
Abstract
Background
The number of fibrous roots that develop into storage roots determines sweetpotato yield. The aim of the present study was to identify the molecular mechanisms involved in the initiation of storage root formation, by performing a detailed transcriptomic analysis of initiating storage roots using next-generation sequencing platforms. A two-step approach was undertaken: (1) generating a database for the sweetpotato root transcriptome using 454-Roche sequencing of a cDNA library created from pooled samples of two root types: fibrous and initiating storage roots; (2) comparing the expression profiles of initiating storage roots and fibrous roots, using the Illumina Genome Analyzer to sequence cDNA libraries of the two root types and map the data onto the root transcriptome database.
Results
Use of the 454-Roche platform generated a total of 524,607 reads, 85.6% of which were clustered into 55,296 contigs that matched 40,278 known genes. The reads, generated by the Illumina Genome Analyzer, were found to map to 31,284 contigs out of the 55,296 contigs serving as the database. A total of 8,353 contigs were found to exhibit differential expression between the two root types (at least 2.5-fold change). The Illumina-based differential expression results were validated for nine putative genes using quantitative real-time PCR. The differential expression profiles indicated down-regulation of classical root functions, such as transport, as well as down-regulation of lignin biosynthesis in initiating storage roots, and up-regulation of carbohydrate metabolism and starch biosynthesis. In addition, data indicated delicate control of regulators of meristematic tissue identity and maintenance, associated with the initiation of storage root formation.
Conclusions
This study adds a valuable resource of sweetpotato root transcript sequences to available data, facilitating the identification of genes of interest. This resource enabled us to identify genes that are involved in the earliest stage of storage root formation, highlighting the reduction in carbon flow toward phenylpropanoid biosynthesis and its delivery into carbohydrate metabolism and starch biosynthesis, as major events involved in storage root initiation. The novel transcripts related to storage root initiation identified in this study provide a starting point for further investigation into the molecular mechanisms underlying this process.
Similar content being viewed by others


Background
Sweetpotato [Ipomoea batatas (L.) Lam.] is the seventh most important food crop in the world [1]. It has a worldwide production of approximately 107 million metric tons and is the third most important root/tuber crop after potato (Solanum tuberosum L.) and cassava (Manihot esculenta Crantz). World production is centered in Southeast Asia, with China being the largest producer, while Sub-Saharan Africa ranks second [2]. Sweetpotato is used as a source of starch, ethanol, and animal fodder in most of Asia while it is considered a subsistence crop in Africa. The USA, Japan, Australia, New Zealand, South Africa and Israel are among the few producing countries that grow sweetpotato as a vegetable to market in developed economies. Global demand for sweetpotato is on the rise due to its high nutritional value. The Nutrition Action Health Letter from the Center for Science in the Public Interest, USA [3], rates sweetpotatoes as the highest ranking vegetable for human nutrition.
The most economically important physiological process in sweetpotato production is storage root (SR) development. Sweetpotatoes are commercially propagated through vegetative cuttings. These cuttings produce adventitious roots that give rise to the SRs [4–7]. Adventitious roots originate from primordia located on the nodes, as well as from the cut ends, i.e., “wound” roots [4, 6]. Initially, white fibrous roots (FRs) develop and some of these subsequently develop into SRs. Depending on the number of FRs induced to form SRs, sweetpotato plants will yield either a high (4 to 8 per plant) or low number of SRs that may even be reduced to one very large SR per plant [7]. Togari [4] described the sequence of anatomical events leading to SR initiation in varieties Okinawan and Beinakzi and reported that the regular vascular cambium layer first appears 20 days after transplanting (DAT), followed by the initial development of secondary anomalous cambium (AC) features at 25 DAT. Togari [4] also documented the incidence of stele lignification and proposed that lignification prevents SR initiation. Wilson and Lowe [5] also suggested that only the appearance of AC can prevent stele lignification.
Recently, we demonstrated in both Georgia Jet and Beauregard sweetpotato varieties (the leading Israeli variety and an important USA variety, respectively), that the period spanning 5 to 35 DAT is critical in determining whether adventitious roots become lignified or initiate as SRs, and that the appearance of AC marks the initial phase of SR formation [7]. The molecular mechanisms underlying the induction of adventitious roots to become SRs are, however, poorly understood. Expression studies have been used in an effort to elucidate factors involved in SR formation. You et al. [8] constructed a cDNA library of early-stage SRs, and identified 22 genes differentially expressed between FRs (non-storage and storage stage) and SRs. Among them were a no apical meristem (NAM)-like and a MADS-box (MCM1, AGAMOUS, DEFICIENS and SRF) protein gene, both of which were down-regulated in SRs. McGregor [9] found several NAC family transcriptional regulator proteins that were down regulated in storage roots, similar to the NAM-like protein described by You et al. [8]. McGregor [9] also identified up-regulated expression of two NAM-like genes, as well as sporamin genes and genes involved in starch biosynthesis, in storage roots that developed six weeks after planting compared to fibrous roots. Several additional MADS-box genes (IbMADS1, IbMADS3, IbMADS4, IbMADS10, IbMADS79, IbAGL17, IbAGL20 and SRD1) expressed in root tissues have been isolated from sweetpotato, and their possible roles in root development have been deduced [10–15]. Tanaka et al. [16] identified 10 genes with differential expression among FRs, thick roots, and SRs. One of the genes, SRF6, encoded a receptor-like kinase with high expression around the primary cambium and xylem meristem. In addition, Tanaka et al. [17] suggested three sweetpotato class 1 knotted1-like homeobox (KNOX1) genes as possible regulators of cytokinin levels in SRs.
De-novo assembly of transcript sequences produced by next-generation sequencing (NGS) technologies offers a rapid approach to obtaining expressed gene sequences for non-model organisms. Indeed, most recently, NGS was used by several research groups to obtain leaf, stem and root transcriptome data for different sweetpotato cultivars [18–20]. Tao et al. [20] used Illumina NGS, employing a combination of different tissues at different developmental stages, to generate 51,736 annotated transcripts, and identified differentially expressed transcripts in different tissues, including roots. Xie et al. [21] analyzed the transcriptome of a purple sweetpotato, obtaining a total of 58,800 unigenes, and suggested UDP-glucose-flavonoid 3-O-glucosyltransferase as one of the key enzymes in anthocyanin biosynthesis and that anthocyanin-3-glucoside might be one of the major components for anthocyanin pigments in the purple sweetpotato.
The presently described study focused on the identification of the molecular mechanisms involved in the initiation of SR formation in the leading sweetpotato variety in Israel, Georgia Jet, by performing a detailed transcriptomic analysis of initiating SRs (ISRs) using NGS platforms. A two-step approach was undertaken: (1) generating a database for the sweetpotato root transcriptome using 454-Roche sequencing of a cDNA library created from pooled samples of two root types: FRs and ISRs; (2) comparing the expression profiles of ISRs and FRs, using the Illumina Genome Analyzer to sequence non-normalized cDNA libraries of the two root types and mapping the data onto the root transcriptome database. Use of the 454-Roche platform generated a total of 524,607 reads, 85.6% of which were clustered into 55,296 contigs that matched 40,278 known genes. The differential expression profiles between the two root types obtained by the Illumina platform indicated down-regulation of classical root functions, such as transport and response to the environment, and of lignin biosynthesis in ISRs, along with up-regulation of carbohydrate metabolism and starch biosynthesis. In addition, the data suggest delicate control of stem cell maintenance and differentiation in sweetpotato vascular development associated with the initiation of SR formation.
Results and discussion
Insight into the transcriptome of sweetpotato roots
Defining the transcriptome using 454 sequencing and de-novo assembly
To obtain insight into the molecular mechanisms involved in the initiation of SR formation in sweetpotato, and to identify candidate genes involved in this process, a two-stage approach was adopted. First, a database of the sweetpotato (var. Georgia Jet) root transcriptome was generated, using 454-Roche sequencing of a cDNA library created from pooled RNA samples of two root types: ISRs and non-initiating FRs. Roots were divided into either ISRs or FRs following microscopic analysis, as shown in Figure 1. Second, the expression profiles of ISRs and FRs were compared, using the Illumina Genome Analyzer, to sequence non-normalized cDNA libraries of the two root types and the data were mapped onto the root transcriptome database.

Microscopic view of adventitious root cross sections 26 days after transplanting. A. Fibrous root (FR) cross section. B. Initiated storage root (ISR) cross section. Tissue sections were taken at 3 cm from the proximal root part and stained with toluidine blue. LC – lignified cells; AC – anomalous cambium; RVC – regular vascular cambium. Scale bars = 0.1 mm.
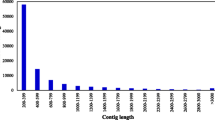
A total of 524,607 high-quality reads were generated with an average read length of 310 bp. The total number of bases (without keys, tags or bad-quality bases) was 1.63E + 08. The MIRA assembly clustered 85.6% (449,282 sequences) of the 454 sequence reads into 55,296 contigs. The sequence length distribution is illustrated in Figure 2. The average length of the contigs was 519 ± 245 bases. The remaining 75,325 (orphan) sequences were retained as singletons. The clustered contig data are available via a web page connected to The Volcani Center, Agricultural Research Organization web page at http://batata.agri.gov.il/.

Sequence length distributions of contig sequences in the sweetpotato root transcriptome.
Functional annotation by sequence comparison with public databases
The transcriptomic data were used to query public genomic databases [i.e. NCBI non-redundant (Nr)] using BLASTX. Of the 55,296 contigs, 40,278 (73%) matched known genes at a cut-off E value ≤ 1.0E-3. Annotations of the two best hits for each contig are given at http://batata.agri.gov.il/ and in Additional file 1. E-value distribution for the top BLAST result for each sequence is given in Figure 3. The E-value distribution of the top hits in the Nr database revealed that 99.5% of the mapped sequences show significant homology (less than 1.0E-5), and 22% of the sequences showed greater than 80% similarity. These results indicated a high level of homology between our sequences and those found in the BLAST database. Similarity distribution of the contigs to their BLAST results is illustrated in Figure 4. Species distribution of the BLAST results is given in Additional file 2, demonstrating that most sweetpotato sequences exhibited similarity to Vitis vinifera, Ricinus communis and Populus trichocarpa sequences (43%, 24% and 18%, respectively), as well as to members of the Solanaceae family. Similarity to sequences of Arabidopsis thaliana was less than 10%. The relatively low number of hits detected with Ipomoea batatas (sweetpotato) may be attributed to the low number of publicly available sequences in the database. The sweetpotato root transcript sequences generated in this study thus add to the recently accumulated sweetpotato sequences [18–21] which can be used for the discovery of new genes involved in root development and functioning and in the initiation of SR formation.

E-value distribution of contig sequences’ BLAST results in the sweetpotato root transcriptome.

Similarity distributions of the contigs to their BLAST results.
Functional classification by gene ontology (GO) and by the Kyoto encyclopedia of genes and genomes (KEGG)
To assess whether the sweetpotato root transcriptomic data were indeed representative of roots and SRs, the annotated contigs were assigned to molecular functions using GO. BlastoGO [22] was used to obtain BLAST results for our contigs against the NCBI Nr database and then again to obtain GO annotations for the BLAST results. Ontologizer [23] was used to perform the GO functional classification for the contigs. Of the 40,278 contigs that matched known genes, 34,308 sequences could be grouped into 4,776 different GO categories, and all parental GO terms [i.e. level 2: Biological Process (BP), Molecular Function (MF), Cellular Component (CC)] were assigned (data not shown). Of the GO annotations, 55.3% were associated with BP, 34.5% were associated with MF, and 10.2% were associated with CC (2640, 1648 and 487, respectively). The contigs were further classified using GOSlim [24] and results are presented in Figure 5 and Additional file 3. There was a high representation of cellular process, metabolic process, primary metabolic process, biosynthetic process, macromolecular metabolic process, response to stimulus, localization and transport in the parental category BP, high representation of binding (including nucleotide, protein, carbohydrate and lipid binding), catalytic, transporter and transcription regulator activities in the MF parental category, and high representation of cell, intracellular, cytoplasm, organelle and membrane-bound organelle in the CC category. Similar results have been recently demonstrated for sweetpotato [19–21] and other root transcriptomes, including rice [25], wild rice [26] and Avena barbata[27], supporting the quality of our Georgia Jet sweetpotato root transcriptome data.

Gene ontology classification of assembled contigs.
To assign the detected contigs to biological pathways, the 55,296 contig sequences were compared using BLASTX with an E-value cutoff of <10E-3 against the KEGG biological pathways database. The contigs were mapped to 140 KEGG pathways, as demonstrated in Additional file 4. Figure 6 summarizes the top 20 represented biological pathways including at least 200 contigs. The highly represented pathways contained starch and sucrose metabolism, purine metabolism, methane metabolism, T-cell receptor signaling pathway, glycolysis, amino sugar and nucleotide sugar metabolism, oxidative phosphorylation, phenylalanine metabolism and phenylpropanoid biosynthesis.

Kyoto Encyclopedia of Genes and Genomes classification of assembled contigs.
Taken together, the generated database of sweetpotato root transcripts (based upon a mixture of FRs and ISRs) contains genes involved in: (a) classical root functions, such as binding and transport (including transmembrane transport and vesicle-mediated transport) as well as responses to the environment (including response to stimulus), in addition to metabolic processes with high representation of oxidation-reduction processes; (b) metabolic processes and regulation of metabolic processes as well as functions related to development. These results demonstrate the value of the generated transcriptomic data to serve as a database and a reference for continued study aimed at detecting early transcriptome changes in sweetpotato adventitious roots upon initiation of SR formation, as detailed below.
Early transcriptome changes in sweetpotato adventitious roots upon initiation of SR formation
High-throughput sequencing at the cDNA level, using Illumina GA-IIx technology, was carried out to compare the transcription profiles of two bar-coded samples: (1) a pooled sample of FRs and (2) a pooled sample, of the same age, of ISRs; 17,703,982 and 14,780,229 reads (1 × 100-bp long) were obtained for the FRs and ISRs samples, respectively. The reads were mapped against the set of 55,296 FLX contigs described above, as detailed in ‘Methods’. To enable a direct comparison between the two samples, the read count per EST contig was normalized as described in ‘Methods’. The results of total normalized read counts in the FR and ISR samples are presented in Additional file 5 and are available via our sweetpotato database http://batata.agri.gov.il/. The reads were found to map to 31,284 contigs out of the 55,296 contigs that served as the database, i.e. 56.6% (Additional file 5).
A contig was considered differentially expressed between the ISR and FR samples if two conditions were met: (1) at least 10 reads were mapped to this contig in at least one of the samples and (2) no reads were mapped to the contig in the other sample, or the fold change between read number in each sample was at least 2.5. Thus, a total of 8,353 contigs were found to exhibit differential expression between the two samples. Of these, 4,075 (48.8%) contigs were up-regulated in ISRs compared to FRs and 4,278 (51.2%) contigs were up-regulated in FRs compared to ISRs (Additional file 6); 803 contigs were found to exhibit at least 10-fold higher read number in ISRs compared to FRs and 1,457 contigs were found to exhibit at least 10-fold higher read number in FRs compared to ISR (including contigs with at least 10 mapped reads in one of the samples and no reads in the other sample; Additional file 6 and http://batata.agri.gov.il/).
Genes involved in sporamin accumulation and carbohydrate metabolism are highly up-regulated during initiation of SR formation
The list of 70 contigs exhibiting highest differential expression (fold-change) in ISRs compared to FRs is summarized in Table 1. In this list, expression values in the ISR sample, expressed as normalized read number, range between 49 and 4,080 reads per contig and the fold-change in expression compared to the FR sample ranges between 46- and 1,467-fold (Table 1). It should be noted that, for contigs that had no reads in the FR sample, read number was changed from zero to one to enable ‘fold-change’ calculation (Table 1). Highest fold-change expression levels were obtained for contigs representing various members of the sporamin and β-amylase genes, exhibiting 174- to 1,467-fold change in expression. The products of these two genes are known to accumulate to high levels in sweetpotato SRs, with sporamin and β-amylase accounting for about 60% and 5% of total soluble proteins in sweetpotato SRs, respectively [28, 29]. Up-regulation of a contig representing an expansin gene in the ISR sample was apparent (contig S_PBL_c3870) exhibiting 98 fold-change in expression. Expansins are known as cell wall-loosening proteins affecting cell expansion in plants and were shown to be involved in root development [[30] and references therein]. In the present work, 15 contigs representing expansin gene sequences were detected by Illumina cDNA sequencing (Additional file 5). Of these, 13 exhibited higher expression in ISRs compared with FRs, including 4 contigs (S_PBL_c1016, S_PBL_c6536, S_PBL_c1824, S_PBL_c6257) that showed high read number (4362, 3066, 1547 and 1446 reads, respectively; Table 1). Our data thus suggest the involvement of expansin in initiation of SR formation and contradict previous findings by Noh et al. [31] showing that down-regulation of the IbEXP1 expansin gene (exhibiting high homology to contigs S_PBL_c6536 and S_PBL_c1824) in Ipomoea batatas cv. Yulmi enhanced SR development of sweetpotato.
In addition, included in this list are genes involved in starch biosynthesis, coding for α and β subunits of ADP glucose pyrophosphorylase (AGPase; exhibiting high expression levels: 4,080 and 1,394 reads for specific β and α members, respectively) and phosphoglucomutase. This list includes a significant number of contigs without annotation (30 contigs; 43%). Looking into the expression levels (number of Illumina-generated reads) of additional starch biosynthesis genes not included in Table 1, high expression of contigs representing several members of granule-bound starch synthase was detected (in the range of 498 to 4,757 reads, exhibiting 15- to 23-fold higher expression in ISRs compared to FRs, for contigs S_PBL_c3042, S_PBL_c9881, S_PBL_c4145; Additional file 5 and http://batata.agri.gov.il/). The high expression level (83-fold higher in ISR compared to FR) of a contig exhibiting homology to ferredoxin suggests its involvement in the redox modulation and activation of AGPase [32].
To follow changes in starch levels in the roots of Georgia Jet during the first 4 weeks after transplanting, spanning the timing of SR initiation [7], we evaluated starch concentrations in the root system at several time points after transplanting (Figure 7). Samples were taken from the entire root system, since up to 3 weeks after transplanting SR initiation could not be distinguished by microscopic analysis of root cross sections. The data indicated a peak of starch accumulation (an over fivefold increase) at the initiation of SR formation, between 3 and 4 weeks after transplanting (Figure 7). Starch levels of non-initiated, 2-month-old FRs were found to be relatively low (0.9 mg g fw-1; Figure 7), suggesting that the increased starch in the pooled root sample was contributed by the ISRs. These data are in agreement with the elevated expression of genes involved in starch biosynthesis detected by our Illumina sequencing (Table 1). To the best of our knowledge, the accumulated results represent novel data with respect to early elevation in expression of starch biosynthesis genes as well as starch accumulation, marking the initial phase of SR development.

Starch levels in developing adventitious roots around the timing of storage root initiation. Root samples of Georgia Jet were pooled from five to seven plants at 1, 2, 3 and 4 weeks after transplanting, spanning the period of storage root (SR) initiation. Data represent an average of three biological replicates ± SE. “Fibrous roots” represent fibrous root samples derived from plants at 8 weeks after transplanting (at least 4 weeks past the period of SR initiation).
Regulators of meristematic tissue identity and maintenance, as well as of cell division, are up-regulated in ISRs
A contig representing a class I knotted1-like protein (S_PBL_c10411) homologous to Ipomoea nil PKn2 (knotted-like gene) mRNA (AB016000.1) and Ibkn4 of Ipomoea batatas (AB016000.1; class-I knotted1-like) showed 60-fold higher expression in ISRs compared to FRs (Table 1). Knotted1-like homeobox (KNOX) transcription factors are regulators involved in the establishment and maintenance of plant meristems, such as the shoot apical meristem [[33] and references therein]. The cambium is a stem-like tissue [34]; the term cambium refers to one or several layers of initials, analogous to the stem cells proposed for other meristems [[35] and references therein]. Divisions of these initials then produce phloem or xylem mother cells, which in turn can undergo several rounds of cell division before differentiating [35]. Schrader et al. [35] identified molecular markers that characterize the cambial zone in poplar, including genes that regulate meristem identity and mark the cambium initials, and genes that control cell division and mark xylem mother cells. Of these genes, KNOX genes such as the poplar PttKNOX1, PttKNOX2 and PttKNOX6 showed high expression in cambial samples [35].
As indicated previously by Wilson and Lowe [5], and as demonstrated by us [7] for both the leading Israeli sweetpotato variety Georgia Jet and an important USA variety, Beauregard, initiation of SR formation is marked by the development of AC cells adjacent to xylem elements, starting 3 to 4 weeks after transplanting. Repeated division of these cambium cells leads to the formation of rows of thin-walled parenchyma cells that will form the storage tissue of the SR [7]. Several contigs homologous to KNOX genes were detected by us in the sweetpotato root transcriptome, with specific members exhibiting higher expression in ISRs compared to FRs (Table 2). It is interesting to note that all identified members of the class I knotted1-like proteins exhibited at least twofold higher expression in ISRs compared to FRs, while genes belonging to the class II knotted1-like family exhibited a versatile expression pattern (Table 2). KNOXI genes have been previously shown to be involved in the development of sweetpotato SRs [17, 36], regulating cytokinin levels in that organ. Tanaka et al. [17] identified three different KNOXI gene fragments—ibkn1, ibkn2 and ibkn3—in sweetpotato SR. Phylogenetic analysis of putative amino acid sequences showed that ibkn1 is homologous to the SHOOT MERISTEMLESS (STM) gene of Arabidopsis thaliana, while ibkn2 and ibkn3 are homologous to the BP gene. Expressions of ibkn1, ibkn2 and ibkn3 were faint or undetectable in fibrous, non-storage roots [17]. Mele eta al. [37] suggested in Arabidopsis that the BP gene regulates the lignin pathway, thus repressing premature cell differentiation. The class I knotted1-like genes found by us to be up-regulated in ISRs included ibkn2 and ibkn3 homologues as well as additional family members (Table 2).
Almost all of the cell-division-regulating genes that were detected in this work showed higher expression in ISRs compared to FRs, including genes encoding cyclin A-like and cyclin D-like proteins and five cyclin-dependent kinases (Table 2 and Additional file 5). These results are in agreement with the observed increase in the number of AC cells in sweetpotato ISR tissue sections (Figure 1) and with reports of accelerated cell division upon SR initiation in other sweetpotato varieties [36]. These results are novel and additional work is needed to characterize the spatial expression of these genes in root sections at different time points during and following SR initiation.
Genes down-regulated during initiation of SR formation
To identify root functions and processes that are down-regulated during the development of FRs into SRs, we looked into genes represented by contigs that exhibit significantly higher fold-change expression in FRs compared to ISRs. The list of 70 contigs exhibiting highest differential expression in FRs compared to ISRs is summarized in Table 3. This list contains a relatively large number of non-annotated contigs, in addition to contigs that represent genes involved in root development and function as well as defense, such as a metallothionein-like protein which has been shown in rice to be involved in root formation [38] and was found to exhibit a high read number (Table 3). Metallothioneins are cysteine-rich metal-binding proteins of low molecular mass that are mainly involved in maintaining metal homeostasis, metal detoxification and stress/defense responses [38]. Additional genes known to be involved in stress response (including to drought and salt stresses) and found to exhibit down-regulated expression in ISRs compared to FRs included an osmotin-like protein (a member of the class 5 pathogenesis-related proteins) and pathogenesis-related protein PR10a (Table 3; [39]), as well as a cysteine protease shown to be involved in senescence and the osmotic stress response [40].
In addition, contig S_PBL_c6142, which showed an over 100-fold higher read number in FRs compared with SRs, was found to exhibit significant homology to peroxidase 10-like mRNA (EC 1.11.1.7) of Vitis vinifera (3E-81) and Ricinus communis (2E-79), involved in the phenylpropanoid and lignin biosynthesis pathways. Angiosperm lignins are complex phenolic polymers that consist mostly of guaiacyl and syringyl units, together with small or trace amounts of p hydroxyphenyl units. Monolignols are synthesized in the cytosol and transported to the cell wall, where their oxidation generates lignins [41]. From a functional point of view, lignins impart strength to cell walls, facilitate water transport, and impede the degradation of wall polysaccharides, thus acting as a major line of defense against pathogens, insects, and other herbivores [42]. In sweetpotato, Togari [4] proposed a direct link between lignification and SR initiation, suggesting that lignification inhibits SR development. The relationship between stele lignification and inability of adventitious roots to develop into SRs has also been observed by Wilson and Lowe [5], Belehu et al. [6] and Villordon et al. [7]. Togari [4] suggested that genetic and environmental factors influence the balance between cambium development and lignification, which in turn determines to a large degree the final SR yield. Indeed, looking into the expression levels of contigs representing additional lignin biosynthesis genes not included in Table 3, such as S_PBL_c158, S_PBL_c20480 and S_PBL_lrc53688, representing coumaroyl-CoA synthase, caffeoyl-CoA O-methyltransferase and cinnamyl alcohol dehydrogenase, respectively, more than sevenfold reduced expression in ISRs compared to FRs was detected (Additional files 5 and 6). This gene-expression pattern parallels the reduced lignification observed in tissue sections of ISRs compared to FRs (Figure 1; [7] and data not shown). In this context of potential cross talk between establishment and development of the cambium meristem on the one hand, and lignification on the other, it is interesting to note that data from studies on a specific peach class I knotted-like gene (knope1) suggest that KNOPE1 prevents cell lignification by repressing lignin genes during peach stem primary growth [43].
Validation of differential expression between ISRs and FRs revealed by the use of Illumina-based sequencing at the cDNA level
The validity of the expression differences detected by Illumina-based sequencing was examined at the RNA level, using on-line quantitative (q) RT-PCR. Results for nine selected contigs are presented (Figure 8), representing five groups of genes that were found to be differentially expressed by the Illumina results: a gene encoding sporamin, a sweetpotato storage protein accounting for about 60% of total soluble proteins, genes encoding enzymes involved in starch biosynthesis (AGPase and granule-bound starch synthase 1—GBBS1), genes regulating meristem identity (homologous to class-I knotted-like homeobox proteins Ibkn2 and Ibkn3), a gene involved in cell division regulation (cyclin-dependent kinase—CDK) and genes encoding enzymes of lignin biosynthesis (4 coumaroyl-CoA synthase 1—4CL; caffeoyl-CoA O-methyltransferase 2—CCoAOMT; cinnamyl alcohol dehydrogenase—CAD). Results are presented as relative expression values of the respective genes in the ISR sample relative to their expression in the FR sample. In all cases tested, the qRT-PCR analyses confirmed the results of the Illumina analyses (Figure 8), namely elevated expression of gene sequences involved in starch biosynthesis and sporamin accumulation, of the gene encoding CDK and of genes belonging to the class-I knotted-like homeobox protein family, and down-regulation of genes encoding enzymes of lignin biosynthesis in ISRs. However, some of the relative expression values differed between the two expression analysis methods. For example, relative expression values detected by the Illumina and qRT-PCR methods for CDK, 4CL and CAD were 5.6, 0.07, 0.08 and 44.8, 0.4 and 0.2, respectively. It should be noted that the Illumina-based expression results were derived from one biological replicate of the ISR sample and one biological replicate of the FR sample (each sample consisting of cDNA representing pooled root tissue of 30 plants), while the qRT-PCR results were derived from at least four biological replicates (each sample consisting of cDNA representing pooled root tissue of 30 plants). Overall, the qRT-PCR results validated the Illumina-based cDNA sequencing results, allowing for the interpretation of global gene expression trends. Independent validations are still required to accurately measure the expression of each gene of interest.

Validation by quantitative on-line RT-PCR analyses of the differential expression between sweetpotato initiating storage and fibrous roots revealed by the use of Illumina-based sequencing at the cDNA level. Expression levels were measured in initiating storage root (ISR) and fibrous root (FR) samples of Georgia Jet sweetpotato variety, using at least four biological replicates. Each replicate consisted of cDNA representing pooled root tissue from 30 plants. Quantitative RT-PCR was performed and values were normalized relative to the expression levels of 18S rRNA in the same cDNA sample. Expression data are the means (± SE) of at least four replicates and are presented as relative expression values of the respective gene in the ISR sample relative to its expression in the FR sample (ISR/FR). The Y axis has a logarithmic scale. ADP glucose pyrophosphorylase – AGPase; coumaroyl-CoA synthase – 4CL; caffeoyl-CoA O-methyltransferase – CCoAOMT; cinnamyl alcohol dehydrogenase – CAD.
Analysis of over-represented GO terms in the subset of differentially expressed contigs, in either ISRs or FRs, relative to the root transcriptome database
The differentially expressed contigs in ISRs and FRs were analyzed for GO-category enrichment relative to the root transcriptome database using AgriGO [44]. The results for the up-regulated contigs in ISRs are presented in Figure 9 and Additional files 7 and 8. The up-regulated contigs in ISRs were found to contain 21 significantly enriched (FDR ≤ 0.05) GO functional terms in the biology process category, including three ‘level 3’ and four ‘level 4’ terms (“carbohydrate metabolic process”, GO:0005975; “alcohol metabolic process”, GO:0006066; “lipid localization”, GO:0010876 as well as “photosynthesis”, GO:0015979; “lipid transport”, GO:0006869; “cellular carbohydrate metabolic process”, GO:0044262; “nodulation”, GO:0009877, respectively). The highly enriched (FDR = 0.0003) “carbohydrate metabolic process” term included several contigs of starch phosphorylase, phosphoglucomutase, starch branching enzyme and 1,3-β-glucosidase, and its daughter terms included “monosaccharide metabolic process” (GO:0005996) and “starch biosynthetic process” (GO:0019252; including several contigs of ADP glucose pyrophosphorylase and starch synthase). The results thus point to enrichment in ISRs of carbohydrate metabolism and starch biosynthesis functions, in accordance with the biochemical analysis results indicating starch accumulation at the time of SR initiation (Figure 7). An additional and interesting enriched term was “protein folding” (GO:0006457), a ‘level 7’ term that included contigs of cyclophilin, peptidyl-prolyl cis/trans isomerase, calnexin CONSTANS interacting protein 3, heat-shock protein and chaperones, and was connected to the term “protein thiol-disulfide exchange” (GO:0006467).

GO-term enrichment in the initiating storage root sample (‘sample’) relative to the root transcriptome database (‘reference’). Initiating storage root sample histograms are indicated in black, while the ‘reference’ histograms are indicated in yellow.
As for molecular function, the up-regulated contigs belonged to 13 significantly enriched GO terms, including the ‘level 1’ term “nutrient reservoir activity” (GO:0045735), containing several contigs of sporamin A and sporamin B precursors, the ‘level 2’ terms “lipid binding” (GO:0008289), “enzyme inhibitor activity” (GO:0004857), and the ‘level 3’ terms “transferase activity, transferring sulfur-containing groups” (GO:0016782) and “peptidase inhibitor activity” (GO:0030414). The most significantly enriched term was the ‘level 6’ term “phosphofructokinase activity” (GO:0008443). “Glucose-1-phosphate adenylyltransferase (GO:0008878), containing several contigs of ADP-glucose pyrophosphorylase subunits α and β, was also significantly enriched (FDR = 0.028). The GO terms of “plastid” (GO:0009536) and “chloroplast” (GO:0009507) and their daughter terms were the most highly enriched in the cellular component category (with FDR values of 4.03E-26 and 4.02E-25, respectively), in accordance with involvement of amyloplasts in SR starch accumulation. The “apoplast” term (GO:0048046) was also found to be significantly enriched in the ISR sample (Figure 9).
The up-regulated contigs in FRs were found to represent a larger number of different enriched GO functional categories (FDR ≤ 0.05) compared to ISRs (Figure 10 and Additional files 9 and 10). Among the enriched terms represented by the up-regulated contigs in FRs, a total of 74 terms were included in the BP category, containing the ‘level 2’ terms “oxidation reduction” (GO:0055114), “small molecule metabolic process” (GO:0044281), “response to stress” (GO:0006950), “response to biotic stimulus” (GO:0009607), “response to chemical stimulus” (GO:0042221) and “secondary metabolic process” (GO:0019748), and the ‘level 3’ terms “response to endogenous stimulus” (GO: GO:0009719) and “transport” (GO:0006810), which in turn included “transmembrane transport” (GO:0055085). The enriched “secondary metabolic process” term exhibited high enrichment in “phenylpropanoid metabolic process” (GO:0009698; containing several contigs of coumaroyl CoA synthase and phenylalanine ammonia lyase), “phenylpropanoid biosynthetic process” (GO:0009699), “coumarin metabolic process” (GO:0009804), “coumarin biosynthetic process” (GO:0009805; containing contigs of p-coumaroyl quinate/shikimate 3′-hydroxylase, caffeoyl coenzyme A 3-o-methtyl transferase), “lignin metabolic process” (GO:0009808), “phenylpropanoid catabolic process” (GO:0046271 see Figure 10) and “lignin catabolic process” (GO:0046274). An interesting group of enriched functional terms (FDR ≤ 0.032) included: “meristem determinacy” (GO:0010022), “meristem maintenance” (GO:0010073) and “inflorescence meristem growth” (GO:0010450), which contained several contigs representing at least three ultrapetala 1-like proteins. Ultrapetala 1-like protein is a putative transcription factor that acts as a key negative regulator of cell accumulation in Arabidopsis shoot and floral meristems [45]. The higher-plant shoot apical meristem is a dynamic structure that continuously produces cells which become incorporated into new leaves, stems and flowers. The maintenance of a constant flow of cells through the meristem depends on coordination of two antagonistic processes: self-renewal of the stem cell population and initiation of the lateral organs. This coordination is stringently controlled by gene networks that contain both positive and negative components. Carles et al. [45, 46] defined the ULTRAPETALA1 (ULT1) gene as a key negative regulator of cell accumulation in Arabidopsis shoot and floral meristems, because mutations in ULT1 caused enlargement of inflorescence and floral meristems, the production of supernumerary flowers and floral organs, and a delay in floral meristem termination.

GO-term enrichment in the fibrous root sample (‘sample’) relative to the root transcriptome database (‘reference’). Fibrous root sample histograms are indicated in black, while the ‘reference’ histograms are indicated in yellow. A. Biological process (BP). B. GO terms included in the ‘Secondary metabolic process’ BP category. C. Molecular function (MF) and cellular component (CC) categories.
In the MF category, the up-regulated contigs belonged to 78 significantly enriched GO terms, including electron carrier, transporter, antioxidant and catalytic activities, and binding. Catalytic activity included “oxidoreductase activity” (GO:0016491), which in turn included “oxidoreductase activity, acting on diphenols and related substances as donors, oxygen as acceptor” (GO:0016682), “oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, NADH or NADPH as one donor, and incorporation of one atom of oxygen” (GO:0016709) and “trans-cinnamate 4 monooxygenase activity” (GO:0016710), as well as “oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, 2-oxoglutarate as one donor, and incorporation of one atom each of oxygen into both donors” (GO:0016706), “naringenin 3-dioxygenase activity” (GO:0045486) and “leucocyanidin oxygenase activity” (GO:0050589). Catalytic activity also included transferase activity which, in turn, included “glucosyltransferase activity” (GO:0046527), “anthocyanidin 3-O-glucosyltransferase activity” (GO:0047213) and “transferase activity, transferring acyl groups other than amino-acyl groups” (GO:0016747) and “hydroxycinnamoyltransferase activity” (GO:0050734). Binding included “heme binding” (GO:0020037), “cofactor binding” (GO:0048037), including “FAD binding” (GO:0050660) as well as “transcription factor activity” (GO:0003700).
It is interesting to note that the enriched term “transcription factor activity” included several ethylene-responsive factors, such as a homologue of tomato ethylene responsive transcriptional coactivator (MBF1), AP2/ERF domain-containing contigs, homologues of dehydration-responsive element binding protein, and ethylene-responsive transcription factor 2 (ERF2) and ERF5, indicating involvement of ethylene in sweetpotato root development. The growth of secondary xylem and phloem depends on the division of cells in the vascular cambium and results in an increase in the diameter of the root. Results of genome-wide expression profiling of xylem and phloem cambium isolated from the root hypocotyl of Arabidopsis suggested a role for several members of the AP2 as well as MYB transcription factor families, in addition to other transcription factors, as regulators of xylem or phloem cell differentiation and activity [47]. This “transcription factor activity” GO term also contained R2R3 MYB class transcription factor homologues, several GRAS2 and GRAS10 homologues, two BEL1 homeotic protein homologues and a CONSTANS-like protein homologue. Members of the GRAS family, such as the short-root protein, are key regulators of root radial patterning [[20] and references therein], meristem maintenance and asymmetric cell division [[20] and references therein], and have been found to be differentially expressed in SRs of the sweetpotato cv. Xushu[20].
The GO terms “large ribosomal subunit” (GO:0015934), “extracellular region” (GO:0005576) and “intrinsic to membrane” (GO:0031224) were found to be significantly enriched in the CC category (Figure 10C).
Analysis of KEGG pathway enrichment in differentially expressed contigs of either ISRs or FRs, relative to the root transcriptome database
The differentially expressed contigs in ISRs and FRs were analyzed for KEGG pathway enrichment, relative to the rest of the root transcriptome database, using Fisher’s Exact Test and FDR correction [48], and the results are presented in Table 4. Four and fifteen pathways were found to be significantly enriched (FDR ≤ 0.05) in ISR and FR, respectively.
Looking into KEGG-enriched pathways in ISRs, enrichment of sulfur metabolism was apparent, with 10 enzymes exhibiting up-regulated expression in ISRs compared to FRs (Additional file 11), including contigs representing sulfite reductase (ferredoxin). Interestingly, sulfite reductase has been recently suggested as a candidate gene with potential function in cassava SR formation [49]. Reduced levels of sulfite reductase in the Arabidopsis mutant sir1 with decreased activity of the enzyme led to decreased hexose and starch contents [50]. It is thus tempting to suggest that sulfite reductase may also have a role in the control of SR starch accumulation during SR initiation. The glycolysis/gluconeogenesis pathway was also found to be significantly enriched in ISRs (FDR = 0.051; Table 4). In cassava, regulatory changes in the glycolysis/gluconeogenesis pathway were demonstrated following SR development [51]. Down-regulation of enolase, L-lactate dehydrogenase and aldehyde dehydrogenase was suggested in cassava to slow down the entry of carbon into the citrate cycle, pyruvate metabolism and propanoate metabolism, leading to less glucose-6P converted to glycerate-3P and glucose-1P, and in most of the glucose-6P being transported into the amyloplast for starch and sucrose synthesis [51]. In sweetpotato, over twofold down-regulation of several contigs of pyruvate decarboxylase and lactate dehydrogenase was detected following initiation of SR formation (Additional file 5), as well as more than twofold up-regulation of contigs of phosphoglucomutase (catalyzing the reversible interconversion between glucose-6P and glucose-1P, the latter serving as a substrate for ADP-Glc pyrophosphorylase, the first committed step in the starch biosynthesis pathway), indicating the possibility of similar regulation.
Among the significantly enriched pathways in FRs, high enrichment was observed for the phenylpropanoid biosynthesis pathway (Table 4). The first three biosynthetic reactions in this pathway, referred to as the general phenylpropanoid pathway, produce p-coumaroyl CoA, which is a major branch-point metabolite between the production of the flavonoids and the pathway that produces monolignols, lignans and hydroxy-cinnamate conjugates [[52] and references therein]. The first of these reactions is the deamination of phenylalanine by phenylalanine ammonia-lyase (PAL) to generate trans-cinnamic acid. Cinnamic acid is then para-hydroxylated by cinnamate 4-hydroxylase (C4H) to produce p-coumaric acid [[52] and references therein], which is then activated to its corresponding CoA thioester by 4-coumarate CoA ligase (4 coumaroyl-CoA synthase; 4CL). All phenylalanine-derived units destined to be incorporated into the lignin polymer must be hydroxylated by C4H, because the p-hydroxy group is required for the activation of monolignols to their corresponding free radicals, and for polymerization into lignin. The phenylpropanoid biosynthesis pathway map is presented in Figure 11, with the enzymes exhibiting up-regulated expression in ISRs and FRs marked in green and red, respectively. Over twofold up-regulation in FRs of contigs representing C4H and 4CL, as well as of contigs of coniferyl-alcohol glucosyltransferase was apparent (Figure 11 and Table 5). In addition, high expression of PAL was detected in the FR sample, whereas an over fourfold reduction in read number was observed in the ISR sample (Additional file 5). The presence of several contigs representing these enzymes may indicate the presence of isoenzymes. 4CL catalyzes the formation of CoA esters of caffeic acid, ferulic acid, 5-hydroxyferulic acid, and sinapic acid, in addition to p-coumaric acid [[53] and references therein]. The plethora of additional potential substrates may explain why there are many 4CL isoenzymes in most plants. In addition to the different substrate specificities, the genes may have a distinct spatiotemporal expression pattern [[53] and references therein]. Looking into the read number of contigs representing genes of the lignin pathway, such as cinnamyl alcohol dehydrogenase (CAD), more than fivefold lower expression was detected in the ISR vs. FR sample (Table 5 and Additional file 5). Taken together, the results indicate down-regulation in the expression of key genes of the phenylpropanoid biosynthesis pathway upon the change in root fate from FR to a storage organ, which may be responsible for the significant reduction in lignin levels (Figure 1), representing novel data not previously described in sweetpotato. Indeed, it has been demonstrated in Arabidopsis and tobacco that down-regulating 4CL results in reduced lignin content [54–56]. Hu et al. [57] showed that down-regulating the expression of 4CL in transgenic aspen (Populus tremuloides Michx.) by antisense inhibition causes up to 45% reduction in lignin. Reductions in lignin content in Arabidopsis plants carrying a mutation in the second enzyme of this pathway, C4H, were shown to accumulate decreased levels of several different classes of phenylpropanoid end products and to exhibit reduced lignin deposition, altered lignin monomer content and a collapsed xylem phenotype [52].

Changes in the phenylpropanoid biosynthesis pathway map between fibrous roots (FRs) and initiating storage roots (ISRs). Enzymes exhibiting up-regulated expression in ISRs and FRs are marked in green and in red, respectively. Marked in light green are enzymes representing gene sequences that exhibit up-regulated expression in both ISRs and FRs (in most cases, a larger number of contigs exhibited higher expression in FRs compared to ISRs (Table 5)). Marked in white are enzymes representing gene sequences that were not detected in the Illumina-generated transcription profiles (exhibited less than 10 reads) or general enzyme categories representing an enzyme class (such as 4.1.1.-, 2.1.1- and 5.2.1-, representing lyases, methyltransferases and isomerases, respectively). Enzyme annotation was obtained from the sequence annotation and GO classification data.
Cross-talk between primary and secondary metabolism is well documented [58]. To activate the lignin biosynthesis pathway, carbon flow should be delivered from carbohydrate metabolism into the phenylpropanoid pathway by producing sufficient phenylalanine via the shikimate pathway. 3-Deoxy-D-arabino-heptulosonate-7-phosphate synthase (EC 2.5.1.54) catalyzes the first step of the shikimate pathway, using phosphoenolpyruvate and erythrose 4-phosphate derived from glycolysis and the pentose phosphate pathway, respectively. It was thus interesting to note that more than fourfold down-regulation in read number of six contigs (S_PBL_c17757, S_PBL_lrc54453, S_PBL_lrc54372, S_PBL_lrc55028, S_PBL_c7135, S_PBL_c8050) representing this gene, was detected in ISR compared to FR (Additional file 5), suggesting reduction in carbon flow toward phenylpropanoid biosynthesis upon SR initiation.
Conclusions
In the present study, the generation of a database of Georgia Jet sweetpotato root transcriptome, together with a comparison of the expression profiles of ISRs and FRs, enabled the identification of genes involved in the earliest stage of SR formation. Down-regulation in the expression of key genes of the phenylpropanoid biosynthesis pathway upon the change in root fate from FR to a storage organ is indicated, which may be responsible for the significant reduction in lignin levels, representing novel data not previously described in sweetpotato.
The results highlight a reduction in carbon flow toward phenylpropanoid biosynthesis and its delivery into carbohydrate metabolism and starch biosynthesis as major events involved in SR initiation. Specific genes in these pathways were pointed out, providing potential targets for sweetpotato genetic engineering. The results also emphasize the potential importance of delicate control at the level of gene expression of regulators of meristematic tissue identity and maintenance, up-regulation of cell-division regulators and down-regulation of specific GRAS family members, in the SR-initiation process, providing novel data with respect to the specific genes involved. In addition, this study adds a valuable resource of sweetpotato root transcript sequences to the available data, facilitating the identification of genes of interest in this food crop which is among the top seven most important food crops in the world.
Methods
Plant material and RNA extraction
Georgia Jet is the most important sweetpotato variety grown in Israel. Fifty Georgia Jet plants were produced from transplants and grown in 30-cm pots filled with unfertilized washed sand in a greenhouse at The Volcani Center, Israel, maintained between 22 and 28°C with no supplemental light. Roots from 20 plants were sampled 3 to 4 weeks after transplanting as detailed in Villordon et al. [7] to identify the timing of SR initiation (marked by initiation of AC cell development). In short, adventitious roots were sectioned at the proximal 3-cm section of the root and the transverse sections were stained with toluidine blue and observed under the microscope. SR initiation was recorded at 4 weeks after transplanting. For each of the remaining 30 plants at this time point, all adventitious roots were sectioned for microscopic analysis and the adjacent root tissue was immediately frozen by plunging into liquid nitrogen. Following microscopic analysis, roots were divided into either ISRs or non-initiated FRs, as shown in Figure 1, and pooled into ISR and FR samples. Root tissue was ground to a fine powder using liquid nitrogen and sea sand (Merck, Darmstadt, Germany), and total RNA was extracted using the Tri reagent (Sigma-Aldrich, Rehovot, Israel). RNA was treated with TURBO DNase (AB Applied Biosystems, Ambion, CA, USA) according to the manufacturer’s instructions. The two total RNA samples were examined by capillary electrophoresis using a Shimadzu MultiNA microchip electrophoresis system, and used for the preparation of two types of cDNA libraries as detailed below.
Preparation of a normalized random-primed cDNA library for 454 sequencing
For cDNA synthesis, the two RNA samples were pooled in equal amounts to form a pool designated ISR_FR. A normalized cDNA library was constructed (Eurofins MWG operon, Ebersberg, Germany: http://www.eurofinsgenomics.eu/). In brief, from the DNase-treated RNA pool, poly(A) + RNA was isolated and used for cDNA synthesis. First-strand cDNA synthesis was primed with a N6 randomized primer. Then 454 adapters A and B were ligated to the 5′ and 3′ ends of the cDNA. The cDNA was amplified with 11 PCR cycles using a proofreading enzyme (Additional file 12A, N0). Normalization was carried out by one cycle of denaturation and reassociation of the cDNAs, resulting in N1-cDNAs. Reassociated ds-cDNA was separated from the remaining ss-cDNA (normalized cDNA) by passing the mixture over a hydroxylapatite column. The ss-cDNAs were then amplified with nine PCR cycles (Additional file 12A, N1). For titanium sequencing, the 600- to 800-bp cDNAs were eluted from a preparative agarose gel. Aliquots of the size-fractionated cDNAs were analyzed by capillary electrophoresis (Additional file 12B). The 600- to 800-bp ds-cDNA exhibited the structure described in Additional file 13.
Sequencing by Roche GS FLX technology, using titanium series chemistry
Following elution from the preparative gel, this size-selected cDNA was sequenced using a Genome Sequencer™ (GS) FLX Titanium Instrument (Roche Diagnostics) following a standard protocol [59]. The 454 Life Sciences (Roche Diagnostic) software was used for image and signal processing. A file containing the trace, “base-calling” and quality score data was generated and stored in standard flowgram format (SFF) for subsequent bioinformatic analyses. The sequence data was deposited into the European Nucleotide Archive [EMBL-EBI: PRJEB4145; sample accession ERS255740]. An automated, in-silico-assembly pipeline (Eurofins MWG Operon) was used to assemble the sequence data de novo. FASTA and associated files of 524,607 high-quality, base-called and clipped reads were extracted from the SFF-file and contigs assembled de novo using MIRA (version 2.9.45 × 1; http://chevreux.org/projects_mira.html; [60]). Mean lengths ± SD in bases were calculated for particular nucleotide sequence data subsets. MIRA assembly clustered the reads into 55,296 contigs.
Preparation of non-normalized 3’-fragment cDNA libraries from each of the two root samples and Illumina GA-II sequencing
The extracted total RNA from each of the two pooled root samples—ISRs and non-initiated FRs—was used for cDNA synthesis. The cDNA libraries were prepared by Eurofins MWG Operon. In brief, total RNAs were first sheared with ultrasound (10 pulses of 30 s at 4°C), then poly(A) + RNA was isolated followed by treatment of the RNA fragments with polynucleotide kinase. An RNA adapter was then ligated to the 5′-phosphate of the 3’-terminal RNA fragments. First-strand cDNA synthesis was performed using an oligo(dT)-adapter primer and MMLVH- reverse transcriptase. The resulting cDNAs were PCR-amplified (number of cycles indicated in Additional file 14A) using high-fidelity DNA polymerase. Barcode sequences, which were attached to the 5′-ends of the cDNAs, are described in Additional file 14A. The cDNAs in the size range of 200–450 bp were eluted from preparative agarose gels. Aliquots of the size-fractionated cDNAs were analyzed by capillary electrophoresis (Additional file 14B). The resulting, ds-cDNAs were of about 200–450 bp as demonstrated in Additional file 14C and contained adapter sequences (Additional file 14D). Illumina sequencing using the GA-II platform was performed by Eurofins MWG operon according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). A quality score of Q30 was used [Q30 being associated with an error rate (namely the probability that a base is called erroneously) of 0.1%]. The specifications concerning Q30 were as follows: ≥70% of the reads having Q30 for reads longer than 75 bases and ≥75% of the reads having Q30 for reads up to 50 bases.
Illumina read mapping against the 454-generated sweetpotato root transcript sequences
The raw Illumina reads were sorted using their tags (ISR: GAGT and FR: CTTG; Additional file 14A), resulting in the following number of reads per sample (HiSeq 2000, 1 × 100 bp): 14,780,229 and 17,703,982 for ISR and FR, respectively. The sequence data was deposited into the European Nucleotide Archive [EMBL-EBI: PRJEB4145; sample accession numbers ERS255744 and ERS255745 for ISRs and FRs, respectively]. Prior to the read mapping, the four tag bases were removed from its 5′ end. These read sequences were then used as input for read mapping against the set of 55,296 FLX EST contigs. The mapping was conducted using the software BWA 0.5.8c (http://bio-bwa.sourceforge.net). Post-processing of the mapping was conducted with samtools 0.1.12a (http://samtools.sourceforge.net). The total number of mapped reads for each of the two samples (ISR and FR) was quantified. To enable a direct comparison between the two samples, the read count per EST contig was normalized using DESeq [61].
Validation of the Illumina-generated transcription profiles of FR and ISR samples using real-time qRT-PCR analyses
RNA samples were prepared from either FRs or ISRs as described above, using at least four biological replicates. Each replicate contained root tissue derived from 30 plants. RNA samples were cleaned of DNA contamination using RQ1 RNase-free DNase (Promega WI, USA). cDNA was synthesized using 1 μg of total RNA and Superscript II (Invitrogen) reverse transcriptase, according to the manufacturer’s instructions. First-strand cDNA was used for qRT-PCR analyses. Primer3web version 4.0: http://primer3.ut.ee) was used for primer design, and qRT-PCR was performed on the ROTOR-GENE™ 6000 (Corbett LIFE SCIENCE, Qiagen), using SYBR® Green. A total reaction volume of 15 μl was used. The reaction mix included 3 μl template, 0.3 μl reverse primer, 0.3 μl forward primer, 7.5 μl Absolute Blue QPCR SYBR Green ROX Mix (Thermo Fisher Scientific Inc. MA, USA), and 3.9 μl RNA-free water. A qRT-PCR assay was performed using the following conditions: 95°C for 9 min followed by 40 cycles of 95°C for 10 s, 60°C for 10 s and 72°C for 20 s. The 2–ΔΔCT method [62] was used to normalize and calibrate transcript values relative to the 18S ribosomal protein, whose expression did not change across sweetpotato root types or developmental stages. The FR sample was used as the calibrator sample. Primer sequences were designed according to the respective contigs assembled from reads that were obtained from the 454-sequencing results and are described in Additional file 15. Use of the oligonucleotide primers listed in Additional file 15 resulted in approximately equal efficiencies of amplification for the various target and reference genes. Each set of experiments was repeated at least four times and final relative quantification results are given as average ± SE.
Functional annotation and analyses of GO-term and KEGG-pathway enrichment
The assembled transcripts were used to query public genomic databases (i.e. NCBI Nr) using BLASTX (Eurofins MWG Operon) and annotations of the two best hits for each contig were recorded. BlastoGO [22] was used to obtain GO annotations and Ontologizer [23] was used to perform GO functional classification. The contigs were further classified using GOSlim (http://www.geneontology.org/GO.slims.shtml). To assign the detected contigs to biological pathways, sequences were compared using BLASTX with an E-value cutoff of <10E-3 against the KEGG database (http://www.genome.jp/kegg/). The differentially expressed contigs in ISRs and FRs were analyzed for GO-category enrichment relative to the root transcriptome database using AgriGO [44]. The differentially expressed contigs in ISRs and FRs were analyzed for KEGG-pathway enrichment relative to the rest of the root transcriptome database using Fisher’s Exact Test and FDR correction [48].
Starch content analysis
Root samples of Georgia Jet were pooled from five to seven plants at 1, 2, 3 and 4 weeks after transplanting, spanning the period of SR initiation [7]. The tissue was ground in liquid nitrogen using a mortar and pestle and ethanol-suspended root samples were extracted three times in hot 80% (v/v) ethanol. The insoluble residue that remained after ethanolic extraction was resuspended in 30 mM HCl and boiled for 30 min. After cooling, the pH was adjusted to 4.5 with KOH. The gelatinized starch was digested for 60 min at 50°C with approximately 36 U of amyloglucosidase from Aspergillus oryza[63]. Reducing sugars were determined according to Miller [64]. Three biological replicates were used for each analysis.
Abbreviations
- AC:
-
Anomalous cambium
- DAT:
-
Days after transplanting
- FR:
-
Fibrous root
- ISR:
-
Initiating storage root
- LC:
-
Lignified cell
- NGS:
-
Next-generation sequencing
- RVC:
-
Regular vascular cambium
- SR:
-
Storage root.
References
FAOSTAT Data. [http://faostat3.fao.org/home/index.html]
Woolfe J: Sweetpotato, an Untapped Food Source. 1992, Cambridge: Cambridge University Press
The Nutrition Action Health Letter from the Center for Science in the Public Interest. USA, [http://www.cspinet.org/nah/]
Togari Y: A study of tuberous root formation in sweet potato. Bull Natl Agric Exp Statn Tokyo. 1950, 68: 1-96.
Wilson LA, Lowe SB: The anatomy of the root system in West Indian sweet potato [Ipomoea batatas (L.) Lam.] cultivars. Ann Bot (Lond). 1973, 37: 633-643.
Belehu T, Hammes PS, Robbertse PJ: The origin and structure of adventitious roots in sweetpotato (Ipomoea batatas). Aust J Bot. 2004, 52: 551-558. 10.1071/BT03152.
Villordon AQ, La Bonte DR, Firon N, Kfir Y, Schwartz A, Pressman E: Characterization of adventitious root development in sweetpotato. Hortscience. 2009, 44: 651-655.
You MK, Hur CG, Ahn YS, Suh MC, Jeong BC, Shin JS, Bae JM: Identification of genes possibly related to storage root induction in sweetpotato. FEBS Lett. 2003, 53: 101-105.
McGregor C: Differential expression and detection of transcripts in sweetpotato (Ipomoea batatas (L.) LAM.) using cDNA microarrays. PhD thesis. 2006, Louisiana State University and Agricultural and Mechanichal College, http://etd.lsu.edu/docs/available/etd-05092006-145816/,
Kim SH, Mizuno K, Fujimura K: Isolation of MADS-box genes from sweet potato (Ipomoea batatas (L.) Lam.) expressed specifically in vegetative tissues. Plant Cell Physiol. 2002, 43: 314-322. 10.1093/pcp/pcf043.
Kim SH, Hamada T, Otani M, Shimada T: Isolation and characterization of MADS box genes possibly related to root development in sweetpotato (Ipomoea batatas L. Lam.). J Plant Biol. 2005, 48: 387-393. 10.1007/BF03030580.
Kim SH, Hamada T, Otani M, Shimada T: Cloning and characterization of sweetpotato MADS-box gene (IbAGL17) isolated from tuberous root. Plant Biotechnol. 2005, 22: 217-220. 10.5511/plantbiotechnology.22.217.
Lalusin AG, Nishita K, Kim S-H, Ohta M, Fujimura T: A new MADS-box gene (IbMADS10) from sweet potato (Ipomoea batatas (L.) Lam) is involved in the accumulation of anthocyanin. Mol Genet Genom. 2006, 275: 44-54. 10.1007/s00438-005-0080-x.
Ku AT, Huang YS, Wang YS, Ma D, Yeh KW: IbMADS1 (Ipomoea batatas MADS-box 1 gene) is involved in tuberous root initiation in sweet potato (Ipomoea batatas). Ann Bot. 2008, 102: 57-67. 10.1093/aob/mcn067.
Noh SA, Lee HS, Huh EJ, Huh GH, Paek KH, Shin JS, Bae JM: SRD1 is involved in the auxin-mediated initial thickening growth of storage root by enhancing proliferation of metaxylem and cambium cells in sweetpotato (Ipomoea batatas). J Exp Bot. 2010, 61: 1337-1349. 10.1093/jxb/erp399.
Tanaka M, Takahata Y, Nakatani M: Analysis of genes developmentally regulated during storage root formation of sweetpotato. J Plant Physiol. 2005, 162: 91-102. 10.1016/j.jplph.2004.06.003.
Tanaka M, Kato N, Nakayama H, Nakatani M, Takahata Y: Expression of class 1 Knotted1-like homeobox genes in the storage roots of sweetpotato (Ipomoea batatas). J Plant Physiol. 2008, 165: 1726-1735. 10.1016/j.jplph.2007.11.009.
Schafleitner R, Tincopa LR, Palomino O, Rossel G, Robles RF, Alagon R, Rivera C, Quispe C, Rojas L, Pacheco JA, Solis J, Cerna D, Kim JY, Hou J, Simon R: A sweetpotato gene index established by de novo assembly of pyrosequencing and Sanger sequences and mining for gene-based microsatellite markers. BMC Genomics. 2010, 11: 604-10.1186/1471-2164-11-604.
Wang ZY, Fang BP, Chen JY, Zhang XJ, Luo ZX, Huang L, Chen X, Li Y: De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics. 2010, 11: 726-10.1186/1471-2164-11-726.
Tao X, Gu Y-H, Wang H-Y, Zheng W, Li X, Zhao C-W, Zhang YZ: Digital gene expression analysis based on integrated de novo transcriptome assembly of sweetpotato [Ipomoea batatas (L.) Lam.]. PLoS One. 2012, 7: e36234-10.1371/journal.pone.0036234.
Xie F, Burklew CE, Yang Y, Liu M, Xiao P, Zhang B, Qiu D: De novo sequencing and comprehensive analysis of purple sweet potato (Ipomoea batatas L.) transcriptome. Planta. 2012, 236: 101-113. 10.1007/s00425-012-1591-4.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676. 10.1093/bioinformatics/bti610.
Bauer S, Grossmann S, Vingron M, Robinson PN: Ontologizer 2.0 – A multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008, 24: 1650-1651. 10.1093/bioinformatics/btn250.
GO Slim and Subset Guide. [http://www.geneontology.org/GO.slims.shtml]
Kyndt T, Denil S, Haegeman A, Trooskens G, De Meyer T, Van Criekinge W, Gheysen G: Transcriptome analysis of rice mature root tissue and root tips in early development by massive parallel sequencing. J Exp Bot. 2012, 63: 2141-2157. 10.1093/jxb/err435.
Yang H, Hu L, Hurek T, Reinhold-Hurek B: Global characterization of the root transcriptome of a wild species of rice, Oryza longistaminata, by deep sequencing. BMC Genomics. 2010, 11: 705-10.1186/1471-2164-11-705.
Swarbreck SM, Lindquist EA, Ackerly DD, Andersen GL: Analysis of leaf and root transcriptomes of soil-grown Avena barbata plants. Plant Cell Physiol. 2011, 52: 317-332. 10.1093/pcp/pcq188.
Maeshima M, Sasaki T, Asahi T: Characterization of major proteins in sweetpotato tuberous roots. Phytochemistry. 1985, 24: 1899-1902. 10.1016/S0031-9422(00)83088-5.
Nakamura K, Ohto M, Yoshida N, Nakamura K: Sucrose induced accumulation of beta-amylase occurs concomitant with the accumulation of starch and sporamin in leaf-petiole cuttings of sweet potato. Plant Physiol. 1991, 96: 902-909. 10.1104/pp.96.3.902.
Lee DK, Ahn JH, Song SK, Choi YD, Lee JS: Expression of an expansin gene is correlated with root elongation in soybean. Plant Physiol. 2003, 131: 985-997. 10.1104/pp.009902.
Noh SA, Lee HS, Kim YS, Paek KH, Shin JS, Bae JM: Down-regulation of the IbEXP1 gene enhanced storage root development in sweetpotato. J Exp Bot. 2012, 64: 129-142.
Tiessen A, Hendriks JHM, Stitt M, Branscheid A, Gibon Y, Farré EM, Geigenberger P: Starch synthesis in potato tubers is regulated by post-translational redox modification of ADP-glucose pyrophosphorylase. A novel regulatory mechanism linking starch synthesis to the sucrose supply. Plant Cell. 2002, 14: 2191-2213. 10.1105/tpc.003640.
Osnato M, Stile MR, Wang Y, Meynard D, Curiale S, Guiderdoni E, Liu Y, Horner DS, Ouwerkerk PBF, Pozzi C, Mueller KJ, Salamini F, Laura Rossini L: Cross talk between the KNOX and ethylene pathways is mediated by intron-binding transcription factors in barley. Plant Physiol. 2010, 154: 1616-1632. 10.1104/pp.110.161984.
Agusti J, Lichtenberger R, Schwarz M, Nehlin L, Greb T: Characterization of transcriptome remodeling during cambium formation identifies MOL1 and RUL1 as opposing regulators of secondary growth. PLoS Genet. 2011, 7 (2): e1001312-10.1371/journal.pgen.1001312.
Schrader J, Nilsson J, Mellerowicz E, Berglund A, Nilsson P, Hertzberg M, Sandberga G: A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell. 2004, 16: 2278-2292. 10.1105/tpc.104.024190.
Ravi V, Naskar SK, Makeshkumar T, Babu B, Krishnan BSP: Molecular physiology of storage root formation and development in sweet potato (Ipomoea batatas (L.) Lam.). J Root Crops. 2009, 35: 1-27.
Mele G, Ori N, Sato Y, Hake S: The knotted1-like homeobox gene BREVIPEDICELLUS regulates cell differentiation by modulating metabolic pathways. Genes Dev. 2013, 17: 2088-2093.
Yuan J, Chen D, Ren Y, Zhang X, Zhao J: Characteristic and expression analysis of a metallothionein gene, OsMT2b, down-regulated by cytokinin suggests functions in root development and seed embryo germination of rice. Plant Physiol. 2008, 146: 1637-1650. 10.1104/pp.107.110304.
Van Loon LC, Van Strien EA: The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol Mol Plant Pathol. 1999, 55: 85-97. 10.1006/pmpp.1999.0213.
Chen HJ, Su CT, Lin CH, Huang GJ, Lin YH: Expression of sweet potato cysteine protease SPCP2 altered developmental characteristics and stress responses in transgenic Arabidopsis plants. J Plant Physiol. 2010, 167: 838-847. 10.1016/j.jplph.2010.01.005.
Vanholme R, Morreel K, Ralph J, Boerjan W: Lignin engineering. Curr Opin Plant Biol. 2008, 11: 278-285. 10.1016/j.pbi.2008.03.005.
Hatfield R, Vermerris W: Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol. 2001, 126: 1351-1357. 10.1104/pp.126.4.1351.
Testone G, Condello E, Verde I, Nicolodi C, Caboni E, Dettori MT, Vendramin E, Bruno L, Bitonti MB, Mele G, Giannino D: The peach (Prunus persica L. Batsch) genome harbours 10 KNOX genes, which are differentially expressed in stem development, and the class 1 KNOPE1 regulates elongation and lignification during primary growth. J Exp Bot. 2012, 63: 5417-5435. 10.1093/jxb/ers194.
Du Z, Zhou X, Ling Y, Zhang Z, Su Z: AgriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38: W64-W70. 10.1093/nar/gkq310.
Carles CC, Choffnes-Inada D, Reville K, Lertpiriyapong K, Fletcher JC: ULTRAPETALA1 encodes a SAND domain putative transcriptional regulator that controls shoot and floral meristem activity in Arabidopsis. Development. 2005, 132: 897-911. 10.1242/dev.01642.
Carles CC, Lertpiriyapong K, Reville K, Fletcher JC: The ULTRAPETALA1 gene functions early in Arabidopsis development to restrict shoot apical meristem activity, and acts through WUSCHEL to regulate floral meristem determinacy. Genetics. 2004, 167: 1893-1903. 10.1534/genetics.104.028787.
Zhao C, Craig JC, Petzold HE, Dickerman AW, Beers EP: The xylem and phloem transcriptomes from secondary tissues of the Arabidopsis root-hypocotyl. Plant Physiol. 2005, 138: 803-818. 10.1104/pp.105.060202.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995, 57: 289-300.
Sojikul P, Kongsawadworakul P, Viboonjun U, Thaiprasit J, Intawong B, Narangajavana J, Svasti MR: AFLP-based transcript profiling for cassava genome-wide expression analysis in the onset of storage root formation. Physiol Plant. 2010, 140: 189-198. 10.1111/j.1399-3054.2010.01389.x.
Khan MS, Haas FH, Samami AA, Gholami AM, Bauer A, Fellenberg K, Reichelt M, Hänsch R, Mendel RR, Meyer AJ, Wirtz M, Hell R: Sulfite reductase defines a newly discovered bottleneck for assimilatory sulfate reduction and is essential for growth and development in Arabidopsis thaliana. Plant Cell. 2010, 22: 1216-1231. 10.1105/tpc.110.074088.
Yang J, An D, Zhang P: Expression profiling of cassava storage roots reveals an active process of glycolysis/gluconeogenesis. J Integr Plant Biol. 2011, 53: 193-211. 10.1111/j.1744-7909.2010.01018.x.
Schilmiller AL, Stout J, Weng JK, Humphreys J, Ruegger MO, Chapple C: Mutations in the cinnamate 4-hydroxylase gene impact metabolism, growth and development in Arabidopsis. Plant J. 2009, 60: 771-782. 10.1111/j.1365-313X.2009.03996.x.
Raes J, Rohde A, Christensen JH, Van de Peer Y, Boerjan W: Genome-wide characterization of the lignification toolbox in Arabidopsis. Plant Physiol. 2003, 133: 1051-1071. 10.1104/pp.103.026484.
Kajita S, Katayama Y, Omori S: Alterations in the biosynthesis of lignin in transgenic plants with chimeric genes for 4-coumarate:coenzyme A ligase. Plant Cell Physiol. 1996, 37: 957-965. 10.1093/oxfordjournals.pcp.a029045.
Kajita S, Hishiyama S, Tomimura Y, Katayama Y, Omori S: Structural characterization of modified lignin in transgenic tobacco plants in which the activity of 4-coumarate:coenzyme A ligase is depressed. Plant Physiol. 1997, 114: 871-879.
Lee D, Meyer K, Chapple C, Douglas CJ: Antisense suppression of 4-coumarate: coenzyme A ligase activity in Arabidopsis leads to altered lignin subunit composition. Plant Cell. 1997, 9: 1985-1998.
Hu WJ, Harding SA, Lung J, Popko JL, Ralph J, Stokke DD, Tsai CJ, Chiang VL: Repression of lignin biosynthesis promotes cellulose accumulation and growth in transgenic trees. Nat Biotechnol. 1999, 17: 808-812. 10.1038/11758.
Aharoni A, Galili G: Metabolic engineering of the plant primary-secondary metabolism interface. Curr Opin Biotechnol. 2011, 22: 239-244. 10.1016/j.copbio.2010.11.004.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380.
Chevreux B, Wetter T, Suhai S: Genome sequence assembly using trace signals and additional sequence information. Comput Sci Biol: Proc. German Conference on Bioinformatics GCB’99 GCB. 1990, 45-56.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-10.1186/gb-2010-11-10-r106.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2-[delta][delta]CT method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Hubbard NL, Pharr DM, Huber SC: Role of sucrose phosphate synthase in sucrose biosynthesis in ripening bananas and its relationship to the respiratory climacteric. Plant Physiol. 1990, 94: 201-208. 10.1104/pp.94.1.201.
Miller GL: Use of dinitrosalicylic acid reagent for determination of reducing sugars. Anal Chem. 1959, 31: 426-428. 10.1021/ac60147a030.
Acknowledgements
Support for this research was provided by a grant from The USA-Israel Binational Agricultural Research and Development grant number US-4015-07.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The study was conceived by NF, DL and AV. NF drafted the manuscript. YK designed the experimental setup, together with NF, DL and AV, and performed the experiments, including tissue sections, RNA extractions and starch content analyses. EL performed the quantitative RT-PCR analyses. JS contributed to the experiment performance and data analysis. TSP performed the bioinformatics analyses, including functional annotation of the database. ADF participated in the bioinformatics analyses, including GO category and KEGG pathway enrichment. AH generated the sweetpotato transcriptome web page. L Althan contributed to the plant material preparation and maintenance. L Adani Nadir participated in RNA extractions. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5183_MOESM1_ESM.xls
Additional file 1: Annotations of the two best hits for each contig (E value ≤ 1.0E-3). Annotations are listed in boxes I2 – I61,668. (XLS 13 MB)
12864_2013_5183_MOESM2_ESM.pdf
Additional file 2: Species distribution of the BLAST results. Presented data were extracted from the detailed data of the number of BLAST results matching each plant species. For each contig, the best BLAST result was used. (PDF 39 KB)
12864_2013_5183_MOESM3_ESM.xls
Additional file 3: Functional annotations of sweetpotato root transcripts (combined initiating storage and fibrous root samples) using GOSlim. The three GO functional categories (BP, MF, CC) were segregated into separate worksheets. Whenever relevant, contigs were assigned to more than one GO term. (XLS 822 KB)
12864_2013_5183_MOESM4_ESM.xls
Additional file 4: Functional classification of sweetpotato root transcripts (combined initiating storage and fibrous root samples) to biological pathways using KEGG.(XLS 38 KB)
12864_2013_5183_MOESM5_ESM.xls
Additional file 5: Total normalized read counts in the fibrous root and initiating storage root samples. Illumina GA-IIx technology was carried out to compare the transcription profiles of a pooled sample of fibrous roots and a pooled sample of initiating storage roots and reads were mapped against our database of 55,296 FLX contigs. Normalization was performed as described in ‘Methods’. One annotation is given for each contig. Annotations of the two best hits for each contig (E value ≤ 1.0E-3) are available in Additional file 1. (XLS 5 MB)
12864_2013_5183_MOESM6_ESM.xls
Additional file 6: Differentially expressed contigs between initiating storage roots (ISRs) and fibrous roots (FRs). A contig was considered as differentially expressed if two conditions were met: (1) at least 10 reads were mapped to this contig in at least one of the samples and (2) no reads were mapped to this contig in the other sample, or the fold-change between its read number in each sample was at least 2.5. The up-regulated contigs in ISR and FR are segregated into separate worksheets. Fold-change expression was calculated relative to the read number in the ISR sample. One annotation is given for each contig. Annotations of the two best hits for each contig (E value ≤ 1.0E-3) are available in Additional file 1. (XLS 1 MB)
12864_2013_5183_MOESM7_ESM.xls
Additional file 7: GO-term enrichment in the initiating storage root sample relative to the root transcriptome database. The specific contigs for each GO term in categories biological process (BP), molecular function (MF) and cellular component (CC) are given on separate sheets. ISR – initiating storage root sample. (XLS 190 KB)
12864_2013_5183_MOESM8_ESM.ppt
Additional file 8: Tree representation of enriched GO terms in the initiating storage root sample. A more intense color indicates a higher number of contigs. ISR – initiating storage root sample. (PPT 780 KB)
12864_2013_5183_MOESM9_ESM.xls
Additional file 9: GO-term enrichment in the fibrous root sample relative to the root transcriptome database.vThe specific contigs for each GO term in categories biological process (BP), molecular function (MF) and cellular component (CC) are given on separate sheets. FR – fibrous root sample. (XLS 860 KB)
12864_2013_5183_MOESM10_ESM.ppt
Additional file 10: Tree representation of enriched GO terms in the fibrous root sample. A more intense color indicates a higher number of contigs. FR – fibrous root sample. (PPT 628 KB)
12864_2013_5183_MOESM11_ESM.xls
Additional file 11: Read number of contigs representing the initiating storage root-enriched sulfur metabolism in the KEGG pathway.(XLS 23 KB)
12864_2013_5183_MOESM12_ESM.pdf
Additional file 12: Analysis of the PCR-amplified cDNAs on a Shimadzu MultiNA microchip electrophoresis system. A. Analysis of the PCR-amplified N0 and N1 cDNAs. B. Analysis of the size-fractionated N1 cDNAs. M – 100 bp ladder. (PDF 34 KB)
12864_2013_5183_MOESM13_ESM.pdf
Additional file 13: Description of the cDNA used for 454 sequencing. The 454 adapter sequences are underlined. The first four bases of adapter primers A1 and B1 represent phosphorothioate-modified bases as specified by Roche. In addition, primer B1 is 5′-biotinylated. The barcode sequence is: CACACG. (PDF 15 KB)
12864_2013_5183_MOESM14_ESM.pdf
Additional file 14: Properties of the non-normalized cDNA samples. A. PCR amplification of the cDNA samples. B. Analysis of the PCR-amplified 3′-fragment cDNAs on the Shimadzu MultiNA microchip electrophoresis system. C. Analysis of the size-fractionated cDNAs on the Shimadzu MultiNA microchip electrophoresis system. D. Description of cDNA for Illumina sequencing. Illumina adapter sequences are underlined. M – 100 bp ladder; ISR – initiating storage root sample; FR – fibrous root sample. (PDF 83 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Firon, N., LaBonte, D., Villordon, A. et al. Transcriptional profiling of sweetpotato (Ipomoea batatas) roots indicates down-regulation of lignin biosynthesis and up-regulation of starch biosynthesis at an early stage of storage root formation. BMC Genomics 14, 460 (2013). https://doi.org/10.1186/1471-2164-14-460
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-460