
Abstract
Background
The large amount of data produced by high-throughput sequencing poses new computational challenges. In the last decade, several tools have been developed for the identification of transcription and splicing factor binding sites.
Results
Here, we introduce the SeAMotE (Sequence Analysis of Motifs Enrichment) algorithm for discovery of regulatory regions in nucleic acid sequences. SeAMotE provides (i) a robust analysis of high-throughput sequence sets, (ii) a motif search based on pattern occurrences and (iii) an easy-to-use web-server interface. We applied our method to recently published data including 351 chromatin immunoprecipitation (ChIP) and 13 crosslinking immunoprecipitation (CLIP) experiments and compared our results with those of other well-established motif discovery tools. SeAMotE shows an average accuracy of 80% in finding discriminative motifs and outperforms other methods available in literature.
Conclusions
SeAMotE is a fast, accurate and flexible algorithm for the identification of sequence patterns involved in protein-DNA and protein-RNA recognition. The server can be freely accessed at http://s.tartaglialab.com/new_submission/seamote.
Similar content being viewed by others

Background
Transcriptional and post-transcriptional events involve the interplay between protein effectors and nucleic acid targets, whose physical interaction is guided by sequence motifs and specific structural elements [1–3]. Motifs are usually defined as short nucleotide sequence patterns of length k (k-mers) and represented with matrices containing the probabilities to find nucleotides in specific positions (position weighted matrices PWMs). In the past decade, the advancement of high-throughput technologies contributed to the generation of a large amount of genomic data [4], promoting development of computational methods to detect regulatory elements such as transcription and splicing factor binding sites [5]. One fundamental requirement of methods for large-scale analysis is that relevant features (e.g., recognition motifs) are identified with good accuracy and in reasonable time [6, 7]. Very importantly, algorithms should be as comprehensive as possible to provide insights into the nature of regulatory elements in their real genomic context, which requires analysis of different biological sets [8].
As discussed by Ma et al.[9] and Weirauch et al.[10], there are several algorithms for de novomotif discovery, but only few are capable of performing a discriminative analysis (i.e., comparison between two sets) on high-throughput datasets:
-
DREME [11] restricts the search for sequence motifs to a simplified form of “regular expression” (RE) words over the IUPAC alphabet, which consists of 11 wildcard characters in addition to the standard DNA alphabet (ACGT). To save computation time, DREME estimates the significance of RE candidates by a heuristic search without scanning the whole input sequences [11];
-
CMF (Contrast Motif Finder) [12] is designed to discriminate between two sets of DNA sequences through non-discrete PWMs. The method takes into account false positive sites when updating PWMs and related variables;
-
DECOD (DECOnvolved Discriminative motif discovery) [13] uses k-mer counts. To compensate for errors introduced from ignoring the context of the k-mer, DECOD uses a deconvolution method that accounts for the higher rates of k-mers containing subsets of the true motif;
-
XXmotif (eXhaustive, weight matriX-based motif discovery) [14] consists of i) a masking stage, where repeat regions, compositionally biased segments and homologous segment pairs are identified; ii) a pattern stage, where p-value enrichments are calculated for seed patterns using all 5-mers (with up to two degenerate IUPAC characters); iii) and a PWM stage, where thousands of candidate PWMs are merged.
Despite the variety of motif discrimination approaches, knowledge of programming languages [8, 15] and acquaintance with web-based bioinformatics platforms [7, 16] often limit their use among non-specialists.
In this article, we introduce SeAMotE, a web-server to perform de novo discriminative motif discovery in nucleic acid datasets. We present an approach that enables the exhaustive search of distinctive patterns in large sets of sequences, in a reasonable amount of computational time and with an easy-to-use user interface.
Methods
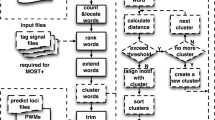
SeAMotE is based on the generation of a pool of nucleotide seeds followed by “zero or one occurrence per sequence” (ZOOPs) model testing [17] coupled with pattern extension and refinement [8]. SeAMotE includes a number of unique features that make the algorithm simple to run and very accurate. The user can i) set a coverage threshold that is employed in the selection of enriched motifs in the positive set (foreground), ii) choose among multiple reference (background) set options and iii) include a redundancy removal step to increase the variability of discovered motifs. As shown in Figure 1, SeAMotE workflow comprises a series of steps that can be summarized as follows:

SeAMotE workflow. Illustration of the method pipeline: red boxes indicate the coverage calculation and seed extension loop; dashed arrows and the blue box represent conditional steps that depend on the user-definable variables, such as providing or selecting a specific background set or filtering out patterns that are closely related.
-
1.
Generation of a pool of k-mers seed motifs using the IUPAC alphabet;
-
2.
Evaluation of the coverage of each pattern in the positive and reference sets;
-
3.
Determination of enriched (Fisher’s exact test) and differentially represented (Youden’s index = Sensitivity + Specificity - 1) motifs;
-
4.
Extension of selected seeds by adding a IUPAC letter in the k + 1 position;
-
5.
Re-iteration of steps 2-4 until the enrichement of at least one pattern remains above the coverage threshold in the positive set;
-
6.
Calculation of motif significance (Fisher’s exact test) and redundancy removal (Hamming distance);
-
7.
Generation of the positon weighted matrices and logo for each motif.
Web-server usage
The SeAMotE server presents a submission page that allows the upload of nucleic acid sequences and selection of parameters. Default parameters (e.g. reference set, coverage threshold, etc.) are defined according to best settings estimated using cross-validation (section “Cross-validation on the CLIP-seq data” in Result and Discussion). However, most of the parameters can be modified by the user, which adds flexibility to the web-service. Detailed descriptions of the submission and output variables are provided in the on-line tutorial (see http://service.tartaglialab.com/static_files/shared/tutorial_seamote.html, tutorial sections “Submission form” and “Interpreting the output”, respectively).
-
1.
At least one input set (FASTA format file) should be provided for the analysis. Currently, the number of sequences is limited to 104, with a maximal length of 15 ·103 nucleotides per sequence;
-
2.
A reference set is required to estimate the significance of the discovered motifs. This can be:
-
Provided by the user (FASTA format file), having the same size restrictions as the input set.
-
Automatically generated as a shuffle set, where the foreground set composition (i.e., individual nucleotide alphabet frequencies) and dimensions (i.e., number of sequences and lengths) are kept constant;
-
Automatically generated as a random set, where the foreground set dimensions are preserved but the internal composition is based on letter frequencies obtained from the human transcriptome/genome;
-
-
3.
The coverage threshold (i.e. the percentage of sequences matching the searched pattern) represents a parameter that the algorithm uses internally to select the most abundant motifs in the two datasets (speed of calculation increases at low coverage threshold).
Optionally, the user can assign a job name for each submission and request for an e-mail notification upon completion (not required to run the server).The output summary contains detailed information about the submission (e.g., job identifier, downloadable datasets) as well as an interactive table (Figure 2). The latter item displays discovered motifs (IUPAC and RE formats), logo representations and statistics used to estimate their significance: motif coverage for positive and reference sets, discrimination factor (Youden’s index) and p-value (Fisher’s exact test) associated with each pattern. In addition, it is possible to retrieve the list of motifs tested (txt format), as well as their individual sequence logo (png format) and positional weighted matrix (txt format) using the links provided in the output page (Figure 2).

SeAMotE output summary. Example of output table showing the list of motifs (IUPAC and RegEx) that better discriminate the input sets along with their logo representation and positional weighted matrix download button, positive and reference coverage (as percentage of sequences containing at least one pattern occurrence), discrimination (Youden’s index) and p-value (Fisher’s exact test). By clicking on the logo, it is possible to retrieve the image file (png format) of the associated motif.
Implementation
SeAMotE operations are executed by C programs, whereas significance estimation, pattern filtering and sequence logo design are computed using R scripts and the Biostrings, stringdist and seqLogo packages. The web-server is implemented in Python, HTML and JavaScript, which provides a convenient framework for the pipeline control and the presentation of the output data. User-provided data are validated by Python scripts and passed to the Amazon Web Services (AWS), which manages the queue system, performs the redistribution of the work on our local machines and, once the job is completed successfully, forwards the user to the output page. Depending on the size datasets and the server load, computations take from between 2–3 and 30–40 minutes (Additional file 1: Figure S1).
Datasets for motif finding
Nucleic acids sequences were collected from ChIP-seq and CLIP-seq experiments available in the public domain [18, 19]. ChIP-seq data comprises 351 ENCODE datasets obtained from three groups, Haib_Tfbs by HudsonAlpha (141 sets), Sydh_Tfbs by Yale and UCD (164 sets), and Uw_Tfbs by University of Washington (46 sets). This collection covers 90 unique transcription factors (TFs) and more than 50 cell-types under different treatments. Same number of low and high intensity peaks (1000 sequences) was used to select negative and positive datasets, respectively [20]. CLIP-seq dataset contains 13 doRINA [18] datasets of 10 RNA-binding proteins (RBPs) [21–28]. Sequences with doRINA scores in the top 5 percentile were considered as positives (bound transcripts; more details on the definition of peaks and the calculation of associated scores can be found in doRINA paper [18]). For each positive set, we selected same amount of sequences in the bottom 5 percentile of doRINA scores to build the negative set (unbound transcripts).
Documentation
The documentation/tutorial of the SeAMotE algorithm is available online, and it can be accessed using the links in the menu at the top of every server page. It contains a brief description of the method, a tutorial and information on the benchmark. Additionally, the web interface in the output page provides help-notes (accessible also through the “mouse-over” function) for table variables and download buttons. Online documentation and “Frequently Asked Questions” (FAQs) sections updates will be provided on a regular basis according to method improvements and users’ inquiries,respectively.
Results and discussion
Identification of TF annotated motifs
To assess the performance of our method for discriminative motif discovery, we run SeAMotE on a collection of 351 ChIP-seq datasets and compared our results with those obtained using other discriminative motif finders. Specifically, we restricted the comparison to methods such as CMF, DECOD, DREME and XXmotif that can be run in batch on large sets of sequences. All methods were run on the same sets of foreground and background sequences under default settings. For each algorithm, we selected up to 5 top-scored motifs. To evaluate the ability of the different methods to recognise sequence patterns reported in literature, we collected TF motifs present in Jaspar [29] and Jolma et al.[30]. We then compared the PWMs generated by CMF, DECOD, DREME, SeAMotE and XXmotif with the motifs available in the reference databases. We considered successful the prediction in which the annotated motif was reported as TOMTOM [31] match. Figures 3A and 3B report the E-value (i.e., the p-value multiplied by twice the number of target motifs) and q-value (the minimal false discovery rate at which the observed similarity would be considered significant) obtained from the analyses with TOMTOM. As shown in Figure 3C and Table 1, SeAMotE succeeded in finding the consensus motifs in 282 (80.3%) cases out of the 351 ChIP datasets with annotated motifs for the TFs. CMF found annotated motifs in 276 (78.6%), DECOD in 248 (70.6%), DREME in 277 (78.9%) and XXmotif in 243 (69.2%) cases (Figure 1C and Table 1). SeAMotE was able to identify annotated motifs in 74.6% of the cases even when considering only the top-ranked PWM (other methods recognized <67% of experimental cases; Figure 1C and Table 1).

Annotated motifs performance comparison. Using 351 ChIP-seq datasets from ENCODE [19], we compared CMF [12], DECOD [13], DREME [11], XXmotif [14] and SeAMotE performances; A) E-values and B) q-values associated with the 5 top-ranked motifs for CMF, DECOD, DREME, SeAMotE and XXmotif. C) Proportion of transcription factors for which annotated motifs were succesfully identified is plotted against the number of top-ranked motifs employed for the TOMTOM search [31].
In 69 out of 351 cases (i.e. 20% of the dataset), SeAMotE identified motifs that are different from those reported in Jaspar[29] and Jolma et al.[30] databases. CMF and DREME identified different patterns in 74% and 67% of such cases (i.e., 51 out of 69 and 46 out of 69, respectively), which suggests that this group of TFs might display diverse binding modes. Indeed, with respect to the 282 successful hits, these motifs are predicted with significantly lower discrimination (p-value = 5.88e−5; Mann-Whitney U test on discrimination). Thus, it is possible that the discrepancy with literature data arises from lower sequence specificity of the TFs, which makes the foreground and background sets more similar and, therefore, less informative. It should be also mentioned that the 69 misassigned cases correspond to 42 TFs, and for 28 of them(66.7%) SeAMotE was able to correctly recognise the annotated binding pattern in at least one cell-type or specific treatment [19]. We also observe that some of the unassigned patterns can be correctly attributed to literature motifs if other comparison tools are employed instead of TOMTOM. In an additional calculation, we used Matlign [32] to compare the similarity between literature patterns and the top-ranked motif identified by SeAMotE. In 36 out of 69 cases, we found that SeAMotE motifs have higher propensity to cluster with those of the same TF family [29, 30]. Intriguingly, we observe that in 54 out of the 69 cases (78.3%) the top-ranked motif is associated with one PWM of an interacting TF, indicating that TF binding could be mediated by other proteins.
Identification of RBP recognition motifs
To demonstrate the flexibility of our method for different types of nucleic acids, we assessed SeAMotE ability to identify significantly enriched motifs in transcripts studied by CLIP-seq technology [33]. In each case analysed, we compared RNAs bound to a specific protein (foreground set) with same amount of unbound transcripts (background set). Since CMF does not allow the discriminative motif discovery on specific nucleic acid strand, we excluded the algorithm from the study and used the other tools for comparison. In our analysis (Figure 3C) we noticed that SeAMotE and DREME show best performances in finding discriminative motifs, followed by DECOD and XXmotif. This result was confirmed also in the analysis of RBP targets (Figure 3C). Indeed, SeAMotE and DREME outperform DECOD and XXmotif in finding sequence patterns (Figure 4A). Compared to DREME, SeAMotE achieves significantly higher discrimination (p-value = 1.36e−14; Mann-Whitney U test), which is reflected in the ability to better separate foreground from background sets (Figure 4A), and significance, denoted by lower p-values associated with each sequence pattern identified (Figure 4B). In addition, SeAMoTe also shows very high sensitivity (∼89%) and accuracy (∼81%) (Table 2). Statistical measures of the performance are also reported for DECOD and XXmotif (Additional file 2: Table S1; p-value for SeAMotE - DECOD comparison: 3.95e−16; p-value for SeAMotE - XXmotif comparison: 1.09e−07; Mann-Whitney U test).

RNA-binding protein motifs performance comparison. Using 13 CLIP-seq experiments available in the public domain [18], we compared DECOD [13], DREME [11], XXmotif [14] and SeAMotE performances. The ability to identify sequence elements that maximize the separation between positive and reference sets is reported for each motif identified using A) discrimination (Youden’s index) and B) significance (FisherŠs exact test). CMF [12] was excluded from the analysis because it does not allow motif discovery on a nucleic acid specific strand.
Cross-validation of the CLIP-seq data
Finally, we assessed SeAMotE performances using a 3-fold cross-validation approach introduced by Patel and Stormo [34]: CLIP-seq sets of positive and negative sequences were randomly divided into three sets of similar sizes (P1, P2, P3) and (N1, N2, N3); two of the three were combined to form a training set and the remaining one was used as test set. By this means, three training (TR1, TR2 and TR3) and three test sets (TE1, TE2 and TE3) were generated. We then compared the most significant motifs found in the training with those present in the test set using TOMTOM [31] (p-value <0.01). SeAMotE was able to correctly reproduce the most enriched motifs using training and testing sets, thus confirming the robustness of our approach (Additional file 3: Table S2).
Conclusions
Algorithms for discriminative motif discovery are useful to identify regulatory elements in DNA and RNA sequences. Comparisons between different sets provide relevant information to rationalize sequence determinants of physical interactions and can be exploited for future experimental design. In this work, we introduced the SeAMotE algorithm for analysis of large-scale nucleic acid datasets. Through an easy-to-use interface, the SeAMotE web-server offers key features such as fast discrimination based on pattern occurrence, choice of multiple reference backgrounds (shuffle, random or custom) and identification of significant motifs in the whole span of tested pattern widths, which provides a range of practical solutions for analysis of experimental data. Indeed, as reported in recent studies, inter-positional sequence patterns and variable binding sites information are key features to identify regulatory motifs and will be used in future computational developments [35]. We demonstrated the powerfulness of SeAMotE for a large number of TF targets, correctly reproducing the results available in literature and showing better performances than other available tools. We also proved the flexibility and robustness of the algorithm by assessing its ability to identify enriched sequence patterns in CLIP experiments and using a three-fold cross-validation. We anticipate that the use of SeAMotE and its integration into DNA/RNA-protein interaction predictors, such as catRAPID [36, 37], would greatly enhance the ability to recognise physical associations.
Availability and requirements
● Project name: SeAMotE
● Project home page:http://s.tartaglialab.com/new_submission/seamote
● Operating system(s): Platform independent
● Programming language: C and R scripts
● Other requirements: Web browser (e.g. Safari, Firefox, Explorer or Chrome)
● Restrictions: No login requirement; users behind a proxy might experience slow-down issues
References
Coulon A, Chow CC, Singer RH, Larson DR: Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet. 2013, 14 (8): 572-584. 10.1038/nrg3484. doi:10.1038/nrg3484. PMID: 23835438 PMCID: PMC3807637.,
Janga SC: From specific to global analysis of posttranscriptional regulation in eukaryotes: posttranscriptional regulatory networks. Brief Funct Genomics. 2012, 11 (6): 505-521. 10.1093/bfgp/els046. doi:10.1093/bfgp/els046. PMID: 23124862.,
Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, Willis AEE: RNA binding protein/RNA element interactions and the control of translation. Curr Protein Peptide Sci. 2012, 13 (4): 294-304. 10.2174/138920312801619475. PMID: 22708490 PMCID: PMC3431537.,
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER: The next-generation sequencing revolution and its impact on genomics. Cell. 2013, 155 (1): 27-38. 10.1016/j.cell.2013.09.006. doi:10.1016/j.cell.2013.09.006. PMID: 24074859.,
Dassi E, Quattrone A: Tuning the engine: an introduction to resources on post-transcriptional regulation of gene expression. RNA Biol. 2012, 9 (10): 1224-1232. 10.4161/rna.22035. doi:10.4161/rna.22035. PMID: 22995832 PMCID: PMC3583852.,
Sinha S: Discriminative motifs. J Comput Biol: J Comput Mol Cell Biol. 2003, 10 (3–4): 599-615. doi:10.1089/10665270360688219. PMID: 12935347.,
Grau J, Posch S, Grosse I, Keilwagen J: A general approach for discriminative de novo motif discovery from high-throughput data. Nucleic Acids Res. 2013, 41 (21): 197-10.1093/nar/gkt831. doi:10.1093/nar/gkt831. PMID: 24057214 PMCID: PMC3834837.,
Yao Z, Macquarrie KL, Fong AP, Tapscott SJ, Ruzzo WL, Gentleman RC: Discriminative motif analysis of high-throughput dataset. Bioinformatics (Oxford, England). 2014, 30 (6): 775-783. 10.1093/bioinformatics/btt615. doi:10.1093/bioinformatics/btt615.,
Ma X, Kulkarni A, Zhang Z, Xuan Z, Serfling R, Zhang MQ: A highly efficient and effective motif discovery method for ChIP-seq/ChIP-chip data using positional information. Nucleic Acids Res. 2012, 40 (7): 50-10.1093/nar/gkr1135. doi:10.1093/nar/gkr1135. PMID: 22228832 PMCID: PMC3326300.,
Weirauch MT, Cote A, Norel R, Annala M, Zhao Y, Riley TR, Saez-Rodriguez J, Cokelaer T, Vedenko A, Talukder S, Bussemaker HJ, Morris QD, Bulyk ML, Stolovitzky G, Hughes TR, DREAM5 Consortium: Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol. 2013, 31 (2): 126-134. 10.1038/nbt.2486. doi:10.1038/nbt.2486. PMID: 23354101 PMCID: PMC3687085.,
Bailey TL: DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics (Oxford, England). 2011, 27 (12): 1653-1659. 10.1093/bioinformatics/btr261. doi:10.1093/bioinformatics/btr261. PMID: 21543442 PMCID: PMC3106199.,
Mason MJ, Plath K, Zhou Q: Identification of context-dependent motifs by contrasting ChIP, binding data. Bioinformatics (Oxford, England). 2010, 26 (22): 2826-2832. 10.1093/bioinformatics/btq546. doi:10.1093/bioinformatics/btq546. PMID: 20870645 PMCID: PMC2971577.,
Huggins P, Zhong S, Shiff I, Beckerman R, Laptenko O, Prives C, Schulz MH, Simon I, Bar-Joseph Z: DECOD: fast and accurate discriminative DNA motif finding. Bioinformatics (Oxford, England). 2011, 27 (17): 2361-2367. 10.1093/bioinformatics/btr412. doi:10.1093/bioinformatics/btr412. PMID: 21752801 PMCID: PMC3157928.,
Luehr S, Hartmann H, Söding J: The XXmotif web server for eXhaustive, weight matriX-based motif discovery in nucleotide sequences. Nucleic Acids Res. 2012, 40 (Web Server issue): 104-109. doi:10.1093/nar/gks602. PMID: 22693218 PMCID: PMC3394272.,
Fauteux F, Blanchette M, Strömvik MV: Seeder: discriminative seeding DNA motif discovery. Bioinformatics. 2008, 24 (20): 2303-2307. 10.1093/bioinformatics/btn444. doi:10.1093/bioinformatics/btn444. PMID: 18718942.,
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A: Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005, 15 (10): 1451-1455. 10.1101/gr.4086505. doi:10.1101/gr.4086505. PMID: 16169926 PMCID: PMC1240089.,
Bailey TL, Elkan C: The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol; ISMB. Int Conf Intell Syst Mol Biol. 1995, 3: 21-29. PMID: 7584439.,
Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C: doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012, 40 (Database issue): 180-186. doi:10.1093/nar/gkr1007. PMID: 22086949 PMCID: PMC3245013.,
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, et al: GENCODE: the reference human genome annotation for the ENCODE, project. Genome Res. 2012, 22 (9): 1760-1774. 10.1101/gr.135350.111. doi:10.1101/gr.135350.111. PMID: 22955987 PMCID: PMC3431492.,
Euskirchen GM, Rozowsky JS, Wei C-L, Lee WH, Zhang ZD, Hartman S, Emanuelsson O, Stolc V, Weissman S, Gerstein MB, Ruan Y, Snyder M: Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007, 17 (6): 898-909. 10.1101/gr.5583007. doi:10.1101/gr.5583007. PMID: 17568005 PMCID: PMC1891348.,
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T: PAR-CliP–a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Visualized Exper: JoVE. 2010, 41: doi:10.3791/2034.. PMID: 20644507 PMCID: PMC3156069.,
Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N: Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011, 43 (3): 340-352. 10.1016/j.molcel.2011.06.008. doi:10.1016/j.molcel.2011.06.008. PMID: 21723171.,
Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M: A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011, 8 (7): 559-564. 10.1038/nmeth.1608. doi:10.1038/nmeth.1608. PMID: 21572407.,
Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano JM, Tuschl T, Ohler U, Keene JD: Integrative regulatory mapping indicates that the RNA-binding, protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011, 43 (3): 327-339. 10.1016/j.molcel.2011.06.007. doi:10.1016/j.molcel.2011.06.007. PMID: 21723170 PMCID: PMC3220597.,
Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T: RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011, 18 (12): 1428-1431. 10.1038/nsmb.2163. doi:10.1038/nsmb.2163. PMID: 22081015 PMCID: PMC3230689.,
Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, Mooney SD, Edenberg HJ, Liu Y: Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009, 19 (3): 381-394. doi:10.1101/gr.082503.108. PMID: 19116412 PMCID: PMC2661799.,
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J: Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011, 14 (4): 452-458. 10.1038/nn.2778. doi:10.1038/nn.2778. PMID: 21358640 PMCID: PMC3108889.,
Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, Ule J: iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 2010, 8 (10): 1000530-10.1371/journal.pbio.1000530. doi:10.1371/journal.pbio.1000530. PMID: 21048981 PMCID: PMC2964331.,
Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C, Chou A, Ienasescu H, Lim J, Shyr C, Tan G, Zhou M, Lenhard B, Sandelin A, Wasserman WW: JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014, 42 (D1): 142-147. 10.1093/nar/gkt997. doi:10.1093/nar/gkt997.,
Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, Palin K, Vaquerizas JM, Vincentelli R, Luscombe NM, Hughes TR, Lemaire P, Ukkonen E, Kivioja T, Taipale J: DNA-binding specificities of human transcription factors. Cell. 2013, 152 (1–2): 327-339. doi:10.1016/j.cell.2012.12.009. PMID: 23332764.,
Tanaka E, Bailey T, Grant CE, Noble WS, Keich U: Improved similarity scores for comparing motifs. Bioinformatics (Oxford, England). 2011, 27 (12): 1603-1609. 10.1093/bioinformatics/btr257. doi:10.1093/bioinformatics/btr257. PMID: 21543443 PMCID: PMC3106196.,
Kankainen M, Löytynoja A: MATLIGN: a motif clustering, comparison and matching tool. BMC Bioinformatics. 2007, 8: 189-10.1186/1471-2105-8-189. doi:10.1186/1471-2105-8-189.,
Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB: CLIP identifies nova-regulated RNA networks in the brain. Science (New York, N.Y.). 2003, 302 (5648): 1212-1215. 10.1126/science.1090095. doi:10.1126/science.1090095. PMID: 14615540.,
Patel RY, Stormo GD: Discriminative motif optimization based on perceptron training. Bioinformatics (Oxford, England). 2014, 30 (7): 941-948. 10.1093/bioinformatics/btt748. doi:10.1093/bioinformatics/btt748. PMID: 24369152 PMCID: PMC3967114.,
Mathelier A, Wasserman WW: The next generation of transcription factor binding site prediction. PLoS Comput Biol. 2013, 9 (9): 1003214-10.1371/journal.pcbi.1003214. doi:10.1371/journal.pcbi.1003214.,
Bellucci M, Agostini F, Masin M, Tartaglia GG: Predicting protein associations with long noncoding RNAs. Nat Methods. 2011, 8 (6): 444-445. 10.1038/nmeth.1611. doi:10.1038/nmeth.1611. PMID: 21623348.,
Agostini F, Zanzoni A, Klus P, Marchese D, Cirillo D, Tartaglia GG: catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics (Oxford, England). 2013, 29 (22): 2928-2930. 10.1093/bioinformatics/btt495. doi:10.1093/bioinformatics/btt495.,
Acknowledgements
The authors would like to thank Roderic Guigó (CRG), Guillaume Filion (CRG), Andreas Zanzoni (Inserm, U1090), Giovanni Bussotti (EMBL-EBI) and Samuel Francis Reid (CRG) for stimulating discussions.
Funding
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007-2013), through the European Research Council, under grant agreement RIBOMYLOME_309545, and from the Spanish Ministry of Economy and Competitiveness (SAF2011-26211). We also acknowledge support from the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013–2017’ (SEV-2012-0208).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GGT and FA conceived the study. FA designed the core algorithm and set up the web-server. DC, RDP and FA performed the computational analysis and analysed the data. FA and GGT wrote the manuscript. All authors read and approved the final version of the manuscript.
Electronic supplementary material
12864_2014_6626_MOESM1_ESM.png
Additional file 1: Figure S1. Motif discovery time performance. Motif discovery runtimes of CMF [12], DECOD [13], DREME [11], XXmotif [14] and SeAMotE algorithms are plotted for each ChIP-seq data set [19]. The cumulative distribution function represents the percentage of annotated TF motifs that are recovered using the corresponding method. (PNG 369 KB)
12864_2014_6626_MOESM2_ESM.png
Additional file 2: Table S1. DECOD [13] and XXmotifs [14] statistical measures. Sensitivity (True Positive Rate, TPR), specificity (SPC), precision (Positive Predictive Value, PPV), false discovery rate (FDR) and accuracy (ACC) achieved by the two methods on the CLIP-seq experimental datasets. Cases in which XXmotif was not able to find any motif in the range of 3-7-mers are indicate with NA. (PNG 535 KB)
12864_2014_6626_MOESM3_ESM.png
Additional file 3: Table S2. Cross-validation on RBPs. The table shows the 3-fold cross-validation performance of the SeAMotE approach on the CLIP data sets [18]. Training sets (TR1, TR2, TR3) are composed by two positive and two negative subsets, while the training sets (TE1, TE2, TE3) are represented by the positive and negative subsets that have not been used in the training. Datasets size, motifs identified along with their matches and coverage in the positive sets are reported for both training and testing analyses. The P-value associated with each training-testing pair of motifs, as calculated with TOMTOM [31], is shown in the last column. (PNG 954 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Agostini, F., Cirillo, D., Ponti, R.D. et al. SeAMotE: a method for high-throughput motif discovery in nucleic acid sequences. BMC Genomics 15, 925 (2014). https://doi.org/10.1186/1471-2164-15-925
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-925