
Abstract
Background
The identification of genetic markers associated with complex traits that are expensive to record such as feed intake or feed efficiency would allow these traits to be included in selection programs. To identify large-effect QTL, we performed a series of genome-wide association studies and functional analyses using 50 K and 770 K SNP genotypes scored in 5,133 animals from 4 independent beef cattle populations (Cycle VII, Angus, Hereford and Simmental × Angus) with phenotypes for average daily gain, dry matter intake, metabolic mid-test body weight and residual feed intake.
Results
A total of 5, 6, 11 and 10 significant QTL (defined as 1-Mb genome windows with Bonferroni-corrected P-value <0.05) were identified for average daily gain, dry matter intake, metabolic mid-test body weight and residual feed intake, respectively. The identified QTL were population-specific and had little overlap across the 4 populations. The pleiotropic or closely linked QTL on BTA 7 at 23 Mb identified in the Angus population harbours a promising candidate gene ACSL6 (acyl-CoA synthetase long-chain family member 6), and was the largest effect QTL associated with dry matter intake and mid-test body weight explaining 10.39% and 14.25% of the additive genetic variance, respectively. Pleiotropic or closely linked QTL associated with average daily gain and mid-test body weight were detected on BTA 6 at 38 Mb and BTA 7 at 93 Mb confirming previous reports. No QTL for residual feed intake explained more than 2.5% of the additive genetic variance in any population. Marker-based estimates of heritability ranged from 0.21 to 0.49 for residual feed intake across the 4 populations.
Conclusions
This GWAS study, which is the largest performed for feed efficiency and its component traits in beef cattle to date, identified several large-effect QTL that cumulatively explained a significant percentage of additive genetic variance within each population. Differences in the QTL identified among the different populations may be due to differences in power to detect QTL, environmental variation, or differences in the genetic architecture of trait variation among breeds. These results enhance our understanding of the biology of growth, feed intake and utilisation in beef cattle.
Similar content being viewed by others

Background
Feed costs comprise the majority of beef production costs and the efficiency of feed utilisation has long been recognised for its economic importance in beef cattle [1, 2]. Improvements in the efficiency of feed utilisation would lead to increased economic returns that would influence the entire beef cattle production system [3]. Feed efficiency is also important for social reasons due to environmental concerns about the methane emissions of cattle and because of the perceived competition in land use for producing crops for direct human consumption or for corn- and soybean-based biofuels. It has been reported that more efficient cattle emit lower amounts of methane [4].
The most commonly used measure of feed efficiency has traditionally been feed conversion ratio, which is the ratio of feed consumed to body weight gain. Selection to improve feed conversion ratio has the potential to increase growth rate in young animals because the two traits are genetically correlated [3]. This could, as a correlated response, produce larger females which are more expensive to maintain in the breeding herd [3]. Residual feed intake (RFI) is an alternate measure of feed efficiency, defined as the difference between an animal’s actual and expected feed intake based on its body weight and growth rate during the feeding period [1, 3]. RFI is considered by some to be a preferred method of measuring feed efficiency because of its phenotypic independence from the traits used to calculate RFI [5].
The estimated heritability of RFI in cattle populations is moderate to high, ranging from 0.08 to 0.46 [6–8]. Based on these heritability estimates and substantial phenotypic variation, RFI has the potential for inclusion in selection criteria to improve feed efficiency and the profitability of beef production [3, 9, 10]. It has also been experimentally demonstrated that direct selection on RFI can improve the feed efficiency of cattle [11]. However, individual feed intake measurements are needed for direct selection and these are expensive to obtain. The cost and logistics associated with recording feed intake have historically been the primary limitations to population-wide selection to improve feed efficiency in livestock. This problem could be ameliorated if genetic markers predictive of feed intake or RFI were available. Consequently, there has been considerable recent research to develop genetic markers that can be used to select animals for improved feed efficiency.
Opportunities to identify trait-associated genetic markers have been advanced by the availability of genome-wide high-density panels of single nucleotide polymorphism (SNP) markers including the Illumina BovineSNP50 BeadChip (50 K) [12] and BovineHD BeadChip (770 K) (Illumina Inc., San Diego, CA; [13]). Genome-wide association studies (GWAS) have now identified SNPs associated with economically important traits in both beef and dairy cattle [14–17]. Marker associations with RFI have previously been reported in beef cattle [18–20] and putative quantitative trait loci (QTL) have been mapped to BTA 1, 2, 5, 7, 8, 12, 14, 16, 17, 18, 19, 20, 21, 24, 26, 28 and 29 [19]. Of 8,786 polymorphic SNPs genotyped in 189 Australian beef cattle sampled for either high or low RFI, 161 were trait-associated (P <0.01) [18]. However, only two of these SNPs remained significant when evaluated in a larger multi-breed sample of animals [18].
The simplest model for performing GWAS is linear regression, where the association between markers and a trait of interest is tested one marker at a time. This type of analysis has been used for GWAS in humans [21] and in animal populations [22] where the extent of linkage disequilibrium among markers is considerably greater [23, 24] due to the small effective population sizes of most livestock breeds. On the other hand, Bayesian variable selection models facilitate the simultaneous fitting of all markers in the model and have been used for GWAS in livestock [25–31] to improve the precision of QTL mapping [32]. Among several Bayesian variable selection models, BayesB [33] has been shown by simulation to map QTL more precisely than other methods [34]. BayesB has also been shown to implicitly account for the population stratification resulting from pedigree relationships [35].
Although several QTL associated with feed efficiency traits in beef cattle have been reported, not all of the genetic variation in these traits has been captured because of inadequate sample size or studies limited to a single population. The extent of genetic variation for feed efficiency traits among different beef cattle populations remains unexplored. The objectives of this study were to map QTL associated with feedlot RFI and its growth and feed intake components; specifically, average daily gain on feed (ADG; kg/d), average daily dry matter intake (DMI; kg/d) and mid-test metabolic body weight (MBW; kg0.75) in a relatively large sample of animals (N =5,133) from 4 different beef cattle populations (Cycle VII, Angus, Hereford and Simmental × Angus, see Methods for more details). A BayesB model was used to simultaneously analyse SNP markers and identify QTL by characterising the proportion of additive genetic variation explained by every non-overlapping 1-Mb region within the genome.
Results and Discussion
Posterior means of additive genetic and residual variances and heritability
For each trait, the analysis generates an estimate of the proportion of phenotypic variation that can be explained by the use of SNP markers to represent identity by state among individuals, which is similar to the heritability estimated when pedigree information is used to represent identity by descent among individuals. The posterior means of heritability, additive genetic and residual variances for ADG, DMI, MBW and RFI in each of the 4 populations (Cycle VII, Angus, Hereford and Simmental × Angus) are in Table 1. Estimates of heritability ranged from 0.19 to 0.30 for ADG, from 0.27 to 0.41 for DMI, from 0.38 to 0.50 for MBW, and from 0.21 to 0.49 for RFI and are similar to those reported in the literature [16, 36, 37]. Heritability estimates in the Cycle VII population reported by [37] using a BayesC model were 0.24, 0.41, 0.58 and 0.57 for ADG, DMI, MBW and RFI, respectively which are similar to those produced in this study by applying a BayesB model to the same population (Table 1). The highest estimates of heritability were obtained for the Cycle VII and Hereford populations. The lower heritability estimates obtained for the other populations is likely due to the nutritional trials that were superimposed on animals during the feeding period that resulted in much larger numbers of contemporary groups (see Methods). The moderate to high heritabilities estimated for RFI indicates a significant potential for the identification of QTL considering the available sample sizes in this study.
Genome wide association – general results
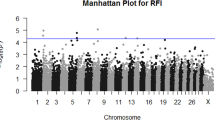
Manhattan plots of the posterior means of the additive genetic variance explained by each 1-Mb window across the genome for RFI, DMI, ADG and MBW are in Figures 1, 2, 3 and 4, respectively. The numbers of chromosome segments shown in Figures 1, 2, 3 and 4 (the X-axis) are not the same across the different populations as different SNP genotyping platforms were used and slightly different SNP filtering criteria were utilised in the different populations (See Methods). Some 1-Mb windows with a Bonferroni-corrected P-value less than 0.05 were identified as significant QTL and are summarised in Tables 2, 3, 4 and 5 for RFI, DMI, ADG, and MBW, respectively. The identity of the most strongly associated SNP (denoted throughout as ‘lead-SNP’ and defined as the SNP with the highest posterior probability of inclusion (sPPI) within the significant 1-Mb window) is also reported for each QTL.

The residual feed intake QTL. Proportion of additive genetic variance explained by SNPs within each 1-Mb window across the genome for residual feed intake in 4 different beef populations: a) Cycle VII, b) Angus, c) Hereford and d) Simmental × Angus. P: Pseudo autosomal region on BTAX, MT: Mitochondrial DNA. Based on UMD3.1 and Y chromosome assembly from Btau4.6.1.

The feedlot dry matter intake QTL. Proportion of additive genetic variance explained by SNPs within each 1-Mb window across the genome for feedlot dry matter intake in 4 different beef populations: a) Cycle VII, b) Angus, c) Hereford and d) Simmental × Angus. P: Pseudo autosomal region on BTAX, MT: Mitochondrial DNA. Based on UMD3.1 and Y chromosome assembly from Btau4.6.1.

The feedlot average daily gain QTL. Proportion of additive genetic variance explained by SNPs within each 1-Mb window across the genome for feedlot average daily gain in 4 different beef populations: a) Cycle VII, b) Angus, c) Hereford and d) Simmental × Angus. P: Pseudo autosomal region on BTAX, MT: Mitochondrial DNA. Based on UMD3.1 and Y chromosome assembly from Btau4.6.1.

The mid-test metabolic body weight QTL. Proportion of additive genetic variance explained by SNPs within each 1-Mb window across the genome for mid-test metabolic body weight in 4 different beef populations: a) Cycle VII, b) Angus, c) Hereford and d) Simmental × Angus. P: Pseudo autosomal region on BTAX, MT: Mitochondrial DNA. Based on UMD3.1 and Y chromosome assembly from Btau4.6.1.
The significant QTL were generally population-specific and had little overlap in genomic location across the 4 beef cattle populations. Although this result could be due to different genetic architectures underlying trait variation among these populations, it may also be due to differences in the power to detect QTL due to the larger numbers of contemporary groups in some populations (202 in Simmental × Angus and 102 in Angus versus 10 and 15 in Hereford and Cycle VII, respectively) or due to gene-by-environment or epistatic interactions. The populations exposed to the large number of different nutritional trials (such as forage feeding, concentrate rations or amino acid and mineral supplementation) were bred in a diverse geographical area (throughout the Midwest United States) in several different years. While genotype-by-environment interactions have been extensively detected using classical quantitative genetic approaches, little has been done to study this phenomenon at the level of the genome. We were not able to test for the existence of gene-by-environment interactions because of a lack of suitable connections between individuals in different contemporary groups across the different populations. Further studies employing special experimental designs are needed to investigate the existence of gene-by-environment interactions for feed efficiency traits in beef cattle.
The QTL on BTA 14 at 24 Mb (associated with MBW in Simmental × Angus, Table 5) was the only QTL that was identified as a suggestive QTL in another population (Cycle VII animals with a nominal P-value of 9.91E−5). Two different lead-SNPs (rs42646660 and rs134751608) were model-selected to tag this QTL. The rs42646660 SNP is located within an intron of XKR4 (XK, Kell blood group complex subunit-related family, member 4) and rs134751608 is 0.06 Mb centromeric of XKR4. Significant associations have previously been reported between XKR4 variants and subcutaneous rump fat thickness in cattle [16, 38]. The PLAG1 (pleiomorphic adenoma gene 1) gene which is located near the 24–25 Mb window boundary on BTA 14 has been shown to have large effects on stature in a Holstein × Jersey F2 cross [39] and on carcass weight in Japanese Black cattle [40]. Whether mutations in XKR4 or in nearby PLAG1 cause variation in MBW in the Cycle VII and Simmental × Angus animals warrants further investigation.
Genome wide association results for RFI
Ten significant 1-Mb SNP windows located on 8 different autosomes were detected for RFI (Table 2). The significant QTL separated by 2 Mb on BTA 14 could easily represent the same QTL as the effects were detected in two different populations (Simmental × Angus and Cycle VII). The largest effect 1-Mb SNP window for RFI was detected at 82 Mb on BTA 15 in the Simmental × Angus population and explained 2.40% of the total additive genetic variance with a Bonferroni-corrected P-value threshold of 1.81E−3 (Table 2). No QTL associated with RFI has previously been reported in this region of the cattle genome but several QTL associated with body size and production traits have been reported [41, 42].
Among the model-selected lead-SNPs tagging RFI QTL, rs109988749 located on BTA 19 is approximately 100 bp from the DNAH17 (dynein, axonemal, heavy chain 17) transcription start site and rs137078861 located on BTA 25 is within an intron of C25H16orf72 (chromosome 25 open reading frame, human C16orf72), which encodes an as yet uncharacterised protein. The remaining lead-SNPs are intergenic variants. DNAH17 encodes axonemal dynein [43]. Dyneins are motor protein complexes that use ATP to generate force and movement on microtubules in a wealth of biological processes, including ciliary beating, cell division and intracellular transport [44]. Therefore, mutations which reduce the efficiency of ATP conversion into movement are highly likely to reduce the efficiency of conversion of feed energy intake into maintenance and growth. Furthermore, serious human diseases arise from motor protein dysfunction supporting the potential for large phenotypic effects due to mutations in motor protein genes [45].
Several of the identified QTL possessed pleiotropic effects (Figure 5). In this study, a 1-Mb QTL that was associated with more than one trait was considered to represent a pleiotropic QTL. However, intervals of this size could easily harbour two different closely linked QTL. Further analyses using multivariate models are needed to dissect pleiotropic QTL from closely linked QTL (see [46]). Among these, the QTL on BTA 20 at 4 Mb was the only pleiotropic or closely linked QTL associated with RFI and MBW identified in the Hereford population. While the phenotypic correlations between RFI and its growth and feed intake component traits are expected to be zero, weak genetic correlations exist and pleiotropic loci affecting both traits have previously been reported [17]. We found that the same lead-SNP (rs133032375), which is located within an intron of STC2 (stanniocalcin 2) was selected for both RFI and MBW. The overexpression of human STC2 in transgenic mice reduces intramembranous and endochondral bone development and skeletal muscle growth and results in a dwarf phenotype [47]. STC2 has also been shown to be a potent negative regulator of postnatal growth in wild-type mice [48]. While STC2 is expressed in developing avian striated muscle and joints [49], the physiological roles of STC2 in cattle are unknown.

The QTL network. The genomic locations (BTA_Mb) and the trait(s) associated with each identified QTL. The traits are average daily gain (ADG), dry matter intake (DMI), metabolic mid-test body weight (MBW) and residual feed intake (RFI). QTL identified in Angus, Cycle VII, Hereford and Simmental × Angus are shown with black, blue, red and green arrows.
Large-effect pleiotropic or closely linked QTL for DMI and MBW
Three pleiotropic or closely linked QTL on BTA 21 at 13 Mb and BTA 7 at 0 and 23 Mb were associated with DMI and MBW. Lead-SNP rs134458731 was selected for both DMI and MBW as tagging the QTL on BTA 7 at 0 Mb in Angus (Tables 3 and 5). This SNP lies within an intron of LOC100125913, which encodes an uncharacterised protein. The B allele (from the Illumina A/B calling system) at this SNP was at a frequency of 0.09 and was associated with an increase in both DMI and MBW in Angus. The direction of allelic effects at this locus is consistent with the positive genetic correlation between DMI and MBW in cattle.
The pleiotropic or closely linked QTL on BTA 7 at 23 Mb, identified in Angus, was the largest-effect QTL identified for either DMI or MBW and explained 10.39% and 14.25% of the additive genetic variance in each trait, respectively (Tables 3 and 5). Two different lead-SNPs (rs133232710 and rs136491020) were selected for this QTL, with both being within the largest intron of ACSL6 (acyl-CoA synthetase long-chain family member 6). ACSL6 is a member of the long-chain acyl-CoA synthetase gene family (ACSL). In mammals, ACSL genes are necessary for fatty acid degradation (β-oxidation), phospholipid remodeling, and the production of long-chain acyl-CoA esters that regulate various physiological, metabolism and cell signaling processes [50, 51]. The ACSL enzymes are membrane-bound, act on non-polar hydrophobic substrates (fatty acids) and their water-soluble and powerful detergent products (acyl-CoAs) are important intermediates for de novo lipid synthesis [52]. In the first step of the two-step reaction catalysed by these enzymes, an acyl-AMP intermediate is formed from ATP. AMP is then exchanged with CoA to produce activated acyl-CoA. Once activated, the fatty acid can function as a signalling molecule, be incorporated into phospholipids or triacylglycerides, or undergo β-oxidation in mitochondria for energy generation [52]. While the classical hypothesis in the regulation of feed intake is that a decrease in glucose utilisation registered by specific sensors in the brain leads to hunger, it has also been shown that lipids have an important role through their provision of energy for cell metabolism [53]. Treating mice with fatty acid synthase inhibitors reduces food intake and body weight [54] and increasing neuronal long-chain acyl-CoA in the rat provides a hypothalamic signal of nutrient availability which results in the inhibition of both food intake and glucose production [55]. It has been observed that feeding high fat diets often results in decreased feed and energy intakes in cattle [56], however, the mechanisms that mediate fat-induced depression of feed intake have not been fully investigated in ruminants [57]. Glucose signaling does not effectively regulate feed intake in ruminants [58] because most of the dietary carbohydrates are fermented in the rumen by microorganisms [59]. Consequently, mechanisms other than glucose signaling must control feed intake in cattle and we identify a role for ACSL6 in this process.
ADG and MBW QTL on BTA 6 and 7 localise to genomic regions harbouring previously reported pleiotropic QTL
Two pleiotropic or closely linked QTL associated with ADG and MBW were identified on BTA 6 at 38 Mb and BTA 7 at 93 Mb (Figure 5). Many cattle studies have reported QTL on BTA 6 at 38 Mb affecting body weights, growth and carcass traits [60–64], calving ease direct [65], milk traits [66–68], reproductive traits [69–71] and feed efficiency traits [37, 62, 63]. In an analysis of BovineSNP50 genotypes scored in 18,274 animals from 10 US beef cattle breeds with observations for twelve body weights, calving ease and carcass traits, the QTL on BTA 6 at 38 Mb had the largest-effect and was associated with the greatest number of traits in the greatest number of breeds [46]. Three genes have been suggested as positional candidates: LAP3 (leucine aminopeptidase 3) [68], NCAPG (non-SMC condensing I complex, subunit G) [72, 73] and LCORL (ligand dependent nuclear receptor corepressor-like) [74]. Lead-SNP rs109294917, which lies within an intron of LCORL, was selected as tagging the QTL in the analyses of both ADG and MBW.
The pleiotropic QTL on BTA 7 at 93 Mb associated with ADG and MBW was the largest effect QTL identified for ADG and explained 9.18% of the additive genetic variance in Hereford (Table 4). This QTL has previously been shown to be the second largest-effect QTL associated with body weights (birth, weaning, yearling and mature weights) in beef cattle and is segregating in many breeds [46]. Two different intergenic lead-SNPs were selected for this QTL in the analyses of ADG and MBW and both are telomeric of ARRDC3 (arresting domain containing 3). ARRDC3 is a member of the arrestin superfamily that regulates obesity in mice and human males [75, 76]. Arrestins are signalling proteins that control metabolism usually through the desensitisation of beta-adrenergic receptors, which are present on the surface of almost every type of mammalian cell. The oral administration of some beta-adrenergic agonists increases muscle and decreases fat accretion in cattle, pigs, poultry, and sheep [77, 78].
The QTL on BTA 5 at 106 Mb explained 3.13% of the additive genetic variance in ADG in Hereford. This QTL appears to be Hereford-specific and pleiotropic, accounting for 2.6, 2.0, 4.9 and 3.9% of the additive genetic variance in birth, weaning, yearling and mature weights, respectively, in an independent population of 2,779 Herefords [46]. The model selected lead-SNP rs132862617 lies within an intron of CCND2 (cyclin D2), a member of the family of D-type cyclins which are implicated in cell cycle regulation, differentiation, and oncogenic transformation by governing the activity of cyclin-dependent kinases [79, 80]. Overexpression of CCND2 in myeloid cells results in a decrease in the duration of G1 (Gap 1 phase in the cell cycle when cell size increases) and an increase in the percentage of cells in S phase (Synthesis phase when DNA replication occurs) in mammalian cells [81, 82]. Since cell proliferation is an essential element of body growth, CCND2 appears to be a viable candidate gene for this QTL.
Conclusions
Although many QTL associated with feed efficiency traits of beef cattle have been identified, very little of the genetic variation in these traits has been explained by the detected QTL because of small sample sizes and the fact that the majority of variation appears to be due to small-effect loci. In this study, we took advantage of a relatively large sample size (5,133 animals from 4 independent beef cattle populations) to identify novel QTL and improve the resolution of the location of previously mapped QTL. This study is the largest GWAS ever performed to identify markers associated with feed efficiency and its component traits in beef cattle and led us to discover several large-effect QTL that cumulatively account for a significant percentage of additive genetic variance (the percentages in each of Tables 2 through 5 are additive within a population). Our results also suggest that QTL associated with feed efficiency traits tend to be population-specific with little overlap across populations, which could be due to differences in the power to detect QTL, environmental variation, or differences in the genetic architecture of trait variation among populations. These results also suggest candidate genes for the detected large-effect QTL which will improve our understanding of the biology of growth, feed consumption and feed utilisation in beef cattle.
Methods
The US Meat Animal Research Center Animal Care and Use Committee approved the procedures used in the experiment applied on Cycle VII animals. For the other 3 experiments data were either collected by commercial producers or were collected under the approval of the University of Missouri (ACUC Protocol 7505) or University of Illinois at Champaign-Urbana (IACUC Protocols 06091 and 09078) Animal Care and Use Committees.
Animals, phenotypic and genotypic data
Feedlot ADG, daily DMI, and MBW traits were measured in 4 different beef cattle populations. In all cases, average daily gain was estimated as the regression of all available weights on weigh dates and average daily feed intake was estimated for each animal from the daily recorded intake of each animal averaged across the number of days on feed and converted to a dry matter intake based upon the estimated moisture content of the ration. The sampled populations included:
-
1)
Cycle VII: 1,160 F1 × F1 steers derived from Cycle VII of the US Meat Animal Research Center Germplasm Evaluation Project. A description of the breed composition and mating design for these animals is in [37]. Briefly, in Cycle VII, Angus, Hereford, Simmental, Gelbvieh, Limousin, Red Angus, and Charolais sires were mated to Angus, Hereford and MARC III composite (1/4 Angus, 1/4 Hereford, 1/4 Pinzgauer, 1/4 Red Poll) cows. The resulting F1 animals were mated to generate 4-way cross progeny which were individually measured for growth and feed intake. A total of 15 contemporary groups formed using year and season of feeding were represented in these data.
-
2)
Angus: 1,658 Angus steers were produced by breeding registered Angus bulls to commercial cows at the Circle A Ranch in Iberia, MO (N =527), at the MFA Incorporated (N =224), Iowa State University (N =41), or were sourced from producers located throughout Missouri (N =866). Animals were produced over 10 years including in 1999 (N =94), 2000 (N =96), 2001 (N =166), 2003 (N =171), 2004 (N =81), 2005 (N =119), 2008 (N =41), 2010 (N =191), 2011 (N =421) and 2012 (N =278). A total of 173 bulls were identified as having sired 1,057 of the steers with half-sib groups ranging in size from 1 to 81 and averaging 6.1 steers. The remaining 601 steers had unknown sires. Animals were fed commercial concentrate rations at the Circle A Ranch using a Calan Gate feeding system or at the University of Missouri using a GrowSafe system for between 60 and 169 days (60–69 d, N =129; 70–79 d, N =66; 80–89 d, N =695; 108 d, N =89; 112 d, N =94; 120–129 d, N =171; 130–139 d; N =173; 140–149 d, N =191; 169 d, N =50). Weights were taken on a range from 3 to 13 occasions (3, N =445; 4, N =82; 6, N =145; 7, N =102; 8, N =121; 9, N =485; 10, N =228; 13, N =50) during feeding. A series of nutritional trials such as forage feeding, concentrate rations or amino acid and mineral supplementation was imposed on 651 of the steers fed at the University of Missouri. Consequently, there were a total of 102 contemporary groups based upon nutritional trial, farm, year and season of origin represented in these data.
-
3)
Hereford: 870 animals were individually fed a concentrate ration at Olsen Ranches, Inc in Harrisburg, Nebraska using a GrowSafe system. Olsen Ranches is the primary test herd for the American Hereford Association's National Reference Sire Program. Seedstock producers from around the U.S. nominate Hereford bulls for inclusion in the program. Phenotype and DNA samples were collected on 840 steers and 30 heifers born in Spring of 2009 (N =194), 2010 (N =293) and 2011 (N =383) and sired by 40 bulls with half-sib groups ranging in size from 1 to 104 and averaging 21.2 animals. Animals were fed for 70 (N =630), 72 (N =209) or 140 (N =31) days and had 8 (N =456), 9 (N =383) or 16 (N =31) weights recorded while on feed. A total of 10 contemporary groups based upon farm of origin, sex, feeding duration and slaughter date were represented in these data.
-
4)
Simmental × Angus: 1,445 Simmental-sired steers originating from 6 ranches were individually fed concentrate rations at the University of Illinois at Urbana-Champaign using a GrowSafe system in 2005 (N =231), 2006 (N =320), 2007 (N =322), 2008 (N =347) and 2009 (N =225). The steers were produced from 122 registered Simmental bulls with half-sib groups ranging in size from 1 to 113 progeny and averaging 11.7. Steers were fed either 122 (N =225), 134 (N =50), 140–149 (N =270), 160 – 169 (N =274), 170 – 179 (N =417), 183 (N =57) or 195 days (N =152) and live weights were taken on two adjacent days at the beginning and at the end of each feeding period. A series of nutritional trials was imposed on these steers resulting in a total of 202 contemporary groups based upon nutritional trial, ranch and year of origin as well as slaughter group.
The Cycle VII animals were genotyped with the BovineSNP50 assay and data for 48,729 SNPs were analysed for this platform [37]. Animals from the Angus and Hereford populations were genotyped with both the BovineHD and BovineSNP50 assays with missing values imputed to the union of the marker sets using Beagle 4.0 with default parameters [83]. The 1,445 Simmental × Angus animals were genotyped with the BovineSNP50 assay, however, BovineHD data for 467 registered Simmental bulls were also available and were used for genotype imputation.
For the Angus, Hereford and Simmental × Angus datasets: Animals were removed from the dataset if their genotype call rates were less than 0.90, if their autosomal heterozygosity exceeded 45% or if they were predicted to be Klinefelter (XXY) individuals (N =7 Angus, N =2 Hereford). For animals genotyped with the 50 K assay (1,093 Angus, 361 Hereford, 1,445 Simmental × Angus), sex was assigned as male if non-pseudo-autosomal X (paX) locus heterozygosity was <0.03, otherwise the animals were assigned as female. For animals genotyped with the 770 K assay (510 Angus, 491 Hereford), sex was assigned as male if non-paX locus heterozygosity was <0.03 and Y locus call rate was ≥0.5, female if non-paX locus heterozygosity was ≥0.03 and Y locus call rate was <0.5, and Klinefelter if non-paX locus heterozygosity was ≥0.03 and Y locus call rate was ≥0.5. Hereford females (n =23) had BTA X heterozygosities (0.18) that were approximately one-half of their autosomal heterozygosities (0.32). The threshold of 0.03 was used to account for genotyping error rate. Similarly, BTA Y call rates were generally negligible in females and very high in males and the BTA Y threshold of 0.5 successfully identified the presence or absence of a Y chromosome. SNPs were removed from the dataset if they had a call rate <0.85, minor allele frequency <0.001 or Hardy-Weinberg Equilibrium p <3 × 10−9. Non-paX and BTA Y SNPs with heterozygosities >0.03 in males and mitochondrial SNPs with heterozygosities >0.03 were also removed leaving 747 473, 684 458 and 690 184 loci for analysis in the Angus, Hereford and Simmental × Angus populations, respectively.
For Angus and Hereford animals with weekly or biweekly body weight measurements and Simmental × Angus animals with two start and ending weights, ADG and MBW were estimated over the test period by linear regression. The total feed intake of each animal over the test period was averaged and adjusted for moisture content to produce the average daily DMI [7]. Residual feed intake was analysed by including partial linear regressions on ADG and MWT in the model used to analyse DMI.
Statistical analysis
The BayesB method was used to simultaneously analyse whole genome SNPs, using GENSEL software [84]. For each of the 4 populations (Cycle VII, Angus, Hereford and Simmental × Angus), each trait (ADG, DMI, MBW and RFI) was separately analysed, with SNP allele substitution effects fitted as random effects. Systematic environmental effects fitted as fixed effects included cohort groups based on birth herd, year and season of birth and sex, resulting in 15, 102, 10 and 202 contemporary group levels in the Cycle VII, Angus, Hereford and Simmental × Angus populations, respectively. For the Cycle VII animals, linear covariates for breed composition and expected heterosis based upon the breed composition of each animal’s parents were also included in the model. The parameter π, which is the proportion of SNPs assumed to have no effect on the trait was set at 0.99 for the Cycle VII animals (genotyped with 50 K SNPs) and at 0.9995 for the other 3 populations (genotyped or imputed to 800 K SNPs) which corresponded to fitting about 400 markers in each MCMC iteration. Allowing only markers with strong associations to traits to be fitted, motivated the choice of π. MCMC methods with 41,040 iterations were used to generate posterior mean estimates of marker effects and variance components after discarding the first 1,000 samples for burn-in.
Due to linkage disequilibrium, the effect of a QTL may be spread over a number of neighboring SNPs. Therefore, the genome was divided into non-overlapping 1-Mb windows based on the UMD3.1 reference assembly base pair locations of markers and the percentage of genetic variance explained by each window was calculated for each trait. The null hypothesis distribution of the percentage of genetic variance explained by each 1-Mb window was generated for each trait by applying the same model (BayesB with the same parameters) on permuted data in the Angus population (data sets in which the genotypes of individuals are randomly assigned to trait values, which maintain the distributional properties of the trait values and the genotypes under the null hypothesis of no true QTL effects). As for the analysis of the unpermuted data, the genome was divided into the same non-overlapping 1-Mb windows and the percentage of genetic variance explained by each window was calculated. The JMP software [85] was used to fit the model and generate the distributions for the percentage of genetic variance explained by the 1-Mb windows. Next, the estimated parameters from the best-fit models (Table 6) were used to calculate the P-values for each window in the analysis of the unpermuted data. The estimated parameters for RFI were also used for ADG, as the estimated parameters for ADG were dissimilar to those for the other traits (Table 6) and produced spurious significance results. The Bonferroni correction was employed to adjust P-values for multiple test comparisons using the p.adjust package in R [86]. Windows with a Bonferroni-corrected P-value <0.05 were identified as significant QTL. The sfdp algorithm from Graphviz software was used to draw the QTL network [87].
Within each of the significant windows, the SNP with the highest sPPI (percentage of the chains in which the specific SNP is included in the model with non-zero effect) was chosen as the most strongly associated SNP within the 1-Mb QTL window and was denoted the ‘lead-SNP’. Posterior mean residual and additive genetic variances and posterior mean of marker-based heritability were reported for each trait in each population.
Availability of data
The data sets supporting the results of this article are available for non-commercial purposes from JFT following the execution of a materials transfer agreement.
Abbreviations
- ADG:
-
Average daily gain
- BTA:
-
Bos taurus chromosome
- DMI:
-
Dry matter intake
- GWAS:
-
Genome-wide association study
- Mb:
-
Million base pairs
- MBW:
-
Metabolic mid-test body weight
- MCMC:
-
Markov-chain Monte Carlo
- paX:
-
Pseudo-autosomal X chromosome
- sPPI:
-
Posterior probability of model inclusion for a given SNP
- QTL:
-
Quantitative trait loci
- RFI:
-
Residual feed intake
- SNP:
-
Single nucleotide polymorphism
References
Koch RM, Swiger LA, Chambers D, Gregory KE: Efficiency of feed use in beef cattle. J Anim Sci. 1963, 22: 486-494.
Dickerson GE, Kunzi N, Cundiff LV, Koch RM, Arthaud VH, Gregory KE: Selection criteria for efficient beef production. J Anim Sci. 1974, 39: 659-673.
Archer JA, Richardson EC, Herd RM, Arthur PF: Potential for selection to improve efficiency of feed use in beef cattle: a review. Austr J Agric Res. 1999, 50: 147-162. 10.1071/A98075.
Hegarty RS, Goopy JP, Herd RM, McCorkell B: Cattle selected for lower residual feed intake have reduced daily methane production. J Anim Sci. 2007, 85: 1479-1486. 10.2527/jas.2006-236.
Kennedy BW, van der Werf JH, Meuwissen TH: Genetic and statistical properties of residual feed intake. J Anim Sci. 1993, 71: 3239-3250.
Liu MF, Goonewardene LA, Bailey DRC, Basarab JA, Kemp RA, Arthur PF, Okine EK, Makarechian M: A study on the variation of feed efficiency in station tested beef bulls. Can J Anim Sci. 2000, 80: 435-441. 10.4141/A99-030.
Arthur PF, Archer JA, Johnston DJ, Herd RM, Richardson EC, Parnell PF: Genetic and phenotypic variance and covariance components for feed intake, feed efficiency, and other postweaning traits in Angus cattle. J Anim Sci. 2001, 79: 2805-2811.
Crowley JJ, McGee M, Kenny DA, Crews DH, Evans RD, Berry DP: Phenotypic and genetic parameters for different measures of feed efficiency in different breeds of Irish performance-tested beef bulls. J Anim Sci. 2010, 88: 885-894. 10.2527/jas.2009-1852.
Herd RM, Archer JA, Arthur PF: Reducing the cost of beef production through genetic improvement in residual feed intake: opportunity and challenges to application. J Anim Sci. 2003, 81 (E. Suppl. 1): E9-E17.
Crews DH: Genetics of efficient feed utilization and national cattle evaluation: a review. Genet Mol Res. 2005, 4: 152-165.
Arthur PF, Herd RM, Wilkins JF, Archer JA: Maternal productivity of Angus cows divergently selected for post-weaning residual feed intake. Aust J Exp Agr. 2005, 45: 985-993. 10.1071/EA05052.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O’Connell J, Moore SS, Smith TPL, Sonstegard TS, Van Tassell CP: Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009, 4: e5350-10.1371/journal.pone.0005350.
Rincon G, Weber KL, Eenennaam AL, Golden BL, Medrano JF: Hot topic: performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J Dairy Sci. 2011, 94: 6116-6121. 10.3168/jds.2011-4764.
Cole JB, VanRaden PM, O’Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, Taylor JF, Wiggans GR: Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009, 92: 2931-2946. 10.3168/jds.2008-1762.
Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T, Smith TPL, Sonstegard TS, Thallman RM, Bennett GL: Genome-wide association study of growth in crossbred beef cattle. J Anim Sci. 2010, 88: 837-848. 10.2527/jas.2009-2257.
Bolormaa S, Hayes BJ, Savin K, Hawken R, Barendse W, Arthur PF, Herd RM, Goddard ME: Genome-wide association studies for feedlot and growth traits in cattle. J Anim Sci. 2011, 89: 1684-1697. 10.2527/jas.2010-3079.
Lu D, Miller S, Sargolzaei M, Kelly M, Vander Voort G, Caldwell T, Wang Z, Plastow G, Moore S: Genome-wide association analyses for growth and feed efficiency traits in beef cattle. J Anim Sci. 2013, 91: 3612-3633. 10.2527/jas.2012-5716.
Barendse W, Reverter A, Bunch RJ, Harrison BE, Barris W, Thomas MB: A validated whole-genome association study of efficient food conversion in cattle. Genetics. 2007, 176: 1893-1905. 10.1534/genetics.107.072637.
Nkrumah JD, Sherman EL, Li C, Marques E, Crews DH, Bartusiak R, Murdoch B, Wang Z, Basarab JA, Moore SS: Primary genome scan to identify putative quantitative trait loci for feedlot growth rate, feed intake, and feed efficiency of beef cattle. J Anim Sci. 2007, 85: 3170-3181. 10.2527/jas.2007-0234.
Sherman EL, Nkrumah JD, Moore SS: Whole genome single nucleotide polymorphism associations with feed intake and feed efficiency in beef cattle. J Anim Sci. 2010, 88: 16-22. 10.2527/jas.2008-1759.
Stranger BE, Stahl EA, Raj T: Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011, 187: 367-383. 10.1534/genetics.110.120907.
Andersson L: Genetic dissection of phenotypic diversity in farm animals. Nat Rev Genet. 2001, 2: 130-138. 10.1038/35052563.
Farnir F, Coppieters W, Arranz JJ, Berzi P, Cambisano N, Grisart B, Karim L, Marcq F, Moreau L, Mni M, Nezer C, Simon P, Vanmanshoven P, Wagenaar D, Georges M: Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 2000, 10: 220-227. 10.1101/gr.10.2.220.
McRae AF, McEwan JC, Dodds KG, Wilson T, Crawford AM, Slate J: Linkage disequilibrium in domestic sheep. Genetics. 2002, 160: 1113-1122.
Onteru SK, Fan B, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF: A whole-genome association study for pig reproductive traits. Anim Genet. 2012, 43: 18-26. 10.1111/j.1365-2052.2011.02213.x.
Fan B, Onteru SK, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF: Genome-wide association study identifies loci for body composition and structural soundness traits in pigs. PLoS One. 2011, 6: e14726-10.1371/journal.pone.0014726.
Mateescu RG, Garrick DJ, Tait RG, Garmyn AJ, Duan Q, Liu Q, Mayes MS, Van Eenennaam AL, VanOverbeke DL, Hilton GG, Beitz DC, Reecy JM: Genome-wide association study of concentrations of iron and other minerals in longissimus muscle of Angus cattle. J Anim Sci. 2013, 91: 3593-3600. 10.2527/jas.2012-6079.
Saatchi M, McClure MC, McKay SD, Rolf MM, Kim J, Decker JE, Taxis TM, Chapple RH, Ramey HR, Northcutt SL, Bauck S, Woodward B, Dekkers JC, Fernando RL, Schnabel RD, Garrick DJ, Taylor JF: Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet Sel Evol. 2011, 43: 40-10.1186/1297-9686-43-40.
Saatchi M, Schnabel RD, Rolf MM, Taylor JF, Garrick DJ: Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet Sel Evol. 2012, 44: 38-10.1186/1297-9686-44-38.
Saatchi M, Ward J, Garrick DJ: Accuracies of direct genomic breeding values in Hereford beef cattle using national or international training populations. J Anim Sci. 2013, 91: 1538-1551. 10.2527/jas.2012-5593.
Wolc A, Arango J, Settar P, Fulton JE, O'Sullivan NP, Preisinger R, Habier D, Fernando R, Garrick DJ, Hill WG, Dekkers JC: Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim Genet. 2012, 43 (Suppl. 1): 87-96.
Pryce JE, Bolormaa S, Chamberlain AJ, Bowman PJ, Savin K, Goddard ME, Hayes BJ: A validated genome-wide association study in 2 dairy cattle breeds for milk production and fertility traits using variable length haplotypes. J Dairy Sci. 2010, 93: 3331-3345. 10.3168/jds.2009-2893.
Meuwissen TH, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Sun X, Habier D, Fernando RL, Garrick DJ, Dekkers JCM: Genomic breeding value prediction and QTL mapping of QTLMAS2010 data using Bayesian Methods. BMC Proc. 2011, 5 (Suppl. 3): S13-
Toosi A, Fernando RL, Dekkers JCM: Genomic selection in admixed and crossbred populations. J Anim Sci. 2010, 88: 32-46. 10.2527/jas.2009-1975.
Pryce JE, Arias J, Bowman PJ, Davis SR, Macdonald KA, Waghorn GC, Wales WJ, Williams YJ, Spelman RJ, Hayes BJ: Accuracy of genomic predictions of residual feed intake and 250-day body weight in growing heifers using 625,000 single nucleotide polymorphism markers. J Dairy Sci. 2012, 95: 2108-2119. 10.3168/jds.2011-4628.
Snelling WM, Allan MF, Keele JW, Kuehn LA, Thallman RM, Bennett GL, Ferrell CL, Jenkins TG, Freetly HC, Nielsen MK, Rolfe KM: Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle. J Anim Sci. 2011, 89: 1731-1741. 10.2527/jas.2010-3526.
Porto Neto LR, Bunch RJ, Harrison BE, Barendse W: Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim Genet. 2012, 43: 785-789. 10.1111/j.1365-2052.2012.02330.x.
Karim L, Takeda H, Lin L, Druet T, Arias JA, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, Harris BL, Keehan MD, Littlejohn MD, Spelman RJ, Georges M, Coppieters W: Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011, 43: 405-413. 10.1038/ng.814.
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, Sugimoto Y, Takasuga A: Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13: 40-
Ashwell MS, Heyen DW, Weller JI, Ron M, Sonstegard TS, Van Tassell CP, Lewin HA: Detection of quantitative trait loci influencing conformation traits and calving ease in Holstein-Friesian cattle. J Dairy Sci. 2005, 88: 4111-4119. 10.3168/jds.S0022-0302(05)73095-2.
Lillehammer M, Arnyasi M, Lien S, Olsen HG, Sehested E, Ødegård J, Meuwissen TH: A genome scan for quantitative trait locus by environment interactions for production traits. J Dairy Sci. 2007, 90: 3482-3489. 10.3168/jds.2006-834.
Milisav I, Affara NA: A potential human axonemal dynein heavy-chain gene maps to 17q25. Mamm Genome. 1998, 9: 404-407. 10.1007/s003359900782.
Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA: Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol. 2013, 14: 713-726. 10.1038/nrm3667.
Fliegauf M, Benzing T, Omran H: When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007, 8: 880-893. 10.1038/nrm2278.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ: Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014, 15: 442-10.1186/1471-2164-15-442.
Gagliardi AD, Kuo EY, Raulic S, Wagner GF, DiMattia GE: Human stanniocalcin-2 exhibits potent growth-suppressive properties in transgenic mice independently of growth hormone and IGFs. Am J Physiol Endocrinol Metab. 2005, 288: E92-E105.
Chang AC, Hook J, Lemckert FA, McDonald MM, Nguyen MA, Hardeman EC, Little DG, Gunning PW, Reddel RR: The murine stanniocalcin 2 gene is a negative regulator of postnatal growth. Endocrinology. 2008, 149: 2403-2410. 10.1210/en.2007-1219.
Mittapalli VR, Pröls F, Huang R, Christ B, Scaal M: Avian stanniocalcin-2 is expressed in developing striated muscle and joints. Anat Embryol (Berl). 2006, 211: 519-523. 10.1007/s00429-006-0100-6.
Faergeman NJ, Knudsen J: Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem J. 1997, 323 (Pt. 1): 1-12.
Soupene E, Kuypers FA: Mammalian long-chain acyl-CoA synthetases. Exp Biol Med. 2008, 233: 507-521. 10.3181/0710-MR-287.
Soupene E, Dinh NP, Siliakus M, Kuypers FA: Activity of the acyl-CoA synthetase ACSL6 isoforms: role of the fatty acid Gate-domains. BMC Biochem. 2010, 11: 18-10.1186/1471-2091-11-18.
Fantino M: Role of lipids in the control of food intake. Curr Opin Clin Nutr Metab Care. 2011, 14: 138-144. 10.1097/MCO.0b013e3283437b78.
Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP: Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000, 288: 2379-2381. 10.1126/science.288.5475.2379.
Obici S, Feng Z, Arduini A, Conti R, Rossetti L: Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003, 9: 756-761. 10.1038/nm873.
Allen MS: Effects of diet on short-term regulation of feed intake by lactating dairy cattle. J Dairy Sci. 2000, 83: 1598-1624. 10.3168/jds.S0022-0302(00)75030-2.
Choi BR, Palmquist DL, Allen MS: Sodium mercaptoacetate is not a useful probe to study the role of fat in regulation of feed intake in dairy cattle. J Nutr. 1997, 127: 171-176.
Forbes JM: The multifactorial nature of food intake control. J Anim Sci. 2003, 81 (Suppl. 2): E139-E144.
Nafikov RA, Beitz DC: Carbohydrate and lipid metabolism in farm animals. J Nutr. 2007, 137: 702-705.
Gutiérrez-Gil B, Williams JL, Homer D, Burton D, Haley CS, Wiener P: Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J Anim Sci. 2009, 87: 24-36.
Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, Hansen C, Moore SS: Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci. 2004, 82: 3405-3414.
Lindholm-Perry AK, Kuehn LA, Oliver WT, Sexten AK, Miles JR, Rempel LA, Cushman RA, Freetly HC: Adipose and muscle tissue gene expression of two genes (NCAPG and LCORL) located in a chromosomal region associated with cattle feed intake and gain. PLoS One. 2013, 8: e80882-10.1371/journal.pone.0080882.
Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, Wheeler TL, Ferrell CL, Jenkins TG, Snelling WM, Freetly HC: Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA 6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011, 12: 103-
Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, Takasuga A: Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009, 10: 43-
Bongiorni S, Mancini G, Chillemi G, Pariset L, Valentini A: Identification of a short region on chromosome 6 affecting direct calving ease in Piedmontese cattle breed. PLoS One. 2012, 7: e50137-10.1371/journal.pone.0050137.
Olsen HG, Lien S, Gautier M, Nilsen H, Roseth A, Berg PR, Sundsaasen KK, Svendsen M, Meuwissen TH: Mapping of a milk production quantitative trait locus to a 420-kb region on bovine chromosome 6. Genetics. 2005, 169: 275-283.
Schrooten C, Bink MC, Bovenhuis H: Whole genome scan to detect chromosomal regions affecting multiple traits in dairy cattle. J Dairy Sci. 2004, 87: 3550-3560. 10.3168/jds.S0022-0302(04)73492-X.
Zheng X, Ju Z, Wang J, Li Q, Huang J, Zhang A, Zhong J, Wang C: Single nucleotide polymorphisms, haplotypes and combined genotypes of LAP3 gene in bovine and their association with milk production traits. Mol Biol Rep. 2011, 38: 4053-4061. 10.1007/s11033-010-0524-1.
Daetwyler HD, Schenkel FS, Sargolzaei M, Robinson JA: A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J Dairy Sci. 2008, 91: 3225-3236. 10.3168/jds.2007-0333.
Holmberg M, Andersson-Eklund L: Quantitative trait loci affecting fertility and calving traits in Swedish dairy cattle. J Dairy Sci. 2006, 89: 3664-3671. 10.3168/jds.S0022-0302(06)72406-7.
Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A: Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein x Jersey crossbred population. Anim Genet. 2009, 40: 27-34. 10.1111/j.1365-2052.2008.01793.x.
Weikard R, Altmaier E, Suhre K, Weinberger KM, Hammon HM, Albrecht E, Setoguchi K, Takasuga A, Kühn C: Metabolomic profiles indicate distinct physiological pathways affected by two loci with major divergent effect on Bos taurus growth and lipid deposition. Physiol Genomics. 2010, 42A: 79-88. 10.1152/physiolgenomics.00120.2010.
Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, Klopp N, Fürbass R, Weikard R, Kühn C: Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics. 2009, 183: 951-964. 10.1534/genetics.109.106476.
Pryce JE, Hayes BJ, Bolormaa S, Goddard ME: Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011, 187: 981-984. 10.1534/genetics.110.123943.
Patwari P, Emilsson V, Schadt EE, Chutkow WA, Lee S, Marsili A, Zhang Y, Dobrin R, Cohen DE, Larsen PR, Zavacki AM, Fong LG, Young SG, Lee RT: The arrestin domain containing 3 protein regulates body mass and energy expenditure. Cell Metab. 2011, 14: 671-683. 10.1016/j.cmet.2011.08.011.
Patwari P, Lee RT: An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012, 23: 216-222. 10.1016/j.tem.2012.03.003.
Mersmann HJ: Overview of the effects of beta-adrenergic receptor agonists on animal growth including mechanisms of action. J Anim Sci. 1998, 76: 160-172.
Beermann DH: Beta-Adrenergic receptor agonist modulation of skeletal muscle growth. J Anim Sci. 2002, 80: E18-E23.
Meyyappan M, Wong H, Hull C, Riabowol KT: Increased expression of cyclin D2 during multiple states of growth arrest in primary and established cells. Mol Cell Biol. 1998, 18: 3163-3172.
Sweeney KJ, Sarcevic B, Sutherland RL, Musgrove EA: Cyclin D2 activates Cdk2 in preference to Cdk4 in human breast epithelial cells. Oncogene. 1997, 14: 1329-1340. 10.1038/sj.onc.1200951.
Ando K, Ajchenbaum-Cymbalista F, Griffin JD: Regulation of G1/S transition by cyclins D2 and D3 in hematopoietic cells. Proc Natl Acad Sci U S A. 1993, 90: 9571-9575. 10.1073/pnas.90.20.9571.
Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ: Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993, 7: 1559-1571. 10.1101/gad.7.8.1559.
Browning BL, Browning SL: A unified approach to genotype imputation and haplotype phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009, 84: 210-223. 10.1016/j.ajhg.2009.01.005.
Garrick DJ, Fernando RL: Implementing a QTL Detection Study (GWAS) Using Genomic Prediction Methodology. Genome-Wide Association Studies and Genomic Prediction. Edited by: Gondro C, van der Werf J, Hayes B. 2013, New York: Humana Press, 275-298.
JMP® Statistical Discovery Software from SAS. 1989–2007, Cary, NC: SAS Institute Inc, http://www.jmp.com/,
R Development Core Team: R: A Language and Environment for Statistical Computing. 2011, Vienna: R Foundation for Statistical Computing, http://www.r-project.org/,
Ellson J, Gansner ER, Koutsofios E, North SC, Woodhull G: Graphviz and Dynagraph – Static and Dynamic Graph Drawing Tools. Graph Drawing Software. Edited by: Jünger M, Mutzel P. 2004, Berlin Heidelberg: Springer, 127-148.
Acknowledgements
This project was supported by National Research Initiative Competitive Grant number 2011-68004-30214 from the USDA National Institute of Food and Agriculture.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Experimental design was implemented by HCF, SLH, MSK, EJP and DWS. Cattle were fed, data were recorded and tissue samples were collected by RDS, JEB, HCF, SLH, HYI, KAJ, MSK, JWK, DDL, HLN, CMS, DWS, MLS and RLW. DNA was extracted and quantitated by JEB, HYI, JWK and RDS and genotyped by EM. Data management, analysis and interpretation: MS, JED, RDS, DJG and JFT. Manuscript writing: MS, DJG and JFT. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Saatchi, M., Beever, J.E., Decker, J.E. et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genomics 15, 1004 (2014). https://doi.org/10.1186/1471-2164-15-1004
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-1004