
Abstract
Background
Commercial Atlantic halibut (Hippoglossus hippoglossus) farming is restricted by variable oocyte quality, slow growth, and early maturation of male fish. Maternally transferred components regulate early developmental processes; therefore, they have an effect on the future viability of the embryo. Using a newly developed Agilent 10 k custom-made oligonucleotide array, we profiled components of the transcriptome involved in immune defence as well as germline and muscle development during early developmental stages: 8-cell embryos (8CS), germ ring stage (GR), 10-somite stage (10SS), and hatched embryos (HT). In addition, we identified differentially expressed transcripts in low (≤9 ± 3% hatching) and high (≥86 ± 3°% hatching) quality eggs at 8CS to identify potential maternal markers for embryo quality.
Results
Out of 2066 differentially expressed transcripts, 160 were identified as maternal transcripts being specifically expressed at 8CS only. Twenty transcripts were differentially expressed in 8-cell embryos between low and high quality egg groups. Several immune-related transcripts were identified as promising molecular markers of hatching success including interferon regulatory factor 7 and mhc class 2A chain. Differential expression was positively validated with quantitative real-time PCR.
Conclusions
We have demonstrated maternal transfer of innate and adaptive immune system transcripts into Atlantic halibut embryos and their relation with future embryo developmental potential. We identified several transcripts as potential molecular markers of embryo quality. The developed microarray represents a useful resource for improving the commercial production of Atlantic halibut.
Similar content being viewed by others

Background
Although the production of farmed Atlantic halibut (Hippoglossus hippoglossus) has increased over the last ten years it still faces significant bottlenecks. Gametes are obtained by hand-stripping and oocytes are frequently of variable quality because of non-optimal timing of gamete collection and stress to broodstock fish [1–3]. Compared to other farmed marine teleosts, Atlantic halibut embryos are poorly developed at hatching with an associated long yolk-sac resorption time and a requirement for extended feeding on live prey before they can be fed with commercial diets [4]. The long live feeding period makes the larvae vulnerable to bacterial and viral diseases and increases their mortality [5]. Gene expression studies of the Atlantic halibut immune system have so far focused on larvae and juveniles [6–9]. However, both innate and adaptive immune system-relevant factors are maternally transferred into teleost oocytes and present during embryonic development, before hatching [10]. Maternally transmitted immune factors have not yet been characterized in Atlantic halibut.
The relatively slow growth of Atlantic halibut juveniles and its sex-dependent growth dimorphism are the main obstacles to commercially viable Atlantic halibut production during the on-growing phase [11, 12]. The muscle growth characteristics of juvenile and adult stages of teleosts are moderated by environmental conditions at the embryonic stages, particularly temperature [13, 14]. Embryonic gene expression patterns of single myogenic regulatory factors, such as myogenic differentiation 1 (myod1), myogenic differentiation 2 (myod2), and myogenin (myog), as well as structural muscle proteins such as myosin heavy chain (myhc), myosin light chain 2a (mylc2a), and myosin light chain 2b (mylc2b), have been described in Atlantic halibut [15, 16].
All-female halibut populations are preferred in commercial farming because of its sex-dependent growth dimorphism. All-female Atlantic halibut production has been established in Canada and Norway [17, 18]. However, to ensure controlled reproduction, the underlying molecular mechanisms of gamete development in Atlantic halibut need to be better understood. For example, the present knowledge of genes controlling proliferation and migration of primordial germ cells (PGCs), precursors of gametes, is restricted in Atlantic halibut to embryonic expression of askopos (kop) and Tudor domain-containing protein 5 (tdrd5) [19].
Early embryonic development in teleosts is driven until the start of zygotic transcription by maternally supplied mRNAs that are incorporated into the oocyte during oogenesis [20]. Maternal mRNAs are critical to embryonic development since they implement basic biosynthetic processes, direct first mitotic divisions, and specify initial cell fate and embryonic patterning [21]. Hence, maternal mRNAs are potential molecular markers for oocyte quality. In aquaculture, early estimation of oocyte quality can avoid costly and unnecessary incubation of low-quality material and improve production and predictability [22]. Several maternal mRNAs have been identified as molecular markers for oocyte quality in farmed mammals [23–26]. In commercially farmed teleosts, only prohibitin 2 (phb2) in rainbow trout (Oncorhynchus mykiss) has been found to be related to oocyte quality [27]. Previously, we have idenfied three uncharacterized maternal transcripts, correlating significantly with Atlantic halibut embryo quality [19]. In addition, we found indications that the transition from maternal to zygotic transcripts (MZT) in Atlantic halibut takes place between the blastula stage and germ ring stage [19, 28].
A microarray created by Douglas et al. [29] has been used to study gene expression in five developmental stages (hatched to post-metamorphosis) of Atlantic halibut, including larvae introduced to microencapsulated diet and juveniles fed fish meal replacement diets [30, 31]. Since the production of the first Atlantic halibut microarray [29], the number of Atlantic halibut ESTs available from NCBI GenBank (http://www.ncbi.nlm.nih.gov/dbEST) has increased by 40%, including 3670 maternal new ESTs [19, 32]. In the present study, we produced a new 10 k custom oligonucleotide array to profile embryonic expression of transcripts involved in immune system, germline, and muscle development, and to identify maternal transcripts. We also examined differentially expressed maternal transcripts in 8-cell embryos from batches with either high (≥86 ± 3%) or low hatching success (≤9 ± 3%) in order to identify potential molecular markers of embryo quality.
Results
Microarray transcript ontology
Of the 10279 probes designed and printed onto the microarray, 5364 (52%) represented transcripts with putative identification based on sequence similarity by BLASTX searches (cut-off E = 1E−6). GO annotations were obtained for 5009 (49%) probes. Most probes (61%) were classified as representing transcripts involved in cellular (24%), metabolic (23%), and regulatory processes (14%, Additional file 1). Their molecular function was dominated by binding (50%), catalytic activities (31%), and cellular compartments were cell (43%) and organelle (31%). A full list of GO annotation for the three domains is presented in Additional files 2, 3 and 4. After signal processing, data normalization, and filtering, 8533 (83%) probes were kept for further analysis of transcript expression during early embryonic development and 8447 (82%) for analysis of differentially expressed transcripts between high and low quality embryos at 8CS.
Transcript expression during embryonic development
Among the four developmental stages, 2066 transcripts were differentially expressed and 339 (16%) were up-regulated only in one of the four developmental stages (p < 0.05). Out of the 339 up-regulated transcripts, 160 (47%) were strictly maternal transcripts, only expressed at 8CS, and 49 (12%), 54 (16%) 76 (22%) transcripts were only expressed at GR, 10SS and HT, respectively (Additional files 4, 5, 6, 7 and 8). No significantly enriched GO terms were found in any of the four groups of transcripts.
Immune defence
Seven transcripts encoding complement system proteins were identified in Atlantic halibut embryos (Figure 1A). Mannose binding lectin (mbl) was highly expressed at 8CS, and to a lower degree at GR and HT. Complement factor B (cfb) was highly expressed at GR and complement factor C5 (c5) at 10SS. Complement factor C3 (c3), H (cfh) and I (cfi) were highly expressed at 10SS and HT. Complement factor D (cfd) was only expressed at HT. Two types of pattern recognition receptors (PRRs) were expressed: Toll-like receptors (TLRs), and C-type lectin receptors (CLRs). Toll-like receptor 5 (tlr5) was highly expressed at 8CS and GR. C-type lectin domain family 4 member C (clec4c) was expressed at 10SS and HT while C-type lectin domain family 4 member E (clec4e) was only expressed at HT. Among transcripts encoding cytokines or related proteins, interferon regulatory factor 7 (irf7) was expressed at 8CS and GR, interferon-induced protein 35 (ifi35) at 8CS and interferon regulatory factor 6 (ifr6) at GR and 10SS. Interleukin enhancer binding factor 2 (ilf2), interleukin 7 (il7), and interferon-induced protein 56 (ifi56) were only expressed at GR. Interleukin 18 (il18) was expressed from GR to HT. Suppressor of cytokine signaling 1 (socs1) was highly expressed at GR.

Clustering of transcript groups. A: Immune system, B: Germ cells, C: Muscle. Transcripts were clustered using the un-weighted pair-group method (UPGMA) using arithmetic averages with normal Euclidian distance as distance measurements. Developmental stages were: embryos at 8-cell stage (8CS); germ ring stage (GR), 10-somite stage (10SS), and hatched embryos (HT). Data were standardized against the first stage 8CS. Colour bar indicates relative expression in relation to 8CS. High intensity expression is represented in red colours while blue colours represent lower expression intensity (n = 3 batches).
Among the major histocompatibility complex (MHC) receptor sub-chain transcripts, mhc class 1A chain (mhc1a) and mhc class 2B chain (mhc2b) were highly present at GR while mhc class 2A chain (mhc2a) transcript level peaked at 10SS. Transcripts of the T-cell receptor A chain (tcra) and the T-cell surface glycoprotein CD3 E chain (cd3e) were found at 8CS and GR while T-cell receptor D chain (tcrd) was expressed from GR to HT. Three transcripts coding T-cell co-signaling regulators were identified: Lymphocyte antigen 9 (ly9), expressed at 8CS and GR, tumor necrosis factor superfamily member 14 (tnfsf14), expressed from GR to HT, and programmed cell death 1 ligand 1 (pd-1 l) highly expressed at 8CS. Cathepsin S (ctss) was expressed at 8CS and GR.
Primordial germ cells
All selected transcripts involved in primordial germ cell proliferation and migration were found at both 8CS and GR, except for tudor domain containing 3 (tdrd3) being only expressed at 8CS (Figure 1B). Transcript level of tudor domain containing 4 (tdrd4), tudor domain containing 5 (tdrd5), tudor domain containing 7 (tdrd7), nanos homolog 3 (nanos3), dead-end homolog 1 (dnd1), piwi-like 1 (piwil1) and piwi-like 2 (piwil2) was highest at 8CS, while the level of vasa (vasa) and askopos (kop) peaked at GR.
Muscle development
Myosin light chain 1 (mylc1) and 2 (mylc2), myosin heavy chain (myh), fast myotomal troponin T embryonic isoform (eftnt), fast myotomal muscle troponin T1 (ftnt1) and T2 (ftntf2) and creatine kinase mitochondrial 2 (ckmt2) were expressed at 8CS, and increasingly at 10SS and HT (Figure 1C). Capping protein actin filament muscle z-line A (capza) was highly expressed at 10SS and HT, and Capping protein actin filament muscle z-line B (capzb) was predominantly expressed at 10SS. Tropomyosin 3 (tpm3), myosin light chain 3 (mylc3), and myosin binding protein C cardiac (mybpc3) were highly expressed at HT. Parvalbumin (pval) was highly expressed at GR and Tropomyosin 4 (tpm4) at GR, 10SS, and HT.
Differential transcript expression in high and low quality eggs
Twenty transcripts were differentially expressed between high and low quality 8CS embryos (p < 0.05; Figure 2 and Table 1). Gene set enrichment analysis resulted in no significant differences in functional terms. The highest differences in transcript levels were found for irf7 (8.0 ± 0.3-fold lower in low quality 8CS embryos, Table 1) and mhc2A (7.8 ± 0.7-fold higher in low quality 8CS embryos). Nine transcripts (57%) had significant BLAST hits representing genes involved in immune response, metabolism, RNA transcription, protein degradation, cell signalling, and cytoskeleton. Conserved domain searches in the remaining 8 differentially expressed transcripts without significant BLAST hits resulted in identification of HH_90606468 as belonging to the chaperonin-like superfamily of proteins. Two differentially expressed transcripts, eef1a2bp and pd-l1 showed strictly maternal expression and were expressed only before GR (Figure 3 and Additional file 5).

Clustering of differentially expressed transcripts in high (H) and low (L) quality oocytes. Transcripts were clustered using the un-weighted pair-group method (UPGMA) using arithmetic averages with normal Euclidian distance as distance measurement. High intensity expression is represented in red colours while blue colours represent lower expression intensity (n = 3 batches).

Validation of expression patterns by quantitative PCR (qPCR). Relative gene expression (±SD) in Atlantic halibut embryonic developmental stages estimated by microarray (grey bars; n = 3) and qPCR (white bars; n = 5). The embryonic developmental stages (n = 3) were: 8-cell stage (8CS), germ ring (GR), 10-somite stage (10SS), and hatched larvae (HT). qPCR data were normalized with Luc. Data were standardized against the 8CS stage. A: irf7, B: 90603123, C: HH_90988292, D: eef1a2bp, E: cyp2n, F: HH_193889799, G: HH_90607090, H: HH_166851124, I: HH_90606468, J: rnf213, K: HH_Contig436, L: psmb9, M: HH_90602035, N: HH_90599790, O: pd-1 l, P: ms4a8a, Q: HH_Contig207, and R: mhc1a.
Microarray validation
A significant correlation was found between qPCR and microarray results (ϱ = 0.88, p < 0.0001, n = 276; Additional file 9). Significant differences in transcript levels between high and low quality 8CS embryos by microarray were confirmed by qPCR for all 20 differentially expressed transcripts (Table 1). Expression patterns of the differentially expressed transcripts during embryonic development (n = 18) followed the same general trend when estimated by qPCR, with a few exceptions. Microarray relative expression data revealed strictly maternal expression (no expression at GR, 10SS and HT) of two transcripts (Figure 3D and O) while according to qPCR data, four transcripts were strictly maternal (Figure 3D, F, K, and O). Membrane-spanning 4-domains subfamily a member 8a (ms4a8a) was expressed only zygotically (no expression before stage 10SS) according to microarray results, but showed maternal-zygotic expression (expressed at all four developmental stages) according to qPCR expression results (Figure 3P). HH_Contig207 expression was detected at 8CS and HT by microarray and addition in GR by qPCR (Figure 3Q). Mhc class 1A chain (mhc1A) expression was apparent in GR and HT by microarray, but only in GR by qPCR (Figure 3R). For 14 out of 20 tested transcripts, qPCR expression levels were up to 1.5 times higher in one or more developmental stages, compared to the microarray results (Figure 3).
Discussion
In hatched Atlantic halibut embryos, the anterior part of the head kidney is present, but no haematopoietic tissue or cells can be observed. Liver, thymus, and spleen, important for the development of the adaptive immune system, are not present yet [7]. Therefore, the innate immune system is the first line of immune protection. The complement system is a major non-cellular component of the innate immune system [33]. The presence of maternally transferred mbl transcripts, an activator of the lectin pathway (LP), indicates that it is the first complement defense mechanism active in Atlantic halibut embryos (Figure 1A). Between GR and HT, expression of transcripts encoding central complement components (c3 and c5) and transcripts encoding alternative pathway (AP) regulatory proteins (cfb, cfd, cfh, and cfi) increases. As previously found in zebrafish and rainbow trout, the AP seems to be functional as immune defense in Atlantic halibut embryos, before the adaptive system is developed [34, 35]. Despite the lack of IgM-bearing cells and organs of the adaptive immune system in Atlantic halibut embryos, transcripts of MHC receptors (mhc1a, mhc2a, mhc2b), T-cell receptors (TCR, tcra, tcrd), and T-cell co-signaling regulators (ly9, tnfsf14, pd-1 l, ctss) were present. These maternally transferred transcripts of the adaptive immune system add to the immune protection of the developing embryo and larvae. A similar transfer of maternal transcripts involved in the innate and adaptive immune system have been identified in in half-smooth tongue sole (Cyngolossus semilaevis) and rainbow trout embryos [36, 37].
Transcripts of three tdrd genes, tdrd4, tdrd5 and tdrd7, showed similar expression pattern as piwil1 and piwil2, reflecting the close interaction of the corresponding protein products (Figure 1B). TDRD4, TDRD5, and TDRD7, together with TDRD1, TDRD6, TDRD8 and TDRD9 form PIWI-TDRD complexes that are essential in retrotransposon silencing, chromatoid body assembly and spermiogenesis [38]. Tdrd5 maternal expression has previously been identified in Atlantic halibut embryos [19]. Among tdrd transcripts, tdrd3 was the only transcript expressed mainly at 8CS. TDRD3 preferably binds to asymmetric dimethyl arginine marks (aDMAs) in somatic cells acting as a transcriptional co-activator [39]. Expression profiles of nanos3 and dnd1 clustered together with piwil and tdrd transcripts with the exception of increased expression at H. Dnd1 and nanos3 code germ plasm and PGCs specific RNA-binding proteins involved in differentiation and survival of PGCs [40, 41]. Through binding to 3’UTRs of target mRNAs, DND1 counteracts miRNA-mediated posttranscriptional repression in PGCs [40, 42]. Expression of dnd1 and nanos3 in Atlantic halibut embryos was similar to expression patterns found in teleost embryos previously [40, 43, 44]. Expression of kop and vasa peaked at GR in Atlantic halibut, diminishing during later embryonic development and at HT. A similar expression pattern of kop, coding a PGC-specific P-loop protein of unknown function, has previously been identified in zebrafish and Atlantic halibut [19, 45]. Vasa, an ATP-dependent RNA helicase, is mostly known as a PGC marker, but has recently also been identified as a regulator of cell cycle progression in somatic cells [46, 47].
Most muscle related transcripts identified during Atlantic halibut embryonic development represented isoforms of transcripts coding structural muscle proteins members of myosin (myh, mylc1, mylc2, mylc6, and mycl3), troponin (ftnt1, ftnt2, and eftnt), tropomyosin (tpm3 and tpm4) and parvalbumin (pval; Figure 1C). Myosin molecules consist of six subunits, two heavy chains (MYHs) of ~200 kDa and four light chains (MYLs) of ~20 kDa. Myhc expression has previously been identified at the 17-somite stage and onwards in Atlantic halibut [15]. In the same study, two isoforms of mylc2 were identified to be stage-specific (embryonic: mylc2a and larval/juvenile: myl2b). In zebrafish, myl1 and myl2 are expressed at the 10-somite stage, followed by myl3 expression at the 12-somite stage. Here we have identified isoforms of myl1 and myl2 with expression at 8CS, 10SS, and HT, while myl3 was mainly expressed at HT. One embryonic/larval troponin isoform (efTnThh) and two adult isoforms have previously been identified (efTnThh-1 and efTnThh-2) in Atlantic halibut larvae during metamorphosis [48]. In the present study, ftnt1, ftnt2 and eftnt showed similar expression patterns, indicating that they represented embryonic/larval isoforms.
In Atlantic halibut embryos, tpm3 expression started after somite formation (10SS) and was highly expressed at HT. Isoforms of tpm3 have been previously found to be essential for embryonic development in mice, but the underlying mechanism is unknown [49]. Both tpm4 (also named δtpm) and mybpc3 are known to be specifically expressed in cardiac muscle in zebrafish [50, 51]. While tpm4 expression was detected from GR to HT, mybpc3 expression was restricted to HT. In the present study, pval expression peaked earlier, at GR, in Atlantic halibut embryos compared to its expression in zebrafish, where it was first detected in 15-somite stage embryos [52]. As a Ca2+-binding protein, PVAL is usually present in high concentrations in fast muscle cells, to a lower extend in specific neurons of the central and peripheral nervous system, and in cells of endocrine glands [53].
The two maternal transcripts, irf7 and mhc2a, were identified as potential markers for quality in Atlantic halibut due to their high level of expression differences in high and low quality 8CS embryos (Table 1). The three uncharacterized transcripts (HH_90603123, HH_90988292, and HH_90596347) had the same potential, but their functions have to be further characterized. IRF7 is the primary regulator of type I interferon (IFN) production and its absence impairs antiviral innate immunity [54]. The significantly lower levels of irf7 transcripts in low quality 8CS embryos may result in reduced immune response capacity resulting in further poor embryonic and larval development. Most of the transcripts showing elevated expression in low quality embryos are involved in adaptive immune response and protein degradation (Table 1). Their elevated concentrations could be the result of a maternally transferred immune response during the oogenesis triggered by unknown inflammation, infection or immune-activated stress.
Polyadenylation of maternal mRNAs during oocyte maturation usually protects mRNAs from degradation and activates their translation [55]. In contrast, regulatory RNA or protein-mediated deadenylation triggers mRNA degradation and translational repression to allow normal embryonic development after the maternal-zygotic transition (MZT) [56]. In Xenopus tropicalis, oocyte post-ovulatory aging (POA) induced a general decrease in maternal transcripts and a female-specific shortening of maternal mRNAs by deadenylation in oocytes that developed into embryos experiencing high malformation and mortality rates [57]. In contrast, POA induced both a decrease and increase in specific maternal transcripts in rainbow trout oocytes [58, 59]. In the present study, timing of hand-stripping Atlantic halibut females was synchronised to their individual ovulation rhythms to avoid POA. Whether low maternal transcript levels are the result of a lack or sub-optimal polyadenylation during Atlantic halibut oocyte maturation, leading to poor transcript translation and/or degradation in low quality early embryos, requires further investigation. Due to the experimental design, it was not possible to estimate whether the transcript level differences in high and low quality 8CS embryos were based on individual female differences and/or inheritability. However, no major differences in transcript levels were observed within the three oocyte batches tested (Figure 2). To test the potential of irf7 and mhc2a as markers for Atlantic halibut oocyte quality, future studies should nevertheless be performed across a higher number of batches from females of known genetic background.
Both abundance of differentially expressed transcripts in high and low quality oocytes (Table 1) and their expression patterns during embryonic development (Figure 3) were successfully confirmed by qPCR. The qPCR and microarray analysis have inherent technical and data normalization challenges that can lead to variability in results. Following strict quality assessment procedures in both techniques, (RNA quality, cross-hybridisation of microarray probes, microarray spot intensity, qPCR primer design, and PCR efficiency) and data filtering after normalization (cut-offs for fold-changes and low microarray spot intensity) resulted in high correlation between qPCR and microarray data (Additional file 9) [60, 61]. Due to the requirement of designing qPCR primers across exon/introns boundaries, microarray probes and qPCR primer locations varied. This could result in the observed variation between qPCR and microarray data. Compared to the previously created Atlantic halibut microarray by Douglas et al. [29], the new microarray contains a higher number of transcripts corresponding to genes expressed during the embryonic development. This microarray was successfully used to screen maternal transcript expression, and to identify differential transcripts expression in low and high quality 8CS embryos. It has proven to be suitable for future analysis of Atlantic halibut embryonic transcript expression which is likely to advance our understanding of important developmental processes in teleosts.
Conclusions
Using a new Atlantic halibut 10 k custom oligonucleotide array, we have demonstrated maternal transfer of innate and adaptive immune system transcripts into Atlantic halibut embryos and profiled their expression in early developmental stages. We identified several transcripts, including irf7 and mhc2a, as potential molecular markers for embryo quality. Microarray validation did prove the usefulness of the tool for further transcript quantification in Atlantic halibut. Both the established information and microarray provide useful resources to improve commercial production of Atlantic halibut.
Methods
Fish husbandry and sample collection
All procedures of fish husbandry and sample collection were in accordance with the guidelines set by the National Animal Research Authority (Forsøksdyrutvalget, Norway). For transcript expression profiling during early development, embryos were collected at the 8-cell stage (8CS), 8 h post fertilisation (hpf); germ ring stage (GR), 82 hpf; 10-somite stage (10SS) 142 hpf; and hatched larvae (HT), 340 hpf, from eight females (30–40 kg) at a commercial Atlantic halibut farm (Risørfisk AS, Risør, Norway). All oocytes were fertilized in vitro with pooled sperm from two random males (15–20 kg) at the peak of their reproductive season. Females and males were fed EWOS Premix (EWOS, Bergen, Norway) and kept under natural photoperiod conditions. Eggs were incubated in large-scale 280 L incubators at salinity 34 ± 1 ‰ and temperature 6.3 ± 0.1°C. Fertilization and hatching percentage was estimated at 8CS and 340 hpf, respectively. Sampling was performed at 8CS to ensure that only fertilized oocytes were collected. Samples were immediately snap-frozen in liquid nitrogen. Only high quality embryos defined as fertilization success ≥ 90 ± 2% and hatching success ≥ 85 ± 2% were used for studies of normal development. Three separate batches of eggs were used for these experiments.
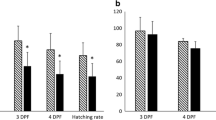
To identify differentially expressed transcripts between high and low quality 8CS embryos, oocytes were collected at the University of Nordland (Bodø, Norway) from 20 females (40–60 kg) kept under natural photoperiod conditions and fed Fish Breed-M (INVE Aquaculture NV, Dendermonde, Belgium). All oocytes were fertilized in vitro with sperm pooled from two random males. Eggs were incubated in 100 × 15 mm Petri dishes in triplicates, approximately 100 eggs per dish, at 5.5 ± 0.5°C in 33 ± 1 ‰ filtered seawater, added 0.5% (v/v) penicillin-streptomycin-neomycin solution (5000 U penicillin, 5 mg streptomycin, and 10 mg neomycin per ml, Sigma, St. Louis, Mo, USA) until hatching at 340 hpf. Samples were snap-frozen in liquid nitrogen. Fertilization and hatching percentages were estimated as described above. Fertilization and hatching percentage was used to categorize collected embryo groups as high and low quality embryos. Embryos with low fertilization rate (≤16 ± 3%) and low hatching percentage (≤7 ± 3%) were defined as low quality embryos (L) and embryos with high fertilization (≥91 ± 2%) and high hatching percentage (≥86 ± 3%) were defined as high quality embryos (H). Three groups of high and three groups of low quality embryos were selected to identify differentially expressed maternal transcripts at the 8CS stage.
Microarray construction and probe design
Approximately 22,000 ESTs were obtained from the NCBI GenBank and subjected to EST pre-processing, clustering and contig assembly using a local installation of ESTExplorer (http://estexplorer.biolinfo.org). In essence, vectors were removed, low quality sequence repeats were masked, and the resulting sequences subjected to clustering and contig assembly using semi-rigid parameters (CAP3 = 80%, 50 ORFs). The resulting 3,105 consensus sequences (contigs) and 7,174 single ESTs (singletons) were subjected to blasting, mapping and annotation by a local installation of Blast2GO (http://www.blast2go.org/) using default parameters with minor modifications. In short, sequences were blasted against the NCBI non-redundant database using BLASTX (E = 10E-3). These results were complemented with a BLASTX against UniProtKB/Swiss-Prot (http://www.ebi.ac.uk/uniprot). Sequences with blast hits were then mapped against the Blast2Go database and resulting mapped sequences annotated in a sequential manner according to decreasing cut-off values (1: E = 10E-6, cut-off: 55, HSP coverage cut-off: 75; 2: E = 10E-6, cut-off: 55, HSP coverage cut-off: 0 and 3: E = 10E-6, cut-off: 60, HSP coverage cut-off: 0, Evidence code weight: ISS = 1.0, IEA = 1.0). Gene ontology (GO) results were enriched by merging Interpro (http://www.ebi.ac.uk/interpro/) annotations to existing GOs as well as GOs augmented by the Blast2Go functionality ANNEX. GO distributions of array probes were displayed after reducing GO complexity using GOslim (generic) [62]. Redundant sequences were removed on the basis of sequence similarity (>70% similarity) and array probe cross-hybridization potential. The resulting 10,279 sequences were subjected to 60-mer probe production by eArray (https://earray.chem.agilent.com/earray/). Probes were printed in quadruples on a 4x44k Agilent custom oligoarray (Agilent Technologies, Santa Clara, US).
RNA extraction
Total RNA from each developmental stage (8CS, GR, 10SS, and HT; n = 3 batches each) and quality type of 8CS embryo (L, H; n = 3 batches each) was extracted according to the Tri reagent method (Sigma, St-Louis, MO, USA) using QIAazol (Qiagen, Nydalen, Sweden). RNA quality was initially checked by gel electrophoresis on a 1% (v/w) agarose gel containing SYBR safe™ DNA gel stain (Invitrogen, Paisley, UK). Genomic DNA was removed by DNAse treatment by Ambion Turbo DNA-free kit (Applied Biosystems, Austin, TX, USA). RNA quality was controlled by photometric analyses (260/230 > 1.8, 260/280 > 1.5) using NanoDrop spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). RNA integrity and quality was then estimated on Agilent 2100 Bioanalyzer and RNA integrity number (RIN) index was calculated for each sample using the Agilent 2100 Expert software. RIN provides a numerical assessment of the integrity of RNA that facilitates the standardization of the quality interpretation; for microarray processing, only RNAs with RIN number > 9.0 were further processed to reduce experimental biases due to poor RNA quality.
Sample labelling and hybridization
For each sample, total RNA (200 ng μl−1) was labelled and amplified with Cy3-dCTP in duplicate using the Agilent Low Input Quick Amp Labelling kit according to the manufacturer’s protocol. Samples were spiked with Agilent One-Color Spike-Mix (1:10). The labelled and amplified cRNA was purified using the Qiagen RNease mini spin kit. Cleaned cRNA was quantified using a NanoDrop spectrophotometer. All samples had cRNA yields > 1.65 and Cy3 specific activity > 6.0. For each developmental stage (8CS, GR, 10SS and HT) and quality type of 8 CS embryo (L, H) three replicates were hybridized at 65°C for 17 h. After hybridization, all arrays were washed according to manufacturer’s protocol followed by a final acetonitrile wash. Slides were immediately scanned using an Agilent High density microarray scanner at 5 μm resolution (Agilent Technologies).
Microarray validation by quantitative real-time PCR
To confirm expression results obtained from microarray analysis, primers were designed for 20 transcripts that were found to be differentially expressed between high and low quality 8CS embryos (Additional file 10). Total RNA was extracted from early embryos at 8CS, GR, 10SS, and HT (n = 5) and low (n = 8) and high (n = 8) quality 8CS embryos as described above and cDNA synthesized using QuantiTect Reverse Transcription kit (Qiagen). Primers were selected close to the 3’ end of the respective EST’s for each transcript. Whenever possible, primers were designed to cross at least one intron/exon border containing both donor and acceptor sites, in order to avoid amplification of any contaminating genomic DNA. Primer pairs for qPCR amplification were designed manually and screened for hairpins, homo- and cross-dimers using Netprimer (http://www.premierbiosoft.com/netprimer/). qPCR was performed as described in Fernandes et al. [28]. Luciferase (Luc, Promega, Madison, WI, US) was used as an external reference to normalize relative gene expression during embryonic development. β-Actin (Actb) and β2-tubulin (Tubb2) were used as reference genes to normalize relative gene expression between high and low quality 8 CS embryos (Additional file 11).
Data analysis
Scanned images were analysed with the Agilent feature extraction Software Version 7.2. Resulting raw data were normalized (75 Quantile, median to baseline of all samples). Features were filtered based on their signal intensity values by satisfying the upper and lower percentile cut-offs 20–100% and outliers were removed with GeneSpring GX 10.0.2 (Agilent Technologies). All data were filtered for missing values and replaced by row mean imputation. A cut-off of ≥ 0.5 was applied to all data in addition to standard background correction to remove features close to the mean low intensity threshold across all arrays (0.38 ± 0.19). Expression analysis, functional profiling and hierarchical clustering were performed using the Babelomics 4.3 analysis suite (http://babelomics.bioinfo.cipf.es). Differentially expressed transcripts in the four embryonic developmental stages and between low and high quality 8CS embryos were estimated using limma with Benjamin and Hochberg false discovery rate (FDR) multiple-test correction (p < 0.05) [63, 64] and a 2-fold-change cut-off. Differentially expressed transcripts during developmental stages were filtered for transcripts up-regulated in only one of the developmental stages. Hierarchical clustering (un-weighted pair-group method with arithmetic averages (UPGMA) with normal Euclidian distance as distance measurement) was performed on selected transcripts involved in immune defence, PGC development, and muscle development among the filtered transcripts up-regulated in only one of the developmental stages. The same hieratchical clustering was performed for all differentially expressed transcripts between low and high quality 8CS embryos. Gene Ontology (GO) term enrichment using FatiGO + (Fishers exact test, two-tailed, Adj. p < 0.05) [65] was performed for transcripts up-regulated in one of the four embryonic stages and differentially expressed in low and high quality oocytes. For microarray validation, qPCR data were log2 transformed to be comparable with the microarray results. Correlation between qPCR and microarray data was estimated by Spearman’s Rho (ϱ). Mann–Whitney U (p < 0.05) was used to determine significant fold-change differences in relative gene expression between high and low quality 8CS embryos obtained by qPCR.
The microarray data have been deposited in the National Center for Biotechnology Information GEO (NCBI GEO; http://www.ncbi.nlm.nih.gov/geo) and are accessible under the GEO series accession number GSE61051.
References
McEvoy L: Ovulatory rhythms and over-ripening of eggs in cultivated turbot, Scophthalmus maximus L. J Fish Biol. 1984, 24: 437-448. 10.1111/j.1095-8649.1984.tb04814.x. doi:10.1111/j.1095-8649.1984.tb04814
Kjorsvik E, Mangor-Jensen A, Holmefjord I: Egg quality in fishes. Adv Mar Biol. 1990, 26: 71-113. doi:10.1023/A:1018400130692
Bromage N, Bruce M, Basavaraja N, Rana K, Shields R, Young C, Dye J, Smith P, Gillespie M, Gamble J: Egg quality determinants in finfish, their role of over-ripening with special reference to the timing of stripping in the Atlantic halibut Hippoglossus hippoglossus. J World Aquacult Soc. 1994, 25: 13-21. 10.1111/j.1749-7345.1994.tb00799.x. doi:10.1111/j.1749-7345.1994.tb00799.x
Falk-Petersen IB: Comparative organ differentiation during early life stages of marine fish. Fish Shellfish Immunol. 2005, 19: 397-412. 10.1016/j.fsi.2005.03.006. doi:10.1016/j.fsi.2005.03.006
Bergh O, Nilsen F, Samuelsen OB: Diseases, prophylaxis and treatment of the Atlantic halibut Hippoglossus hippoglossus: a review. Dis Aquat Organ. 2001, 48: 57-74.
Patel S, Malde K, Lanzén A, Olsen RH, Nerland AH: Identification of immune related genes in Atlantic halibut (Hippoglossus hippoglossus L.) following in vivo antigenic and in vitro mitogenic stimulation. Fish Shellfish Immunol. 2009, 27: 729-738. 10.1016/j.fsi.2009.09.008. doi:10.1016/j.fsi.2009.09.008
Patel S, Sørhus E, Fiksdal IU, Espedal PG, Bergh O, Rødseth OM, Morton HC, Nerland AH: Ontogeny of lymphoid organs and development of IgM-bearing cells in Atlantic halibut (Hippoglossus hippoglossus L.). Fish Shellfish Immunol. 2009, 26: 385-395. 10.1016/j.fsi.2008.11.018. doi:10.1016/j.fsi.2008.11.018
Øvergård A-C, Nerland AH, Patel S: Cloning, characterization, and expression pattern of Atlantic halibut (Hippoglossus hippoglossus L.) CD4-2, Lck, and ZAP-70. Fish Shellfish Immunol. 2010, 29: 987-997. 10.1016/j.fsi.2010.08.011. doi:10.1016/j.fsi.2010.08.011
Øvergård A-C, Fiksdal IU, Nerland AH, Patel S: Expression of T-cell markers during Atlantic halibut (Hippoglossus hippoglossus L.) ontogenesis. Dev Comp Immunol. 2011, 35: 203-213. 10.1016/j.dci.2010.09.009. doi:10.1016/j.dci.2010.09.009
Zhang S, Wang Z, Wang H: Maternal immunity in fish. Dev Comp Immunol. 2013, 39: 72-78. 10.1016/j.dci.2012.02.009. doi:10.1016/j.dci.2012.02.009
Foss A, Imsland AK, Vikingstad E, Stefansson SO, Norberg B, Pedersen S, Sandvik T, Roth B: Compensatory growth in Atlantic halibut: effect of starvation and subsequent feeding on growth, maturation, feed utilization and flesh quality. Aquaculture. 2009, 290: 304-310. 10.1016/j.aquaculture.2009.02.021. doi:10.1016/j.aquaculture.2009.02.021
Imsland AK, Roth B, Foss A, Vikingstad E, Stefansson SO, Pedersen S, Sandvik T, Norberg B: Long-term effect of photoperiod manipulation on growth, maturation and flesh quality in Atlantic halibut. Aquacult Res. 2009, 40: 1260-1269. 10.1111/j.1365-2109.2009.02224.x. doi:10.1111/j.1365-2109.2009.02224
Macqueen DJ, Robb DHF, Olsen T, Melstveit L, Paxton CGM, Johnston IA: Temperature until the “eyed stage” of embryogenesis programmes the growth trajectory and muscle phenotype of adult Atlantic salmon. Biol Lett. 2008, 4 (3): 294-298. 10.1098/rsbl.2007.0620. doi:10.1098/rsbl.2007.0620
Campos C, Valente LMP, Conceição LEC, Engrola S, Sousa V, Rocha E, Fernandes JMO: Incubation temperature induces changes in muscle cellularity and gene expression in Senegalese sole (Solea senegalensis). Gene. 2013, 516 (2): 209-217. 10.1016/j.gene.2012.12.074. doi:10.1016/j.gene.2012.12.074
Galloway TF, Bardal T, Kvam SN, Dahle SW, Nesse G, Randøl M, Kjørsvik E, Andersen O: Somite formation and expression of MyoD, myogenin and myosin in Atlantic halibut (Hippoglossus hippoglossus L.) embryos incubated at different temperatures: transient asymmetric expression of MyoD. J Exp Biol. 2006, 209 (Pt 13): 2432-2441.
Andersen Ø, Dahle SW, van Nes S, Bardal T, Tooming-Klunderud A, Kjørsvik E, Galloway TF: Differential spatio-temporal expression and functional diversification of the myogenic regulatory factors MyoD1 and MyoD2 in Atlantic halibut (Hippoglossus hippoglossus). Comp Biochem Physiol B Biochem Mol Biol. 2009, 154: 93-101. 10.1016/j.cbpb.2009.05.009. doi:10.1016/j.cbpb.2009.05.009
Tvedt HB, Benfey TJ, Martin-Robichaud DJ, McGowan C, Reith M: Gynogenesis and sex determination in Atlantic halibut (Hippoglossus hippoglossus). Aquaculture. 2006, 252: 573-583. 10.1016/j.aquaculture.2005.06.042. doi:10.1016/j.aquaculture.2005.06.042
Babiak J, Babiak I, Van Nes S, Harboe T, Haugen T, Norberg B: Induced sex reversal using an aromatase inhibitor, Fadrozole, in Atlantic halibut (Hippoglossus hippoglossus L.). Aquaculture. 2012, 324–325: 276-280.
Mommens M, Fernandes JM, Bizuayehu TT, Bolla SL, Johnston IA, Babiak I: Maternal gene expression in Atlantic halibut (Hippoglossus hippoglossus L.) and its relation to egg quality. BMC Res Notes. 2010, 3: 138-10.1186/1756-0500-3-138.
Lubzens E, Young G, Bobe J, Cerda J: Oogenesis in teleosts: how fish eggs are formed. Gen Comp Endocr. 2009, 16: 367-389. doi:10.1016/j.ygcen.2009.05.022, doi.org/10.1016/j.ygcen.2009.05.022
Dworkin M, Dworkin-Rastl E: Functions of maternal mRNA in early development. Mol Reprod Dev. 1990, 26: 261-297. 10.1002/mrd.1080260310. doi:10.1002/mrd.1080260310
Bobe J, Labbé C: Egg and sperm quality in fish. Gen Comp Endocrinol. 2010, 165: 535-548. 10.1016/j.ygcen.2009.02.011. doi:10.1016/j.ygcen.2009
Leoni GG, Bebbere D, Succu S, Berlinguer F, Mossa F, Galioto M: Relations between relative mRNA abundance and developmental competence of Ovine Oocytes. Mol Reprod Dev. 2007, 257 (August 2006): 249-257. doi:10.1002/mrd
Hamel M, Dufort I, Robert C, Léveillé M-C, Leader A, Sirard M-A: Genomic assessment of follicular marker genes as pregnancy predictors for human IVF. Mol Hum Reprod. 2010, 16: 87-96. 10.1093/molehr/gap079. doi:10.1093/molehr/gap079
Balboula AZ, Yamanaka K, Sakatani M, Hegab A, Zaabel SM, Takahashi M: Intracellular cathepsin B activity is inversely correlated with the quality and developmental competence of bovine preimplantation embryos. Mol Reprod Dev. 2010, 77: 1031-1039. 10.1002/mrd.21250. doi:10.1002/mrd.21250
Zhang D-X, Park W-J, Sun S-C, Xu Y-N, Li Y-H, Cui X-S, Kim N-H: Regulation of maternal gene expression by MEK/MAPK and MPF signaling in porcine oocytes during in vitro meiotic maturation. J Reprod Dev. 2011, 57: 49-56. 10.1262/jrd.10-087H. doi.org/10.1262/jrd.10-087H
Bonnet E, Fostier A, Bobe J: Microarray-based analysis of fish egg quality after natural or controlled ovulation. BMC Genomics. 2007, 8: 55-10.1186/1471-2164-8-55. doi:10.1186/1471-2164-8-55
Fernandes JMO, Mommens M, Hagen O, Babiak I, Solberg C: Selection of suitable reference genes for real-time PCR studies of Atlantic halibut development. Comp Biochem Physiol B Biochem Mol Biol. 2008, 150: 23-32. 10.1016/j.cbpb.2008.01.003. doi:10.1016/j.cbpb.2008.01.003
Douglas SE, Knickle LC, Williams J, Flight RM, Reith ME: A first generation Atlantic halibut Hippoglossus hippoglossus (L.) microarray: application to developmental studies. J Fish Biol. 2008, 72: 2391-2406. 10.1111/j.1095-8649.2008.01861.x. doi:10.1111/j.1095-8649.2008.01861.x
Murray HM, Lall SP, Rajaselvam R, Boutilier LA, Flight RM, Blanchard B, Colombo S, Mohindra V, Yúfera M, Douglas SE: Effect of early introduction of microencapsulated diet to larval Atlantic halibut, Hippoglossus hippoglossus L. assessed by microarray analysis. Mar Biotechnol (NY). 2010, 12: 214-229. 10.1007/s10126-009-9211-4.
Murray HM, Lall SP, Rajaselvam R, Boutilier LA, Blanchard B, Flight RM, Colombo S, Mohindra V, Douglas SE: A nutrigenomic analysis of intestinal response to partial soybean meal replacement in diets for juvenile Atlantic halibut, Hippoglossus hippoglossus, L. Aquaculture. 2010, 298: 282-293. 10.1016/j.aquaculture.2009.11.001. doi:10.1016/j.aquaculture.2009.11.001
Bai J, Solberg C, Fernandes JMO, Johnston IA: Profiling of maternal and developmental-stage specific mRNA transcripts in Atlantic halibut Hippoglossus hippoglossus. Gene. 2007, 386: 202-210. 10.1016/j.gene.2006.09.012. doi:10.1016/j.gene.2006.09.012
Ricklin D, Hajishengallis G, Yang K, Lambris JD: Complement - a key system for immune surveillance and homeostasis. Nat Immunol. 2011, 11: 785-797. doi:10.1038/ni.1923
Løvoll M, Kilvik T, Boshra H, Bøgwald J, Sunyer JO, Dalmo RA: Maternal transfer of complement components C3-1, C3-3, C3-4, C4, C5, C7, Bf, and Df to offspring in rainbow trout (Oncorhynchus mykiss). Immunogenetics. 2006, 58: 168-179. 10.1007/s00251-006-0096-3. doi:10.1007/s00251-006-0096-3
Wang Z, Zhang S, Wang G, An Y: Complement activity in the egg cytosol of zebrafish Danio rerio: evidence for the defense role of maternal complement components. PLoS One. 2008, 3: e1463-10.1371/journal.pone.0001463. doi:10.1371/journal.pone.0001463
Yu Y, Zhong Q, Li C, Jiang L, Yan F, Wang Z, Zhang Q: Isolation and characterization of Toll-like receptor 9 in half-smooth tongue sole Cynoglossus semilaevis. Fish Shellfish Immun. 2009, 26 (3): 492-499. 10.1016/j.fsi.2009.02.005.
Li M, Russell SK, Lumsden JS, Leatherland JF: The influence of oocyte cortisol on the early ontogeny of intelectin and TLR-5, and changes in lysozyme activity in rainbow trout (Oncorhynchus mykiss) embryos. Comp Biochem Physiol B Biochem Mol Biol. 2011, 160: 159-165. 10.1016/j.cbpb.2011.08.002. doi:10.1016/j.cbpb.2011.08.002
Siomi MC, Mannen T, Siomi H: How does the royal family of Tudor rule the PIWI-interacting RNA pathway?. 2010, 24 (7): 636-646. doi:10.1038/nature07754
Sikorsky T, Hobor F, Krizanova E, Pasulka J, Kubicek K, Stefl R: Recognition of asymmetrically dimethylated arginine by TDRD3. Nucleic Acids Res. 2012, 40 (22): 11748-11755. 10.1093/nar/gks929. doi:10.1093/nar/gks929
Weidinger G, Stebler J, Slanchev K, Dumstrei K, Wise C, Lovell-Badge R, Thisse C, Thisse B, Raz E: dead end, a novel vertebrate germ plasm component, is required for zebrafish primordial germ cell migration and survival. Curr Biol. 2003, 13: 1429-1434. 10.1016/S0960-9822(03)00537-2. doi:10.1016/S0960-9822(03)00537-2
Julaton VTA, Reijo Pera RA: NANOS3 function in human germ cell development. Hum Mol Genet. 2011, 20: 2238-2250. 10.1093/hmg/ddr114. doi:10.1093/hmg/ddr114
Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JAF, Slanchev K, Le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Orom UA, Lund AH, Perrakis A, Raz E, Agami R: RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell. 2007, 131: 1273-1286. 10.1016/j.cell.2007.11.034. doi:10.4161/cc.7.7.5644
Presslauer C, Nagasawa K, Fernandes JMO, Babiak I: Expression of vasa and nanos3 during primordial germ cell formation and migration in Atlantic cod (Gadus morhua L.). Theriogenology. 2012, 78: 1262-1277. 10.1016/j.theriogenology.2012.05.022.
Nagasawa K, Fernandes JMO, Yoshizaki G, Miwa M, Babiak I: Identification and migration of primordial germ cells in Atlantic salmon, Salmo salar: Characterization of vasa, dead end, and lymphocyte antigen 75 genes. Mol Reprod Dev. 2013, 80 (2): 118-131. 10.1002/mrd.22142. doi:10.1002/mrd.22142
Blaser H, Eisenbeiss S, Neumann M, Reichman-Fried M, Thisse B, Thisse C, Raz E: Transition from non-motile behaviour to directed migration during early PGC development in zebrafish. J Cell Sci. 2005, 118 (Pt 17): 4027-4038. doi:10.1242/jcs.02522
Yoon C, Kawakami K, Hopkins N: Zebrafish vasa homologue RNA is localized to the cleavage planes of 2- and 4-cell-stage embryos and is expressed in the primordial germ cells. Development. 1997, 124: 3157-3165. doi:10.1002/mrd.22142
Yajima M, Wessel GM: The multiple hats of Vasa Function and its regulation of cell cycle progression. Mol Reprod Dev. 2012, 78: 861-867. doi:10.1002/mrd.21363
Campinho MA, Silva N, Nowell MA, Llewellyn L, Sweeney GE, Power DM: Troponin T isoform expression is modulated during Atlantic halibut metamorphosis. BMC Dev Biol. 2007, 7: 71-10.1186/1471-213X-7-71. doi:10.1186/1471-213X-7-71
Hook J, Lemckert F, Schevzov G, Fath T, Gunning P: Functional identity of the gamma tropomyosin gene: Implications for embryonic development, reproduction and cell viability. Bioarchitecture. 2011, 1: 49-59. 10.4161/bioa.1.1.15172. doi:10.4161/bioa.1.1.15172
Thisse B, Thisse C: Fast release clones: a high throughput expression analysis 2004, ZFIN direct data submission. http://zfin.org,
Zhao L, Zhao X, Tian T, Lu Q, Skrbo-Larssen N, Wu D, Kuang Z, Zheng X, Han Y, Yang S, Zhang C, Meng A: Heart-specific isoform of tropomyosin4 is essential for heartbeat in Zebrafish Embryos. Cardiovasc Res. 2008, 80: 200-208. 10.1093/cvr/cvn177. doi:10.1093/cvr/cvn177
Xu Y, He J, Wang X, Lim TM, Gong Z: Asynchronous activation of 10 muscle-specific protein (MSP) genes during zebrafish somitogenesis. Dev Dyn. 2000, 219: 201-215. 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1043>3.3.CO;2-9. doi:10.1002/1097-0177(2000)9999:9999
Berchtold MW, Brinkmeier H, Müntener M: Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000, 80: 1215-1265.
Ning S, Pagano JS, Barber GN: IRF7: activation, regulation, modification and function. Genes Immun. 2011, 12: 399-414. 10.1038/gene.2011.21. doi:10.1038/gene.2011.21
Hake LE, Richter JD: Translational regulation of maternal mRNA. Biochim Biophys Acta. 1997, 1332: M31-M38. doi.org/10.1016/S0304-419X(96)00039-X
Tadros W, Lipshitz HD: The maternal-to-zygotic transition: a play in two acts. Development. 2009, 136: 3033-3042. 10.1242/dev.033183. doi:10.1242/dev.033183
Kosubek A, Klein-Hitpass L, Rademacher K, Horsthemke B, Ryffel GU: Aging of Xenopus tropicalis eggs leads to deadenylation of a specific set of maternal mRNAs and loss of developmental potential. PLoS One. 2010, 5: e13532-10.1371/journal.pone.0013532. doi:10.1371/journal.pone.0013532
Aegerter S, Jalabert B, Bobe J: mRNA stockpile and egg quality in rainbow trout (Oncorhynchus mykiss ). Fish Physiol Biochem. 2003, 28: 317-318.
Aegerter S, Jalabert B, Bobe J: Messenger RNA stockpile of cyclin B, insulin-like growth factor I, insulin-like growth factor II, insulin-like growth factor receptor Ib, and p53 in the rainbow trout oocyte in relation with developmental competence. Mol Reprod Dev. 2004, 67: 127-135. 10.1002/mrd.10384. doi:10.1002/mrd.10384
Chuaqui RF, Bonner RF, Best CJM, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, Linehan WM, Knezevic V, Emmert-Buck MR: Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2002, 32: 509-514. 10.1038/ng1034. doi:10.1038/ng1034
Bustin SA, Nolan T: Pitfalls of quantitative real-time REverse-transcription polymerase chain reaction. J Biomol Tech. 2004, 15: 155-166.
Conesa A, Götz S: Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008, 2008: 619832-doi:10.1155/2008/619832
Benjamin Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995, 57: 289-300. doi.org/10.2307/2346101
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3 (1): doi:10.2202/1544-6115.1027
Al-Shahrour F, Díaz-Uriarte R, Dopazo J: FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004, 20: 578-580. 10.1093/bioinformatics/btg455. doi:10.1093/bioinformatics/btg455
Acknowledgements
The authors would like to thank Mr. Kjell Emil Naas and Mr. Yngve Attramadal from RisørFisk AS, and Mr. Tormod Skålsvik and Mr. Bjørnar Eggen from University of Nordland, for their invaluable help during the sampling of Atlantic halibut eggs and larvae and You Song (NIVA) for assistance with the microarray analysis. This study was funded by the Research Council of Norway (Project nr. 182653/V10) and the NIVA SIS project “MolPOP”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM contributed to experimental design, performed microarray, qPCR work and statistical analysis, and wrote the draft. KET designed microarray and performed microarray data processing. IB, IAJ and JMOF conceived the study, designed the experiment, assisted in analysis and drafting the manuscript. All authors have read and approved the manuscript.
Electronic supplementary material
12864_2014_6689_MOESM1_ESM.docx
Additional file 1: GO annotations for 10 k Atlantic halibut microarray. A: Biological processes, B: Molecular functions, C: Cell components. (DOCX 90 KB)
12864_2014_6689_MOESM2_ESM.docx
Additional file 2: Biological process (BP) gene ontology annotations (GOs) for Atlantic halibut 10 k microarray probes.(DOCX 19 KB)
12864_2014_6689_MOESM3_ESM.docx
Additional file 3: Molecular function (MF) gene ontology annotations (GOs) for Atlantic halibut 10 k microarray probes.(DOCX 16 KB)
12864_2014_6689_MOESM4_ESM.docx
Additional file 4: Cellular component (CC) gene ontology annotations (GOs) for Atlantic halibut 10 k microarray probes.(DOCX 16 KB)
12864_2014_6689_MOESM9_ESM.docx
Additional file 9: Microarray validation. Correlation plot of fold-change differences from 20 differentially expressed genes analyses by microarray and qPCR (n = 276). Correlation is given as Spearman’s Rho (ϱ). Abbreviations: H: High quality oocytes, L: Low quality oocytes; 8CS: 8-cell stage, GR: Germ ring, 10SS: 10-somite stage and HT: Hatched embryo. (DOCX 327 KB)
12864_2014_6689_MOESM11_ESM.docx
Additional file 11: Raw cycle threshold (CT) levels (mean ± S.E.) of reference genes for qPCR normalization. A) Actβ and B) Tubb2 in high (H) and low (L) quality Atlantic halibut oocytes (n = 8). C) Luc during early embryonic development of Atlantic halibut (n = 5). 8CS: 8-cell stage; GR: germ ring stage, 10SS: 10-somite stage, and HT: hatched embryo. (DOCX 41 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mommens, M., Fernandes, J.M., Tollefsen, K.E. et al. Profiling of the embryonic Atlantic halibut (Hippoglossus hippoglossus L.) transcriptome reveals maternal transcripts as potential markers of embryo quality. BMC Genomics 15, 829 (2014). https://doi.org/10.1186/1471-2164-15-829
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-829