
Abstract
Background
The antifungal protein AFPNN5353 is a defensin-like protein of Aspergillus giganteus. It belongs to a group of secretory proteins with low molecular mass, cationic character and a high content of cysteine residues. The protein inhibits the germination and growth of filamentous ascomycetes, including important human and plant pathogens and the model organsims Aspergillus nidulans and Aspergillus niger.
Results
We determined an AFPNN5353 hypersensitive phenotype of non-functional A. nidulans mutants in the protein kinase C (Pkc)/mitogen-activated protein kinase (Mpk) signalling pathway and the induction of the α-glucan synthase A (agsA) promoter in a transgenic A. niger strain which point at the activation of the cell wall integrity pathway (CWIP) and the remodelling of the cell wall in response to AFPNN5353. The activation of the CWIP by AFPNN5353, however, operates independently from RhoA which is the central regulator of CWIP signal transduction in fungi.
Furthermore, we provide evidence that calcium (Ca2+) signalling plays an important role in the mechanistic function of this antifungal protein. AFPNN5353 increased about 2-fold the cytosolic free Ca2+ ([Ca2+]c) of a transgenic A. niger strain expressing codon optimized aequorin. Supplementation of the growth medium with CaCl2 counteracted AFPNN5353 toxicity, ameliorated the perturbation of the [Ca2+]c resting level and prevented protein uptake into Aspergillus sp. cells.
Conclusions
The present study contributes new insights into the molecular mechanisms of action of the A. giganteus antifungal protein AFPNN5353. We identified its antifungal activity, initiated the investigation of pathways that determine protein toxicity, namely the CWIP and the Ca2+ signalling cascade, and studied in detail the cellular uptake mechanism in sensitive target fungi. This knowledge contributes to define new potential targets for the development of novel antifungal strategies to prevent and combat infections of filamentous fungi which have severe negative impact in medicine and agriculture.
Similar content being viewed by others


Background
All organisms have evolved several defence systems in order to protect themselves against bacteria, fungi and viruses. Higher organisms have developed a complex network of humoral and cellular responses, called adaptive immunity. A second defence system, the innate immunity, consists of many components, including small peptides with a broad antimicrobial spectrum [1, 2]. The production of such proteins with antimicrobial activity is not limited to higher eukaryotes, but also found in microorganisms, including fungi. The diversity of these proteins is reflected in their mode of action and their species-specificity. Some of them form pores in the membrane, others are known to inhibit cell wall synthesis or interfere with nucleic acids and their synthesis [3, 4]. They can be involved in the inhibition of protein synthesis or interfere with cell cycle control [3, 4]. A relatively new group of antimicrobial proteins secreted by filamentous ascomycetes includes small, cationic and cysteine-rich proteins. So far, only few antifungal proteins have been characterized, namely AFP from Aspergillus giganteus, ANAFP from Aspergillus niger, PAF from Penicillium chrysogenum and NAF from Penicillium nalgiovense [5–8].
The mode of action of these proteins is not fully understood. Nevertheless, there is evidence, that their toxicity is mediated by interaction with distinct molecules or receptors at the outer layers of the cell, e.g. cell wall or plasma membrane. Deleterious effects can then be induced either by transmitting signals from the outer layers into the cell, or by internalization of the protein and interaction with internal molecules [9–15]. Similar to substances that perturb the cell wall, such as caspofungin, congo red or calcofluor white (CFW) [10, 16], the A. giganteus antifungal protein AFP was found to modulate the cell wall composition by enhancing the expression of the α-1,3-glucan synthase A gene (agsA), possibly by the activation of the cell wall integrity pathway (CWIP), and inhibiting chitin synthesis in sensitive fungi [10]. This, however, stands in contrast to the mode of action of the P. chrysogenum antifungal protein PAF which fails to activate the CWIP [9]. However, the central players that trigger cell wall remodelling in AFP-sensitive fungi have not been investigated so far.
Another mechanistic function of antifungal proteins is the interference with ion, especially Ca2+ ion homeostasis and signalling [15, 17, 18]. We could recently show that the P. chrysogenum antifungal protein PAF severely perturbed the Ca2+ homeostasis of Neurospora crassa by rapidly elevating the cytoplasmic Ca2+ [Ca2+]c resting level [17]. Numerous reports indicate that the activity of antifungal proteins can be antagonized by the external addition of Ca2+ ions to the test medium [15, 17–21] pointing towards the induction of adaptive responses which may be triggered by Ca2+ signalling [15, 17].
The aim of this study was to characterize in more detail the mode of action of the A. giganteus AFP variant protein AFPNN5353 and to investigate the pathways that might be affected/modulated by this antifungal protein. Therefore, we focussed our interest on the involvement of the CWIP and the Ca2+ signalling in the toxicity of AFPNN5353. To address these questions, we used the highly AFPNN5353 sensitive model organisms A. nidulans and A. niger for which appropriate mutant strains were available.
Results
In silico analysis of AFPNN5353
CLUSTALW amino acid (aa) sequence analysis of AFPNN5353 with other known antifungal proteins revealed that AFPNN5353 from A. giganteus strain A3274 is a protein homologous to AFP from A. giganteus strain MDH 18894 [8, 22]. AFPNN5353 exhibits > 90% identity with AFP, but only 42% identity with the P. chrysogenum PAF and 27% identity with the A. niger ANAFP. In fact, the secreted mature form of AFPNN5353 consists of 51 aa and differs only in 5 aa from AFP (Figure 1). Three aa exchanges belong to structurally related aa, one aa exhibits weak similarity and one aa is different (position 4). These aa exchanges do not influence the theoretical isoelectric point (pI) of AFPNN5353, which is the same as for AFP (pI 9.3, http://expasy.org/tools/protparam.html). Most importantly, AFPNN5353 still contains the putative chitin-binding domain CKYKAQ present in AFP but not in PAF or ANAFP and also harbors all conserved cysteine residues important for protein stabilization [10, 23].

Clustalw sequence alignment http://www.ebi.ac.uk/Tools/msa/clustalw2/ of the antifungal proteins AFP NN5353 and AFP from A. giganteus , ANAFP from A. niger and PAF from P. chrysogenum. Identical amino acids (aa) are marked with (*), aa with strong similarity are indicated with (:) and aa with weak similarity are marked with (.).
Antifungal activity of the protein AFPNN5353
To investigate the antifungal specificity of AFPNN5353, fifteen filamentous fungi were tested for their susceptibility to the protein. Since antifungal proteins might be useful for biotechnological applications, filamentous human and plant pathogenic fungi were selected as test organisms (e.g. Fusarium oxysporum, Botrytis cinerea, Mucor sp. and A. fumigatus) in addition to the model organisms A. nidulans and A. niger. As shown in Table 1, thirteen out of fifteen tested moulds were found to be sensitive against AFPNN5353. A. nidulans wild type, N. crassa wild type and A. niger wild type were the most sensitive strains to AFPNN5353. The minimal inhibitory concentration (MIC) of AFP (the concentration that completely inhibited conidial germination in liquid growth assays) was 0.2 μg/ml for A. nidulans, 0.5 μg/ml for N. crassa and 1 μg/ml for A. niger. Two strains were unaffected at the protein concentrations tested: M. circenelloides and M. genevensis were insensitive against AFPNN5353 when concentrations up to 500 μg/ml were used.
AFPNN5353 interferes with the cell wall integrity of A. nidulans
It is known that antifungal compounds such as congo red, caffeine, CFW or caspofungin interfere with cell wall biosynthesis and weaken the cell wall in fungi (reviewed by [24]). The remodeling of the cell wall by these antifungal compounds is mediated by the activation of the CWIP. In fungi, extracellular signals are transmitted via the membrane bound small GTPase RhoA to the central regulators Pkc and Mpk, which are regulated by phosphorylation/dephosphorylation. The signal transduction cascade eventually enforces transcription of cell wall synthesis genes, partly via the transcription factor RlmA [16, 25]. Respective loss-of-function or conditional mutants show hypersensitive phenotypes in the presence of cell wall perturbing agents [9, 24–26]. Similar to substances that weaken the cell wall, the A. giganteus antifungal protein AFP modulates the cell wall composition by inhibiting chitin synthesis in sensitive fungi (e.g. A. niger, A. oryzae) and inducing the expression of agsA most likely by the activation of the CWIP [10].
To study the involvement of the CWIP in AFPNN5353 toxicity, we first tested whether the osmotic stabilizer sorbitol counteracts the toxicity of AFPNN5353. In the absence of AFPNN5353 A. nidulans proliferated less well in the presence of 1 M sorbitol and reached only 30% growth compared to the growth in standard medium (100%). Nevertheless, the addition of 1 M sorbitol to the growth medium strongly reduced the activity of AFPNN5353 on A. nidulans wild type. The osmotic stabilizer ameliorated growth in the presence of 0.05 μg/ml AFPNN5353 by 80% compared to a 10% growth rate in the absence of sorbitol (Table 2). This was even more accentuated when 0.1 and 0.2 μg/ml AFPNN5353 were applied, suggesting that AFPNN5353 indeed weakens the cell wall of A. nidulans.
To investigate whether AFPNN5353 induces agsA gene transcription similar to AFP via the Pkc/Mpk signalling pathway, we tested the effect of the antifungal protein on the transgenic A. niger strain RD6.47 which expresses a nuclear-targeted GFP protein fused to the A. niger agsA promoter. RD6.47 germlings were treated with AFPNN5353 (conc. 10 to 100 μg/ml) for 2 h and analyzed microscopically. As shown in Additional file 1, a nuclear signal was clearly detectable in germlings of RD6.47 treated with ≥ 50 μg/ml AFPNN5353, similar to that when exposed to 10 μg/ml caspofungin. In untreated germlings, however, no signal could be observed. These observations perfectly match with the data obtained for AFP [10]. It has to be noted here that antifungal protein concentrations higher than the MIC determined for conidia (> 10-50 fold) are needed to inhibit the growth of germlings or hyphae of sensitive fungi [10, 27] (data not shown).
Next, we tested several A. nidulans mutant strains affected in central players of the CWIP for their susceptibility to AFPNN5353 by determining their radial growth in the presence or absence of the antifungal protein. Since RhoA is an essential protein in A. nidulans, two strains with ectopic copies of the constitutively active rhoAG14V allele and the dominant rhoAE40I allele [28] were tested in comparison to the wild type strain (GR5). The rhoAG14V mutation prevents the hydrolysis of GTP and therefore renders RhoA constantly active [28]. Similarly, the GTP hydrolysis is inhibited in the RhoAE40I strain, but this mutation also perturbs the binding of the GTPase activating protein (GAP) to RhoA and possibly disturbs downstream effectors of RhoA-GAP [28]. The constitutively active RhoAG14V and the dominant RhoAE40I strain exhibited the same sensitivity towards AFPNN5353 as the wild type strain at low protein concentrations (≤ 0.2 μg/ml) (Figure 2A). Interestingly, the dominant RhoAE40I strain was more resistant to AFPNN5353 than the wild type strain or the RhoAG14V strain at higher protein concentrations (1 μg/ml) (Figure 2A). Therefore, we suggest that the toxicity of AFPNN5353 is transmitted by RhoA-GAP targets and not by RhoA itself. These mutants performed similarly when exposed to the orthologous P. chrysogenum antifungal protein PAF [9].

AFP NN5353 susceptibility of A. nidulans mutants RhoAG14V, RhoAE40I, alcA -PkcA and Δ mpkA compared to the respective recipient strains GR5 and R153. (A) A total of 2 × 103 conidia were point inoculated on agar plates (CM for GR5, RhoAG14V, RhoAE40I and ΔmpkA, repressive MM containing 1% glucose according to [26] for R135 and alcA-PkcA) containing the appropriate supplements and 0, 0.2 and 1 μg/ml AFPNN5353 for GR5, RhoAG14V, RhoAE40I, R135 and alcA-PkcA. The ΔmpkA mutant and its reference strain GR5 were exposed to 0, 0.5 and 1 μg/ml AFPNN5353. The plates were incubated at 37°C for 48 h. (B) 1 × 104 conidia/ml of the ΔmpkA mutant and GR5 were treated with 0.05 μg/ml AFPNN5353 or without protein (controls) in a total volume of 200 μl of appropriately supplemented CM in 96-well plates.
In addition, mutants defective in PkcA and MpkA activity were tested for their AFPNN5353 susceptibility. As pkcA is an essential gene in A. nidulans, a conditional alcA-PKC mutant strain was used, where the pkcA gene was put under the control of the alcA promoter, which is repressed by glucose but derepressed by glycerol [26]. Both the conditional alcA-PKC mutant (cultivated under repressive conditions) and a ΔmpkA mutant were hypersensitive to AFPNN5353 compared to their recipient strains R153 and GR5, respectively, indicating that the activity of PkcA and MpkA confers a certain resistance to AFPNN5353 (Figure 2A). The hypersensitive phenotype of the ΔmpkA mutant was also confirmed by liquid growth inhibitory assays. In unchallenged liquid condition, the GR5 and the ΔmpkA mutant showed a comparable proliferation rate (Figure 2B). In the presence of 0.05 μg/ml AFPNN5353, however, the mpkA deletion strain did not germinate whereas the GR5 strain still exhibited 11% growth. Note that growth inhibition in liquid conditions requires less antifungal protein to monitor its toxicity than on solid media probably due to less diffusion in the latter case (data not shown).
From these data we conclude that AFPNN5353 interferes with the cell wall homeostasis of A. nidulans and that this interaction is mediated by PkcA/MpkA signalling, although independently from RhoA.
AFPNN5353 disrupts calcium homeostasis in A. niger
Supplements other than osmotic stabilizers can also antagonize the activity of antifungal proteins from plants and ascomycetes. For example, the addition of cations such as Ca2+ ions to the growth medium reversed the antifungal activity of the P. chrysogenum PAF [17], the A. giganteus AFP [15, 21] and of plant defensins [29, 30] which are usually positively charged due to their high pI. A cation-sensitive antifungal mode of action can for example be associated with the perturbation of the intracellular Ca2+ homeostasis by antifungal peptides [17, 18] but might also result from the interference of cations with antifungal-target interaction(s).
Therefore, we tested to which extend these effects also account for the antifungal activity of AFPNN5353. To this end, we selected A. niger as model organism because this mould was highly sensitive to AFPNN5353 and a transgenic strain was available that expressed the recombinant codon optimized Ca2+-sensitive photoprotein aequorin for measuring the [Ca2+]c resting level in response to AFPNN5353 [31]. First, we tested the activity of AFPNN5353 in Vogels* medium supplemented with 5-20 mM CaCl2 or without CaCl2 as a control (data not shown). Addition of CaCl2 did not influence the growth of A. niger up to a concentration of 20 mM. The growth of A. niger exposed to AFPNN5353, however, ameliorated in the presence of increasing concentrations of CaCl2. 20 mM CaCl2 neutralized the toxicity of 0.5-1.0 μg/ml AFPNN5353 and the treated samples resumed growth to 100% (Table 3).
Next, we determined the influence of AFPNN5353 on the intracellular Ca2+ signature. Before AFPNN5353 addition, the resting level of the intracellular Ca2+ was 0.08 μM. We could show, however, that the [Ca2+]c resting level was significantly increased in twelve h old A. niger cultures that were treated with 20 μg/ml AFPNN5353. The [Ca2+]c resting level rose to a maximum of 0.19 μM within the first 8 min and stayed elevated throughout the time of measurement (60 min), whereas the Ca2+ level of the untreated control remained at 0.08 μM (Figure 3). This indicated that AFPNN5353 indeed disrupts Ca2+ homeostasis in A. niger.

Increase in resting [Ca2+] c of twelve h old A. niger germlings treated with AFP NN5353 or no protein (controls). Measurements were taken every 1.4 minutes. Values represent average of six samples.
To exclude the possibility that the AFPNN5353 induced rise in the [Ca2+]c resting level is due to membrane permeabilization and/or pore formation, we studied the effects of AFPNN5353 on germlings in the presence of CMFDA, a membrane permeant dye that is metabolized by viable cells, and the membrane impermeant dye propidium iodide (PI). Additional file 2 shows that samples treated with 20 μg/ml AFPNN5353 for 10 min metabolized CMFDA but did not take up PI, resulting in green but no red fluorescence, similar to untreated controls. This indicated that the plasma membrane was still intact after 10 min of protein treatment. Samples exposed to ethanol did not metabolize CMFDA but appeared bright red due to PI internalization, indicating that here the membrane was permeabilized. We therefore conclude that the rapid increase in [Ca2+]c within the first 10 min of protein treatment is not the result of uncontrolled Ca2+ influx due to plasma membrane permeabilization.
The calcium chelator BAPTA abrogates the AFPNN5353-induced calcium signature
The increased [Ca2+]c in response to AFPNN5353 treatment could originate from extracellular and/or from intracellular Ca2+ stores, such as mitochondria, vacuoles, endoplasmic reticulum or the Golgi apparatus. To discriminate between the extracellular and intracellular source of the [Ca2+]c increase, we tested the influence of the Ca2+-selective membrane impermeable chelator BAPTA. On its own, BAPTA did not influence the resting level of [Ca2+]c in twelve h old A. niger cultures (Figure 4). However, a pretreatment of the samples with 10 mM BAPTA before the addition of AFPNN5353 inhibited the protein-specific increase in [Ca2+]c resting level (Figure 4). Interestingly, the elevated [Ca2+]c in response to a 40 min AFPNN5353-treatment dropped to the resting level immediately after the addition of 10 mM BAPTA (Figure 4), indicating that the AFPNN5353-induced elevation of the [Ca2+]c resting level requires the continuous influx of extracellular Ca2+ and eventually results in loss of [Ca2+]c homeostasis.

Effect of the extracellular chelator BAPTA on the AFP NN5353 induced [Ca2+] c resting level. 10 mM BAPTA (final conc.) were applied 40 min before or 40 min after treatment with 20 μg/ml AFPNN5353. Samples without supplements were used as controls. SD (n = 6) was less than 10% of the values presented.
Extracellular calcium ameliorates the AFPNN5353-induced rise in [Ca2+]c
To decipher the observation that high external CaCl2 concentrations counteracted AFPNN5353 toxicity (Table 3), we monitored the effect of externally added Ca2+ on the AFPNN5353-induced Ca2+ signature. To this end, A. niger germlings were preincubated with 20 mM CaCl2 for 10 min before 20 μg/ml AFPNN5353 was added and the changes in the [Ca2+]c resting level were monitored over a time course of 60 min. This treatment resulted in a less pronounced rise of the [Ca2+]c resting level compared to samples without preincubation with CaCl2. In contrast, the presence of 20 mM CaCl2 alone had no major effect on the intracellular [Ca2+]c resting level which resembled that of the control without AFPNN5353 (data not shown). The values of the [Ca2+]c resting levels of the last 10 min (50 to 60 min) measurement of AFPNN5353 treatment in the presence or absence of high Ca2+ concentration (20 mM versus 0.7 mM) are summarized in Table 4. The average of the [Ca2+]c of the controls which were not exposed to AFPNN5353 was 0.039 μM in the presence of 0.7 μM CaCl2 (standard condition) and 0.062 μM in the presence of 20 mM CaCl2. When AFPNN5353 was added, there was no significant elevation of the [Ca2+]c in high-Ca2+ medium (20 mM) (0.057 μM) whereas the [Ca2+]c rised to 0.146 μM at standard CaCl2 concentration (0.7 mM). These results suggest that Ca2+ externally added prior to the addition of AFPNN5353 counteracts the AFPNN5353 induced perturbation of the [Ca2+]c and growth inhibitory effect, at least partly, by controlling the [Ca2+]c resting level.
AFPNN5353 decreases the amplitude of the [Ca2+]c response to mechanical perturbation in A. niger
It is known that a range of external stimuli transiently increase [Ca2+]c levels in Aspergilli and other fungi [31, 32]. One of these physiological stimuli is mechanical perturbation, which is achieved by the rapid injection of isotonic medium into the test system. This stimulus results in a unique Ca2+ signature, likely involving different components of the Ca2+-signalling and Ca2+ homeostatic machinery. Changes in this specific Ca2+ signature in the presence of compounds, such as AFPNN5353, can give insights into the mode of action of these compounds. In our study, twelve h old cultures of A. niger were pre-incubated with AFPNN5353 for 60 min and thereafter subjected to mechanical perturbation (rapid injection of 100 μl Vogels medium). The resulting Ca2+ signature, including [Ca2+]c resting level, kinetics and amplitude, were determined and compared with controls that were not exposed to the protein but also subjected to mechanical perturbation. As shown in Figure 5, AFPNN5353 provoked a less pronounced [Ca2+]c amplitude; however, the [Ca2+]c level remained elevated even after the stimulus specific response had stopped.

Effects of AFP NN5353 on the [Ca2+] c response to mechanical perturbation. Twelve h old A. niger cultures were treated with 20 μg/ml AFPNN5353 for 60 min before stimulation by mechanical perturbation (addition of 100 μl Vogels medium). The [Ca2+]c signature was monitored for 5 min. Values represent the average of six samples.
AFPNN5353 binding and uptake are essential for protein toxicity in A. nidulans
To understand the function of antifungal proteins, the identification of the site of action in target organisms is crucial. So far, controversial reports exist of the localization of the homologous A. giganteus AFP protein. AFP has been detected to bind to outer layers, e.g. the cell wall or the plasma membrane of sensitive fungi [20, 21] and a time- and concentration-dependent intracellular localization was reported [20]. In another study, Alexa-labelled AFP was shown to be internalized by the fungal cell and to localize to the nucleus [33].
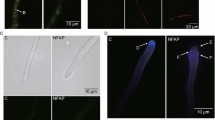
To dissect the uptake and localization of AFPNN5353, we performed indirect immunofluorescence staining with A. nidulans wild type exposed to a sublethal concentration of AFPNN5353 (0.2 μg/ml). We applied a protein amount below the toxic concentration for hyphae to maintain the cellular structure and to avoid apoptotic cell disruption [34]. Our study revealed that the protein was internalized after 90 min of incubation, mostly in hyphal tips, but also within hyphal segments (Figure 6A, B). The protein seemed not to localize to cell compartments, but was distributed in the cytoplasm. Similar results were obtained with A. niger wild type (data not shown). Control experiments proved the specificity of the intracellular immunofluorescent signals: no intracellular fluorescent signals were detected in samples where either AFPNN5353 (Figure 6C, D) or the primary antibody or the secondary antibody was omitted (data not shown).

Indirect immunofluorescence staining of A. nidulans with rabbit anti-AFP NN5353 antibody. Fungi were incubated with 0.2 μg/ml AFPNN5353 (A, E, G) or without antifungal protein (C). 20 μg/ml latrunculin B (E) and 10 mM Ca2+ (G) significantly reduced protein uptake. (B, D, F, H) are the respective light microscopic images of (A, C, E, G). Scale bar 10 μm.
To analyse the AFPNN5353 localization in more detail, A. nidulans was incubated with AFPNN5353 in the presence of latrunculin B, a potent inhibitor of actin polymerization and endocytosis [35–37]. At low latrunculin B concentrations (5 μg/ml), protein uptake was severely reduced compared to the positive control without latrunculin B (data not shown), whereas 20 μg latrunculin B/ml completely inhibited the uptake of 0.2 μg/ml AFPNN5353. The solvent of latrunculin B, DMSO, had no adverse effect on protein uptake (data not shown). This indicates that AFPNN5353 enters the A. nidulans cells by an endocytotic mechanism (Figure 6E, F).
Based on our observation that Ca2+ ions antagonize the growth inhibitory activity of AFPNN5353, we questioned whether Ca2+ prevents actin-mediated internalisation of the antifungal protein. Indeed, the presence of 10 mM CaCl2 inhibited protein uptake (Figure 6G, H). Most interestingly, no specific fluorescent signals were detectable in M. circinelloides when treated with up to 500 μg/ml of antifungal protein (data not shown), indicating that AFPNN5353 does not bind to insensitive strains.
Discussion
In this study we provide important insights into the mechanistic basis of AFPNN5353, a AFP homologous protein.
Species specificity tests revealed that AFPNN5353 is active against a broad range of filamentous fungi, including human and plant pathogens. Although the proteins AFPNN5353 and AFP are almost identical and show a similar toxicity, MICs for AFPNN5353 differed slightly from those reported for AFP [21]. We attribute this discrepancy to differences in the experimental setups, e.g. fungal strains, medium composition, conidial inoculum, incubation times, cultivation temperature etc., rather than to the differences in the primary sequence of both proteins.
It has been reported that the closely related AFP protein interfered with cell wall synthesis [10] and our finding that the osmotic stabilizer sorbitol neutralized AFPNN5353 toxicity further corroborated this assumption. Two A. nidulans mutants, the conditional alcA-PkcA and the mpkA deletion mutant showed a hypersensitive phenotype when exposed to AFPNN5353. This is in agreement to the reported function of cell wall stressing agents, such as CFW or caffeine in S. cerevisiae and A. nidulans [9, 16, 24, 26, 38, 39] and to the Penicillium antifungal protein PAF [9]. Importantly, Mpk function is essential for CWIP activation in both, unicellular and filamentous fungi [10, 16, 40] and triggers the activation of the transcription factors Rlm1p and SBF which regulate the expression of cell cycle regulated genes and genes involved in the synthesis and remodelling of the fungal cell wall in S. cerevisiae [41, 42]. Similarly, RlmA dependent induction of the expression of the ags gene was also reported for aspergilli [25]. Importantly, the activation of the CWIP can occur in a RhoA-dependent, e.g. with CFW [9, 43], or RhoA-independent way, the latter proved for PAF and caffeine [9, 16] and for AFPNN5353 (this study). As proposed by [28] the dominant rhoAE40I allele suffers from a perturbation of its GAP binding domain and downstream effectors of Rho-GAP might be disturbed. Therefore, we hypothesize that Rho-GAP targets might be involved in the toxicity of AFPNN5353 similarly to the mode of action of the P. chrysogenum PAF [9]. Our assumption of the activation of the CWIP by AFPNN5353 was further strengthened by the fact, that AFPNN5353 treatment induced agsA expression in the A. niger reporter strain. This result was consistent with the activity of AFP and caspofungin [10], but differed to the function of PAF, where no CWIP activation and no induction of cell wall biosynthesis genes occurred [9].
Therefore, we conclude that AFPNN5353 triggers cell wall remodeling via Pkc/Mpk signalling. We further deduce from our data that similarities and differences exist in the molecular targets and the mode of action of antifungal proteins from filamentous fungi, e.g. AFPNN5353 and PAF - despite their homology. This phenomenon was also reported for other closely related antifungal proteins, such as the plant defensins MsDef1 and MtDef4 from Medicago spp. [44].
Apart from the activation of the CWIP, the perturbation of the Ca2+ homeostasis represents a major mechanistic function of antifungal proteins in sensitive fungi [17, 18]. The intracellular Ca2+ response to AFPNN5353 in A. niger reflected that of the Penicillium antifungal protein PAF in N. crassa [17]. The rapid and sustained increase of the [Ca2+]c resting level depended on a sustained influx of Ca2+ ions from the external medium. Moreover, the AFPNN5353 induced changes in the Ca2+ signature of mechanically perturbed A. niger cells further underlines the disruption of the Ca2+ response and homeostasis by AFPNN5353. The addition of CaCl2 to the growth medium reduced the susceptibility of A. niger towards the antifungal protein and decreased the AFPNN5353 specific rise in the [Ca2+]c resting level. Both observations point towards an adaptive response which is mediated most probably via Ca2+ signalling. First, high extracellular Ca2+ concentrations trigger chitin synthesis in A. niger and thereby confer increased protection against antifungal proteins as shown for AFP [15]. Second, it primes the Ca2+ homeostatic machinery to better maintain a low [Ca2+]c resting level when challenged with the antifungal protein, e.g. by (i) the increase of the activity of existing Ca2+ pumps/transporters to counteract the AFPNN5353-specific intracellular Ca2+ perturbation, or (ii) the modulation of the expression of Ca2+ channels/pumps/exchangers [17]. The former hypothesis (i) might be supported by the observation that the addition of CaCl2 only 10 min before A. niger was challenged with AFPNN5353 restored the low [Ca2+]c resting level. However, the perturbation of the Ca2+ homeostasis by a sustained elevation of the [Ca2+]c resting level indicates that A. niger is not able to restore the low [Ca2+]c resting level after exposure to AFPNN5353 and this might trigger programmed cell death (PCD) on the long term as it was shown to occur in A. nidulans in response to the P. chrysogenum PAF [34].
Since AFP was shown to cause membrane permeabilization [21], the influx of Ca2+ might be due to changes in membrane permeability for this ion, if not the formation of pores. However, our staining experiments with CMFDA and PI exclude this possibility at least in the first 10 min of exposure to AFPNN5353 when the [Ca2+]c resting level reaches its maximum. This result is further corroborated by the fact that higher external concentrations of Ca2+ reduced the AFPNN5353 specific rise in [Ca2+]c resting level which - in our opinion - would not occur with leaky membranes. However, we do not exclude changes in membrane permeability at longer exposure times to this antifungal protein and more studies are needed to answer this question.
Finally, we observed that the internalization of AFPNN5353 is characteristic for sensitive but not resistant moulds. A lack of binding of AFPNN5353 to insensitive fungi might point towards the absence or inaccessibility of a putative interacting molecule at the cell surface. AFPNN5353 localized to the cytoplasm of target fungi only when actin filaments were formed. This is in agreement with the endocytotic uptake and intracellular localization of the P. chrysogenum antifungal protein PAF in sensitive filamentous fungi [14, 45]. Importantly, we observed that AFPNN5353 was internalized by hyphae even under sub-inhibitory concentrations (0.2 μg/ml for A. nidulans) which suggests that a threshold concentration is required to cause severe growth defects in target fungi.
The presence of high concentrations of extracellular Ca2+ counteracted AFPNN5353 uptake. This finding parallels well with the report of [20] that the presence of cations, such as Ca2+, interfered with the binding of AFP to the surface of F. oxysporum and with our observations made with the Penicillium PAF (unpublished data). One possible explanation might be that extracellular Ca2+ ions compete with AFPNN5353 for the same molecular target on the fungal surface which might represent a first binding receptor or even a "gate" for protein uptake [20, 21] or, alternatively, that the interacting target is repressed under these conditions [17]. An additional explanation might be that the primary cell-surface localized AFPNN5353 target might be masked due to a Ca2+-dependent stimulation of chitin synthesis and cell wall remodeling as recently observed for AFP in A. niger [15]. This further suggests that the activation of the CWIP and the agsA induction does not mediate sufficient resistance to survive the toxic effects of AFPNN5353. Instead, according to the "damage-response framework of AFP-fungal interactions" [15], the chitin response might represent the better strategy for fungi to survive the antifungal attack.
Conclusions
Based on the growth inhibitory activity, antifungal proteins like AFPNN5353 can be well considered as promising candidates for future antimycotic drug developments. However, for biotechnological exploitation, the detailed knowledge on the mode of action is demanded. Our study shows that the detrimental effects caused by the A. giganteus antifungal protein AFPNN5353 in sensitive target aspergilli are based on the interaction of this protein with more than one signalling pathway. In Figure 7, we present a tentative working model. The toxicity of AFPNN5353 is mediated via PkcA/MpkA signalling which occurs independently from RhoA. Instead, so far unidentified RhoA-GAP effector molecules might contribute to AFPNN5353 toxicity. The activation of the CWIP by AFPNN5353 induces the agsA gene expression which is, however, insufficient to counteract toxicity of the protein. Furthermore, AFPNN5353 leads to an immediate and significant increase of the [Ca2+]c resting level in the cell. This sustained perturbation of the Ca2+ homeostasis could lead to PCD [17, 34]. The presence of extracellular Ca2+ neutralizes the toxic effects of AFPNN5353 and improves the resistance of the target organism possibly by decreasing the elevated [Ca2+]c resting level and stimulating the fortification of the cell wall by the induction of chsD expression as shown for AFP [15]. Further investigations are in progress to clarify how these pathways are interconnected and interfere with each other on the molecular level.

Tentative model of the mechanistic function of the A. giganteus antifungal protein AFP NN5353 on Aspergillus sp. The response against AFPNN5353 attack is mediated via PkcA/MpkA signalling and results in increased agsA transcription. However, the activity of the CWIP occurs independently from RhoA and so far unidentified RhoA-GAP effector molecules might contribute to the AFPNN5353 toxicity. Furthermore, AFPNN5353 leads to an immediate and significant increase of the [Ca2+]c resting level in the cell. The sustained perturbation of the Ca2+ homeostasis could lead to PCD [17, 34]. The presence of elevated concentrations of extracellular Ca2+ counteracts the toxic effects of AFPNN5353 and improves the resistance of the target organism by decreasing the elevated [Ca2+]c resting level. Whereas cell wall remodelling via CWIP seems to be insufficient to counteract AFPNN5353 activity, the fortification of the cell wall by the induction of chsD expression might represent an adequate response to increase resistance [15].
Methods
Strains, Media and Chemicals
Fungal strains used in this study are listed in Table 5. All strains were obtained from the culture collections FGSC, ATCC, CBS, from the Institute of Microbiology, Division of Systematics, Taxonomy and Evolutionary Biology at the Leopold Franzens University of Innsbruck, or the strain collection of the Department of Biotechnology, National Institute of Chemistry, Ljubljana, Slovenia. Unless otherwise stated, all fungi were grown in complete medium (CM) [19] with the respective supplements [28, 38]. R153 and alcA-PkcA were grown in defined minimal medium (MM) according to [26]. Ca2+ response experiments were performed in Vogels medium [46]. For experiments with CaCl2 supplementation, the KH2PO4 concentration of the culture media was reduced from 37 mM to 10 mM to avoid precipitation of supplemental Ca2+ and these media were called CM* and Vogels*. Chemicals were purchased from Sigma. AFPNN5353 and polyconal rabbit anti-AFPNN5353 antibody were generous gifts from Mogens T. Hansen, Novozymes, Denmark. The antifungal protein was isolated from A. giganteus strain A3274 (CBS 526.65), purified and analyzed by HPLC as described in the patent application WO94/01459 [47].
Growth inhibition assays
Antifungal activity assays were performed in 96-well plates in CM or Vogels medium inoculated with 1 × 104 conidia/ml and supplemented with various concentrations of AFPNN5353 or with equivalent amounts of buffer (untreated controls). Fungal growth was monitored microscopically with an Olympus CK40 microscope equipped with a Zeiss MRc digital camera and the growth rates were determined spectrophotometrically as described previously [19]. Alternatively, 2 × 103 conidia were spotted in 5 μl aliquots on appropriately supplemented agar plates. The plates were then incubated at 37°C for up to 72 h. Every 24 h, the plates were photographed and the colony diameters were determined. All assays were performed as technical triplicates and biological duplicates.
Analysis of the induction of the agsAexpression by a GFP-based reporter system
The A. niger reporter strain RD6.47 carries the agsA promoter fused to a nucleus-targeted GFP (H2B::eGFP) [27]. Activation of the CWIP can be monitored by the increase in nuclear fluorescence. Analysis of the activation of the agsA promoter by 10-100 μg/ml AFPNN5353 was performed as described in [10]. As a positive control, caspofungin at a concentration of 10 μg/ml was used. Fluorescence images were taken from coverslips observed with an Axioplan 2 microscope (Zeiss) equipped with a Sony DKC-5000 digital camera.
Fluorescence staining
Indirect immunofluorescence staining
A. nidulans was grown over night on glass cover slips at 30°C in CM. They were further incubated for 90 min in the presence or absence (controls) of 0.2 μg/ml AFPNN5353. The samples were stained as described previously [14] and incubated with rabbit-anti-AFPNN5353 antibody (1:2.500, Novozymes, Denmark) for at least 60 min. Immunocomplexes were detected with FITC-conjugated swine-anti-rabbit IgG (1:40, DAKO, Germany). All samples were embedded in Vectashield mounting medium (Vector Laboratories, Burlingame, USA). Microscopy was done with a Zeiss Axioplan fluorescence microscope or a Zeiss confocal laser scanning microscope as described in [14].
For incubation with latrunculin B (Sigma, Austria), samples were treated with 0.2 μg/ml AFPNN5353 and 10 μg/ml latrunculin B for 80 min. As a control, samples were treated with DMSO to exclude artifacts evoked by the dissolvent of latrunculin B.
For detection of AFPNN5353 in the presence of elevated concentrations of CaCl2, fungi were grown in CM* medium and then treated with 0.2 μg/ml AFPNN5353 in the presence of 10 mM CaCl2 for 90 min.
Analysis of membrane permeabilization and cell viability
To determine if AFPNN5353 permeabilized the plasma membrane of A. niger germlings, we used a combination of propidium iodide (PI) and fluorescein diacetate (cell tracker, CMFDA green, Invitrogen) according to [48]. Twelve h old A. niger germlings were grown in Vogels medium and pretreated with the two dyes (final conc. 5 μg/ml each) for 15 min before AFPNN5353 was added to a final concentration of 20 μg/ml. Samples without AFPNN5353 served as controls for positive CMFDA staining, while ethanol (70%) was used to permeabilize the membrane for positive PI staining.
Analysis of the calcium response to AFPNN5353application
105 conidia/ml of the A. niger strain A533 expressing codon optimized aequorin were grown in Vogels* medium containing 10 μM coelenterazine (Biosynth, Switzerland) at 30°C for twelve h in the dark. The [Ca2+]c resting level and mechanical perturbation experiments and the calibration of [Ca2+]c were performed as described in [17].
References
Hancock RE, Scott MG: The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci USA. 2000, 97 (16): 8856-8861. 10.1073/pnas.97.16.8856.
Kamysz W, Okroj M, Lukasiak J: Novel properties of antimicrobial peptides. Acta Biochim Pol. 2003, 50 (2): 461-469.
Aerts AM, Francois IE, Cammue BP, Thevissen K: The mode of antifungal action of plant, insect and human defensins. Cell Mol Life Sci. 2008, 65 (13): 2069-2079. 10.1007/s00018-008-8035-0.
Gupte MD, Kulkarni PR: A study of antifungal antibiotic production by Streptomyces chattanoogensis MTCC 3423 using full factorial design. Lett Appl Microbiol. 2002, 35 (1): 22-26. 10.1046/j.1472-765X.2002.01119.x.
Geisen R: P. nalgiovense carries a gene which is homologous to the paf gene of P. chrysogenum which codes for an antifungal peptide. Int J Food Microbiol. 2000, 62 (1-2): 95-101. 10.1016/S0168-1605(00)00367-6.
Lee GD, Shin SY, Maeng CY, Jin ZZ, Kim KL, Hahm KS: Isolation and characterization of a novel antifungal peptide from Aspergillus niger. Biochem Biophys Res Commun. 1999, 263: 646-651. 10.1006/bbrc.1999.1428.
Marx F, Haas H, Reindl M, Stoffler G, Lottspeich F, Redl B: Cloning, structural organization and regulation of expression of the Penicillium chrysogenum paf gene encoding an abundantly secreted protein with antifungal activity. Gene. 1995, 167 (1-2): 167-171. 10.1016/0378-1119(95)00701-6.
Wnendt S, Ulbrich N, Stahl U: Molecular cloning, sequence analysis and expression of the gene encoding an antifungal-protein from Aspergillus giganteus. Curr Genet. 1994, 25 (6): 519-523. 10.1007/BF00351672.
Binder U, Oberparleiter C, Meyer V, Marx F: The antifungal protein PAF interferes with PKC/MPK and cAMP/PKA signalling of Aspergillus nidulans. Mol Microbiol. 2010, 75 (2): 294-307. 10.1111/j.1365-2958.2009.06936.x.
Hagen S, Marx F, Ram AF, Meyer V: The antifungal protein AFP from Aspergillus giganteus inhibits chitin synthesis in sensitive fungi. Appl Environ Microbiol. 2007, 73 (7): 2128-2134. 10.1128/AEM.02497-06.
Lobo DS, Pereira IB, Fragel-Madeira L, Medeiros LN, Cabral LM, Faria J, Bellio M, Campos RC, Linden R, Kurtenbach E: Antifungal Pisum sativum defensin 1 interacts with Neurospora crassa cyclin F related to the cell cycle. Biochemistry. 2007, 46 (4): 987-996. 10.1021/bi061441j.
Marx F, Binder U, Leiter E, Pocsi I: The Penicillium chrysogenum antifungal protein PAF, a promising tool for the development of new antifungal therapies and fungal cell biology studies. Cell Mol Life Sci. 2008, 65 (3): 445-454. 10.1007/s00018-007-7364-8.
Moreno AB, Penas G, Rufat M, Bravo JM, Estopa M, Messeguer J, San Segundo B: Pathogen-induced production of the antifungal AFP protein from Aspergillus giganteus confers resistance to the blast fungus Magnaporthe grisea in transgenic rice. Mol Plant Microbe Interact. 2005, 18 (9): 960-972. 10.1094/MPMI-18-0960.
Oberparleiter C, Kaiserer L, Haas H, Ladurner P, Andratsch M, Marx F: Active internalization of the Penicillium chrysogenum antifungal protein PAF in sensitive aspergilli. Antimicrob Agents Chemother. 2003, 47 (11): 3598-3601. 10.1128/AAC.47.11.3598-3601.2003.
Ouedraogo JP, Hagen S, Spielvogel A, Engelhardt S, Meyer V: Survival strategies of yeast and filamentous fungi against the antifungal protein AFP. J Biol Chem. 2011, 286 (16): 13859-13868. 10.1074/jbc.M110.203588.
Fujioka T, Mizutani O, Furukawa K, Sato N, Yoshimi A, Yamagata Y, Nakajima T, Abe K: MpkA-Dependent and -independent cell wall integrity signaling in Aspergillus nidulans. Eukaryot Cell. 2007, 6 (8): 1497-1510. 10.1128/EC.00281-06.
Binder U, Chu M, Read ND, Marx F: The antifungal activity of the Penicillium chrysogenum protein PAF disrupts calcium homeostasis in Neurospora crassa. Eukaryot Cell. 2010, 9 (9): 1374-1382. 10.1128/EC.00050-10.
Thevissen K, Ghazi A, De Samblanx GW, Brownlee C, Osborn RW, Broekaert WF: Fungal membrane responses induced by plant defensins and thionins. J Biol Chem. 1996, 271 (25): 15018-15025. 10.1074/jbc.271.25.15018.
Kaiserer L, Oberparleiter C, Weiler-Gorz R, Burgstaller W, Leiter E, Marx F: Characterization of the Penicillium chrysogenum antifungal protein PAF. Arch Microbiol. 2003, 180 (3): 204-210. 10.1007/s00203-003-0578-8.
Martin-Urdiroz M, Martinez-Rocha AL, Di Pietro A, Martinez-del-Pozo A, Roncero MI: Differential toxicity of antifungal protein AFP against mutants of Fusarium oxysporum. Int Microbiol. 2009, 12 (2): 115-121.
Theis T, Wedde M, Meyer V, Stahl U: The antifungal protein from Aspergillus giganteus causes membrane permeabilization. Antimicrob Agents Chemother. 2003, 47 (2): 588-593. 10.1128/AAC.47.2.588-593.2003.
Wnendt S, Felske-Zech H, Henze PP, Ulbrich N, Stahl U: Characterization of the gene encoding alpha-sarcin, a ribosome-inactivating protein secreted by Aspergillus giganteus. Gene. 1993, 124 (2): 239-244. 10.1016/0378-1119(93)90399-N.
Meyer V: A small protein that fights fungi: AFP as a new promising antifungal agent of biotechnological value. Appl Microbiol Biotechnol. 2008, 78 (1): 17-28. 10.1007/s00253-007-1291-3.
Levin DE: Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2005, 69 (2): 262-291. 10.1128/MMBR.69.2.262-291.2005.
Damveld RA, Arentshorst M, Franken A, vanKuyk PA, Klis FM, van den Hondel CA, Ram AF: The Aspergillus niger MADS-box transcription factor RlmA is required for cell wall reinforcement in response to cell wall stress. Mol Microbiol. 2005, 58 (1): 305-319. 10.1111/j.1365-2958.2005.04827.x.
Ronen R, Sharon H, Levdansky E, Romano J, Shadkchan Y, Osherov N: The Aspergillus nidulans pkcA gene is involved in polarized growth, morphogenesis and maintenance of cell wall integrity. Curr Genet. 2007, 51 (5): 321-329. 10.1007/s00294-007-0129-y.
Meyer V, Damveld RA, Arentshorst M, Stahl U, van den Hondel CA, Ram AF: Survival in the presence of antifungals: genome-wide expression profiling of Aspergillus niger in response to sublethal concentrations of caspofungin and fenpropimorph. J Biol Chem. 2007, 282 (45): 32935-32948. 10.1074/jbc.M705856200.
Guest GM, Lin X, Momany M: Aspergillus nidulans RhoA is involved in polar growth, branching, and cell wall synthesis. Fungal Genet Biol. 2004, 41 (1): 13-22. 10.1016/j.fgb.2003.08.006.
Terras FR, Schoofs HM, De Bolle MF, Van Leuven F, Rees SB, Vanderleyden J, Cammue BP, Broekaert WF: Analysis of two novel classes of plant antifungal proteins from radish (Raphanus sativus L.) seeds. J Biol Chem. 1992, 267 (22): 15301-15309.
Terras FR, Torrekens S, Van Leuven F, Osborn RW, Vanderleyden J, Cammue BP, Broekaert WF: A new family of basic cysteine-rich plant antifungal proteins from Brassicaceae species. FEBS Lett. 1993, 316 (3): 233-240. 10.1016/0014-5793(93)81299-F.
Bencina M, Legisa M, Read ND: Cross-talk between cAMP and calcium signalling in Aspergillus niger. Mol Microbiol. 2005, 56 (1): 268-281. 10.1111/j.1365-2958.2005.04541.x.
Nelson G, Kozlova-Zwinderman O, Collis AJ, Knight MR, Fincham JR, Stanger CP, Renwick A, Hessing JG, Punt PJ, van den Hondel CA, Read ND: Calcium measurement in living filamentous fungi expressing codon-optimized aequorin. Mol Microbiol. 2004, 52 (5): 1437-1450. 10.1111/j.1365-2958.2004.04066.x.
Moreno AB, Martinez Del Pozo A, San Segundo B: Biotechnologically relevant enzymes and proteins. Antifungal mechanism of the Aspergillus giganteus AFP against the rice blast fungus Magnaporthe grisea. Appl Microbiol Biotechnol. 2006, 72 (5): 883-895. 10.1007/s00253-006-0362-1.
Leiter E, Szappanos H, Oberparleiter C, Kaiserer L, Csernoch L, Pusztahelyi T, Emri T, Pocsi I, Salvenmoser W, Marx F: Antifungal protein PAF severely affects the integrity of the plasma membrane of Aspergillus nidulans and induces an apoptosis-like phenotype. Antimicrob Agents Chemother. 2005, 49 (6): 2445-2453. 10.1128/AAC.49.6.2445-2453.2005.
Morton WM, Ayscough KR, McLaughlin PJ: Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol. 2000, 2 (6): 376-378. 10.1038/35014075.
Conner SD, Schmid SL: Regulated portals of entry into the cell. Nature. 2003, 422 (6927): 37-44. 10.1038/nature01451.
Lamaze C, Fujimoto LM, Yin HL, Schmid SL: The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997, 272 (33): 20332-20335. 10.1074/jbc.272.33.20332.
Bussink HJ, Osmani SA: A mitogen-activated protein kinase (MPKA) is involved in polarized growth in the filamentous fungus, Aspergillus nidulans. FEMS Microbiol Lett. 1999, 173 (1): 117-125.
Ichinomiya M, Uchida H, Koshi Y, Ohta A, Horiuchi H: A protein kinase C-encoding gene, pkcA, is essential to the viability of the filamentous fungus Aspergillus nidulans. Biosci Biotechnol Biochem. 2007, 71 (11): 2787-2799. 10.1271/bbb.70409.
Reinoso-Martin C, Schuller C, Schuetzer-Muehlbauer M, Kuchler K: The yeast protein kinase C cell integrity pathway mediates tolerance to the antifungal drug caspofungin through activation of Slt2p mitogen-activated protein kinase signaling. Eukaryot Cell. 2003, 2 (6): 1200-1210. 10.1128/EC.2.6.1200-1210.2003.
Igual JC, Johnson AL, Johnston LH: Coordinated regulation of gene expression by the cell cycle transcription factor Swi4 and the protein kinase C MAP kinase pathway for yeast cell integrity. EMBO J. 1996, 15 (18): 5001-5013.
Jung US, Levin DE: Genome-wide analysis of gene expression regulated by the yeast cell wall integrity signalling pathway. Mol Microbiol. 1999, 34 (5): 1049-1057. 10.1046/j.1365-2958.1999.01667.x.
Kuranda K, Leberre V, Sokol S, Palamarczyk G, Francois J: Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol Microbiol. 2006, 61 (5): 1147-1166. 10.1111/j.1365-2958.2006.05300.x.
Ramamoorthy V, Zhao X, Snyder AK, Xu JR, Shah DM: Two mitogen-activated protein kinase signalling cascades mediate basal resistance to antifungal plant defensins in Fusarium graminearum. Cell Microbiol. 2007, 9 (6): 1491-1506. 10.1111/j.1462-5822.2006.00887.x.
Batta G, Barna T, Gaspari Z, Sandor S, Kover KE, Binder U, Sarg B, Kaiserer L, Chhillar AK, Eigentler A, Leiter É, Hgedüs N, Pócsi I, Lindner H, Marx F: Functional aspects of the solution structure and dynamics of PAF - a highly-stable antifungal protein from Penicillium chrysogenum. FEBS J. 2009, 276 (10): 2875-2890. 10.1111/j.1742-4658.2009.07011.x.
Vogel HJA: Convenient growth medium for Neurospora (Medium N). Microbiological Genetics Bulletin. 1956, 13: 42-43.
Pedersen AH, Halkier T, Nielsen BA, Lange L, Mikkelsen JM, Rasmussen G, Hansen MT: A fungicidally active compound. 1994, Novo Nordisk A/S, Denmark; Patent WO 94/01459
Palma-Guerrero J, Huang IC, Jansson HB, Salinas J, Lopez-Llorca LV, Read ND: Chitosan permeabilizes the plasma membrane and kills cells of Neurospora crassa in an energy dependent manner. Fungal Genet Biol. 2009, 46 (8): 585-594. 10.1016/j.fgb.2009.02.010.
Acknowledgements
We thank Mogens T. Hansen (Novozymes, Denmark) for the generous gift of AFPNN5353 and the polyclonal rabbit anti-AFPNN5353 antibody. We gratefully acknowledge Renate Weiler-Görz for technical assistance.
This study was financially supported by the Austrian Science Fund FWF (P19970-B11) and the Österreichischer Austauschdienst ÖAD (Wissenschaftlich-Technische Zusammenarbeit Österreich und Slowenien, SI15/2009).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
UB carried out the growth inhibition assays, the indirect immunofluorescence stainings, the Ca2+ measurements and the calculations to convert the luminescence units into the [Ca2+]c levels. She also performed the statistical analysis and helped to draft the manuscript. MB contributed the A. niger A533 strain, helped with the Ca2+ measurements and participated in the design of the study. AE contributed to the indirect immunofluorescence stainings. VM contributed the A. niger RD6.47 strain and performed the agsA induction assays. FM conceived of the study, participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2011_1499_MOESM1_ESM.TIFF
Additional file 1: The expression of nucleus-targeted GFP under the control of the agsA promoter in A. niger in response to cell wall interfering substances. Differential interfering contrast images and corresponding fluorescence images of A. niger RD6.47 indicate the expression of a nucleus-targeted GFP under the control of the A. niger agsA promoter. Five h old germlings were (A) left untreated (negative control), (B) treated with 50 μg/ml AFPNN5353 and (C) with 10 μg/ml caspofungin (positive control) as described in Materials and Methods. Scale bar, 20 μm. (TIFF 2 MB)
12866_2011_1499_MOESM2_ESM.TIFF
Additional file 2: Viability staining of A. niger germlings after AFP NN5353 exposure. Twelve h old A. niger germlings were stained with fluorescein diacetate (CMFDA, middle pannels) and propidium iodide (right pannels). The left panels show the respective light micrographs. All samples were pretreated with the dyes for 15 min before 20 μg/ml AFPNN5353 was added (B). Controls remained untreated (A) or were exposed to 70% ethanol (C). Scale bar, 50 μm. (TIFF 9 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Binder, U., Bencina, M., Eigentler, A. et al. The Aspergillus giganteus antifungal protein AFPNN5353activates the cell wall integrity pathway and perturbs calcium homeostasis. BMC Microbiol 11, 209 (2011). https://doi.org/10.1186/1471-2180-11-209
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-11-209