995, 177 (9): 2305-2314.
Schwyn B, Neilands JB: Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 1987, 160 (1): 47-56. 10.1016/0003-2697(87)90612-9.
Poole RK, Anjum MF, Membrillo-Hernandez J, Kim SO, Hughes MN, Stewart V: Nitric oxide, nitrite, and Fnr regulation of hmp (flavohemoglobin) gene expression in Escherichia coli K-12. J Bacteriol. 1996, 178 (18): 5487-5492.
Corker H, Poole RK: Nitric oxide formation by Escherichia coli. Dependence on nitrite reductase, the NO-sensing regulator Fnr, and flavohemoglobin Hmp. J Biol Chem. 2003, 278 (34): 31584-31592. 10.1074/jbc.M303282200.
Bang IS, Liu L, Vazquez-Torres A, Crouch ML, Stamler JS, Fang FC: Maintenance of nitric oxide and redox homeostasis by the Salmonella flavohemoglobin hmp. J Biol Chem. 2006, 281 (38): 28039-28047. 10.1074/jbc.M605174200.
Hernandez-Urzua E, Zamorano-Sanchez DS, Ponce-Coria J, Morett E, Grogan S, Poole RK, Membrillo-Hernandez J: Multiple regulators of the Flavohaemoglobin (hmp) gene of Salmonella enterica serovar Typhimurium include RamA, a transcriptional regulator conferring the multidrug resistance phenotype. Arch Microbiol. 2007, 187 (1): 67-77.
Partridge JD, Bodenmiller DM, Humphrys MS, Spiro S: NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol Microbiol. 2009, 73 (4): 680-694. 10.1111/j.1365-2958.2009.06799.x.
Sebastian S, Agarwal S, Murphy JR, Genco CA: The gonococcal fur regulon: identification of additional genes involved in major catabolic, recombination, and secretory pathways. J Bacteriol. 2002, 184 (14): 3965-3974. 10.1128/JB.184.14.3965-3974.2002.
Shaik YB, Grogan S, Davey M, Sebastian S, Goswami S, Szmigielski B, Genco CA: Expression of the iron-activated nspA and secY genes in Neisseria meningitidis group B by Fur-dependent and -independent mechanisms. J Bacteriol. 2007, 189 (2): 663-669. 10.1128/JB.01638-06.
Grinberg I, Shteinberg T, Hassan AQ, Aharonowitz Y, Borovok I, Cohen G: Functional analysis of the Streptomyces coelicolor NrdR ATP-cone domain: role in nucleotide binding, oligomerization, and DNA interactions. J Bacteriol. 2009, 191 (4): 1169-1179. 10.1128/JB.01145-08.
Torrents E, Grinberg I, Gorovitz-Harris B, Lundstrom H, Borovok I, Aharonowitz Y, Sjoberg BM, Cohen G: NrdR controls differential expression of the Escherichia coli ribonucleotide reductase genes. J Bacteriol. 2007, 189 (14): 5012-5021. 10.1128/JB.00440-07.
Borovok I, Kreisberg-Zakarin R, Yanko M, Schreiber R, Myslovati M, Aslund F, Holmgren A, Cohen G, Aharonowitz Y: Streptomyces spp. contain class Ia and class II ribonucleotide reductases: expression analysis of the genes in vegetative growth. Microbiology. 2002, 148 (Pt 2): 391-404.
Panosa A, Roca I, Gibert I: Ribonucleotide reductases of Salmonella typhimurium: transcriptional regulation and differential role in pathogenesis. PLoS One. 2010, 5 (6): e11328-10.1371/journal.pone.0011328.
Naranuntarat A, Jensen LT, Pazicni S, Penner-Hahn JE, Culotta VC: The interaction of mitochondrial iron with manganese superoxide dismutase. J Biol Chem. 2009, 284 (34): 22633-22640. 10.1074/jbc.M109.026773.
Jouihan HA, Cobine PA, Cooksey RC, Hoagland EA, Boudina S, Abel ED, Winge DR, McClain DA: Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol Med. 2008, 14 (3-4): 98-108.
Partridge JD, Sanguinetti G, Dibden DP, Roberts RE, Poole RK, Green J: Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J Biol Chem. 2007, 282 (15): 11230-11237. 10.1074/jbc.M700728200.
Amit R, Oppenheim AB, Stavans J: Increased bending rigidity of single DNA molecules by H-NS, a temperature and osmolarity sensor. Biophys J. 2003, 84 (4): 2467-2473. 10.1016/S0006-3495(03)75051-6.
Dame RT, Luijsterburg MS, Krin E, Bertin PN, Wagner R, Wuite GJ: DNA bridging: a property shared among H-NS-like proteins. J Bacteriol. 2005, 187 (5): 1845-1848. 10.1128/JB.187.5.1845-1848.2005.
Dorman CJ: H-NS: a universal regulator for a dynamic genome. Nat Rev Microbiol. 2004, 2 (5): 391-400. 10.1038/nrmicro883.
Goransson M, Sonden B, Nilsson P, Dagberg B, Forsman K, Emanuelsson K, Uhlin BE: Transcriptional silencing and thermoregulation of gene expression in Escherichia coli. Nature. 1990, 344 (6267): 682-685. 10.1038/344682a0.
Mojica FJ, Higgins CF: In vivo supercoiling of plasmid and chromosomal DNA in an Escherichia coli hns mutant. J Bacteriol. 1997, 179 (11): 3528-3533.
Ueguchi C, Mizuno T: The Escherichia coli nucleoid protein H-NS functions directly as a transcriptional repressor. EMBO J. 1993, 12 (3): 1039-1046.
Crawford MJ, Goldberg DE: Regulation of the Salmonella typhimurium flavohemoglobin gene. A new pathway for bacterial gene expression in response to nitric oxide. J Biol Chem. 1998, 273 (51): 34028-34032. 10.1074/jbc.273.51.34028.
Crawford MJ, Goldberg DE: Regulation of the Salmonella typhimurium flavohemoglobin gene. A NEW PATHWAY FOR BACTERIAL GENE EXPRESSION IN RESPONSE TO NITRIC OXIDE. J Biol Chem. 2006, 281 (6): 3752-
Gilberthorpe NJ, Lee ME, Stevanin TM, Read RC, Poole RK: NsrR: a key regulator circumventing Salmonella enterica serovar Typhimurium oxidative and nitrosative stress in vitro and in IFN-gamma-stimulated J774.2 macrophages. Microbiology. 2007, 153 (Pt 6): 1756-1771.
Joseph B, Przybilla K, Stuhler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W: Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006, 188 (2): 556-568. 10.1128/JB.188.2.556-568.2006.
Stojiljkovic I, Baumler AJ, Heffron F: Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol. 1995, 177 (5): 1357-1366.
Dibden DP, Green J: In vivo cycling of the Escherichia coli transcription factor FNR between active and inactive states. Microbiology. 2005, 151 (Pt 12): 4063-4070.
Jones HM, Gunsalus RP: Regulation of Escherichia coli fumarate reductase (frdABCD) operon expression by respiratory electron acceptors and the fnr gene product. J Bacteriol. 1987, 169 (7): 3340-3349.
Melville SB, Gunsalus RP: Mutations in fnr that alter anaerobic regulation of electron transport-associated genes in Escherichia coli. J Biol Chem. 1990, 265 (31): 18733-18736.
McCrindle SL, Kappler U, McEwan AG: Microbial dimethylsulfoxide and trimethylamine-N-oxide respiration. Adv Microb Physiol. 2005, 50: 147-198.
Privalle CT, Fridovich I: Transcriptional and maturational effects of manganese and iron on the biosynthesis of manganese-superoxide dismutase in Escherichia coli. J Biol Chem. 1992, 267 (13): 9140-9145.
McCollister BD, Bourret TJ, Gill R, Jones-Carson J, Vazquez-Torres A: Repression of SPI2 transcription by nitric oxide-producing, IFNgamma-activated macrophages promotes maturation of Salmonella phagosomes. J Exp Med. 2005, 202 (5): 625-635. 10.1084/jem.20050246.
Foster JW, Bearson B: Acid-sensitive mutants of Salmonella typhimurium identified through a dinitrophenol lethal screening strategy. J Bacteriol. 1994, 176 (9): 2596-2602.
Pettis GS, Brickman TJ, McIntosh MA: Transcriptional mapping and nucleotide sequence of the Escherichia coli fepA-fes enterobactin region. Identification of a unique iron-regulated bidirectional promoter. J Biol Chem. 1988, 263 (35): 18857-18863.
Hunt MD, Pettis GS, McIntosh MA: Promoter and operator determinants for Fur-mediated iron regulation in the bidirectional fepA-fes control region of the Escherichia coli enterobactin gene system. J Bacteriol. 1994, 176 (13): 3944-3955.
Escolar L, Perez-Martin J, de Lorenzo V: Coordinated repression in vitro of the divergent fepA-fes promoters of Escherichia coli by the iron uptake regulation (Fur) protein. J Bacteriol. 1998, 180 (9): 2579-2582.
Brickman TJ, Ozenberger BA, McIntosh MA: Regulation of divergent transcription from the iron-responsive fepB-entC promoter-operator regions in Escherichia coli. J Mol Biol. 1990, 212 (4): 669-682. 10.1016/0022-2836(90)90229-F.
Erickson KD, Detweiler CS: The Rcs phosphorelay system is specific to enteric pathogens/commensals and activates ydeI, a gene important for persistent Salmonella infection of mice. Mol Microbiol. 2006, 62 (3): 883-894. 10.1111/j.1365-2958.2006.05420.x.
Young GM, Postle K: Repression of tonB transcription during anaerobic growth requires Fur binding at the promoter and a second factor binding upstream. Mol Microbiol. 1994, 11 (5): 943-954. 10.1111/j.1365-2958.1994.tb00373.x.
Griggs DW, Konisky J: Mechanism for iron-regulated transcription of the Escherichia coli cir gene: metal-dependent binding of fur protein to the promoters. J Bacteriol. 1989, 171 (2): 1048-1054.
Runyen-Janecky LJ, Reeves SA, Gonzales EG, Payne SM: Contribution of the Shigella flexneri Sit, Iuc, and Feo iron acquisition systems to iron acquisition in vitro and in cultured cells. Infect Immun. 2003, 71 (4): 1919-1928. 10.1128/IAI.71.4.1919-1928.2003.
Chao TC, Becker A, Buhrmester J, Puhler A, Weidner S: The Sinorhizobium meliloti fur gene regulates, with dependence on Mn(II), transcription of the sitABCD operon, encoding a metal-type transporter. J Bacteriol. 2004, 186 (11): 3609-3620. 10.1128/JB.186.11.3609-3620.2004.
Kitphati W, Ngok-Ngam P, Suwanmaneerat S, Sukchawalit R, Mongkolsuk S: Agrobacterium tumefaciens fur has important physiological roles in iron and manganese homeostasis, the oxidative stress response, and full virulence. Appl Environ Microbiol. 2007, 73 (15): 4760-4768. 10.1128/AEM.00531-07.
Platero R, Peixoto L, O'Brian MR, Fabiano E: Fur is involved in manganese-dependent regulation of mntA (sitA) expression in Sinorhizobium meliloti. Appl Environ Microbiol. 2004, 70 (7): 4349-4355. 10.1128/AEM.70.7.4349-4355.2004.
Runyen-Janecky L, Dazenski E, Hawkins S, Warner L: Role and regulation of the Shigella flexneri sit and MntH systems. Infect Immun. 2006, 74 (8): 4666-4672. 10.1128/IAI.00562-06.
Kammler M, Schon C, Hantke K: Characterization of the ferrous iron uptake system of Escherichia coli. J Bacteriol. 1993, 175 (19): 6212-6219.
Aranda J, Cortes P, Garrido ME, Fittipaldi N, Llagostera M, Gottschalk M, Barbe J: Contribution of the FeoB transporter to Streptococcus suis virulence. Int Microbiol. 2009, 12 (2): 137-143.
Boulette ML, Payne SM: Anaerobic regulation of Shigella flexneri virulence: ArcA regulates Fur and iron acquisition genes. J Bacteriol. 2007, 189 (19): 6957-6967. 10.1128/JB.00621-07.
Mihara H, Hidese R, Yamane M, Kurihara T, Esaki N: The iscS gene deficiency affects the expression of pyrimidine metabolism genes. Biochem Biophys Res Commun. 2008, 372 (3): 407-411. 10.1016/j.bbrc.2008.05.019.
Fee JA: Regulation of sod genes in Escherichia coli: relevance to superoxide dismutase function. Mol Microbiol. 1991, 5 (11): 2599-2610. 10.1111/j.1365-2958.1991.tb01968.x.
Niederhoffer EC, Fee JA: Novel effect of aromatic compounds on the iron-dependent expression of the Escherichia coli K12 manganese superoxide dismutase (sodA) gene. Biol Met. 1990, 3 (3-4): 237-241. 10.1007/BF01140586.
Schrum LW, Hassan HM: The effects of fur on the transcriptional and post-transcriptional regulation of MnSOD gene (sodA) in Escherichia coli. Arch Biochem Biophys. 1994, 309 (2): 288-292. 10.1006/abbi.1994.1115.
Tardat B, Touati D: Iron and oxygen regulation of Escherichia coli MnSOD expression: competition between the global regulators Fur and ArcA for binding to DNA. Mol Microbiol. 1993, 9 (1): 53-63. 10.1111/j.1365-2958.1993.tb01668.x.
Hassett DJ, Sokol PA, Howell ML, Ma JF, Schweizer HT, Ochsner U, Vasil ML: Ferric uptake regulator (Fur) mutants of Pseudomonas aeruginosa demonstrate defective siderophore-mediated iron uptake, altered aerobic growth, and decreased superoxide dismutase and catalase activities. J Bacteriol. 1996, 178 (14): 3996-4003.
Hassett DJ, Howell ML, Ochsner UA, Vasil ML, Johnson Z, Dean GE: An operon containingfumC and sodA encoding fumarase C and manganese superoxide dismutase is controlled by the ferric uptake regulator in Pseudomonas aeruginosa fur mutants produce elevated alginate levels. J Bacteriol. 1997, 179 (5): 1452-1459.
Goh EB, Bledsoe PJ, Chen LL, Gyaneshwar P, Stewart V, Igo MM: Hierarchical control of anaerobic gene expression in Escherichia coli K-12: the nitrate-responsive NarX-NarL regulatory system represses synthesis of the fumarate-responsive DcuS-DcuR regulatory system. J Bacteriol. 2005, 187 (14): 4890-4899. 10.1128/JB.187.14.4890-4899.2005.
Overton TW, Griffiths L, Patel MD, Hobman JL, Penn CW, Cole JA, Constantinidou C: Microarray analysis of gene regulation by oxygen, nitrate, nitrite, FNR, NarL and NarP during anaerobic growth of Escherichia coli: new insights into microbial physiology. Biochem Soc Trans. 2006, 34 (Pt 1): 104-107.
Golby P, Kelly DJ, Guest JR, Andrews SC: Transcriptional regulation and organization of the dcuA and dcuB genes, encoding homologous anaerobic C4-dicarboxylate transporters in Escherichia coli. J Bacteriol. 1998, 180 (24): 6586-6596.
Xiong A, Singh VK, Cabrera G, Jayaswal RK: Molecular characterization of the ferric-uptake regulator, fur, from Staphylococcus aureus. Microbiology. 2000, 146 (Pt 3): 659-668.
Muller K, Matzanke BF, Schunemann V, Trautwein AX, Hantke K: FhuF, an iron-regulated protein of Escherichia coli with a new type of [2Fe-2S] center. Eur J Biochem. 1998, 258 (3): 1001-1008. 10.1046/j.1432-1327.1998.2581001.x.
Acknowledgements and Funding
This work was supported in part by the North Carolina Agricultural Research Service (to HMH. BT was supported, in part, by NIH T32 AI060519. MM and SP were supported in part by NIH grants R01AI 083646, R01AI 075093, R21AI 083964, R01AI 07397, R01AI 039557 and R01AI 052237.
We are grateful to Drs. FC Fang, SJ Libby, and A Vazquez-Torres for strains and plasmids. We thank Gabriele Gusmini and Russell Wolfinger for guidance with statistical analysis; and Fred Long and Xiao-Qin Xia for their expert bioinformatics assistance. We thank Dr. M. Evans for reading the manuscript and Dr. Robarge and Kim Hutchison for ICP-OES analysis of metals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
All authors have read and approved this work. BT, RCF, HMH designed and conducted the experiments and contributed to the writing and editing of the manuscript. RCF conducted the microarrays, constructed the Fur Logo, and contributed to the editing of the manuscript. MM and SP constructed and provided the microarray slides and reviewed the manuscript. BT and HMH conceived the research idea, directed the research, and contributed to the writing and editing of the manuscript.
Electronic supplementary material
12866_2011_1522_MOESM2_ESM.XLS
Additional file 2: Table S2. Fur Regulated Genes. This file contains the genes that were differentially expressed between 14028s and Δfur under anaerobic conditions. (XLS 91 KB)
12866_2011_1522_MOESM3_ESM.PDF
Additional file 3: Table S3. Fumarate reductase activity under anaerobic conditions. This file contains the specific activity of fumarate reductase in cell-free extracts isolated from 14028s and Δfur under anaerobic conditions. (PDF 143 KB)
12866_2011_1522_MOESM4_ESM.PDF
Additional file 4: Table S4. Genes regulated by Fur and Fnr under anaerobiosis and contain putative binding sites for both regulators. This file contains genes that were differentially expressed in 14028s, Δfur, and the fnr, which contain a putative binding site for Fur and for Fnr. (PDF 23 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Troxell, B., Fink, R.C., Porwollik, S. et al. The Fur regulon in anaerobically grown Salmonella enterica sv. Typhimurium: identification of new Fur targets. BMC Microbiol 11, 236 (2011). https://doi.org/10.1186/1471-2180-11-236
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-11-236