
Abstract
Background
The gene doublesex (dsx) is known as a key factor regulating genetic sex determination in many organisms. We previously identified two dsx genes (DapmaDsx1 and DapmaDsx2) from a freshwater branchiopod crustacean, Daphnia magna, which are expressed in males but not in females. D. magna produces males by parthenogenesis in response to environmental cues (environmental sex determination) and we showed that DapmaDsx1 expression during embryonic stages is responsible for the male trait development. The D. magna dsx genes are thought to have arisen by a cladoceran-specific duplication; therefore, to investigate evolutionary conservation of sex specific expression of dsx genes and to further assess their functions in the environmental sex determination, we searched for dsx homologs in four closely related cladoceran species.
Results
We identified homologs of both dsx genes from, D. pulex, D. galeata, and Ceriodaphnia dubia, yet only a single dsx gene was found from Moina macrocopa. The deduced amino acid sequences of all 9 dsx homologs contained the DM and oligomerization domains, which are characteristic for all arthropod DSX family members. Molecular phylogenetic analysis suggested that the dsx gene duplication likely occurred prior to the divergence of these cladoceran species, because that of the giant tiger prawn Penaeus monodon is rooted ancestrally to both DSX1 and DSX2 of cladocerans. Therefore, this result also suggested that M. macrocopa lost dsx2 gene secondarily. Furthermore, all dsx genes identified in this study showed male-biased expression levels, yet only half of the putative 5’ upstream regulatory elements are preserved in D. magna and D. pulex.
Conclusions
The all dsx genes of five cladoceran species examined had similar amino acid structure containing highly conserved DM and oligomerization domains, and exhibited sexually dimorphic expression patterns, suggesting that these genes may have similar functions for environmental sex determination in cladocerans.
Similar content being viewed by others


Background
Sex determination is a fundamental developmental process, affecting the sexual differentiation of gonads, and leads to sex-specific differences in behavior, physiology and morphology. Sex-determining systems can be divided into two categories: genotypic sex determination (GSD) and environmental sex determination (ESD) [1–3]. GSD is attributed to the genetic segregation of genes, often residing on sex chromosomes that initiate alternate sex-determining developmental pathways. In contrast, ESD has repeatedly arisen during animal evolution [4] and is initiated by diverse environmental cues, such as temperature, photoperiod, nutrition and population density, that trigger alternative genetic signals, resulting in the regulation of male or female sex-determining genes [5, 6].
Natural selection of rare mutational variants has been suggested to mediate the transitions between GSD and ESD [7, 8]. A previous phylogenetic analysis revealed that there have been at least three independent switches from GSD to ESD in lizards, and six transitions from ESD to GSD in turtles [9]. Moreover, previous experiments using temperature-sensitive mutations created artificially in Caenorhabditis elegans demonstrated how GSD could rapidly evolve into ESD as a consequence of a mutation in key sex determining genes [9]. Orthologs of GSD genes such as dmrt1, sox9 and cyp19a (aromatase) are expressed in the gonads during the temperature-sensitive period in ESD of reptiles [10]. Thus, according to the current interpretation of these data, ESD mechanisms are likely to share many sex-determining components with GSD [5].
Sex determination systems in insects vary considerably in key factors and regulatory mechanisms to develop sex-specific traits. The sex determination mechanism in Drosophila melanogaster is best understood. The ratio of X chromosomes to autosomes (X:A ratio) is thought to provide the initial signal for the activation of sex-lethal (sxl), a master gene of the sex determination cascade. Then, sxl is produced as the sex-specific splicing isoforms. Sxl in female acts on the pre-mRNA of transformer (tra) resulting in reproduction of functional Tra. The functional Tra in the female, in concert with Tra-2, regulates the production of female-specific doublesex (dsx) mRNA. The male-specific splice form of dsx mRNA is the default splice-variant in D. melanogaster. Dsx regulates the various sex-specific traits such as gonads. Recently, sex determination mechanisms have also been demonstrated in various insect lineages such as Diptera (Musca domestica and Ceratitis capitata), Hymenoptera (Apis mellifera and Nasonia vitripennnis) and Coleoptera (Tribolium castaneum). These studies revealed that tra and dsx are highly conserved among insects [11–14]. However, in case of Lepidoptera, Bombyx mori, tra and tra-2 are assumed not to be required for the sex-specific splicing of Bmdsx pre-mRNA, because Bmdsx has no Tra/Tra-2 binding motif. Recently, it has been revealed that binding of the BmPSI, a Bombyx homolog of P-element somatic inhibitor, to the exonic splicing suppressor sequence on expected region is involved in sex-specific splicing of Bmdsx[12]. These data suggest that upstream genetic cascades of dsx might be diverse among insects.
The Cladocera (commonly called water fleas) is an ancient clade of branchiopod crustaceans comprising 16 or 18 family lineages [15, 16] that all reproduce by cyclical parthenogenesis involving ESD [17]. The most well studied species are from the family Daphniidae, particularly of the genus Daphnia. Daphnia inhabit freshwater ponds and lakes on all continents and are known to switch between parthenogenetic and sexual reproduction when environmental conditions for growth and reproduction deteriorate. During normal growing conditions, populations are most often entirely composed of females. However, shortened photoperiod, a lack of food and/or increased population density all lead to the clonal production of males that are genetically identical to their sisters and mothers [18]. First instar male juveniles are easily distinguished from the females [19]. During maturation, daphniids undergo morphological sexual differentiation of various somatic tissues, including the armament of a first thoracic leg with the copulatory hook in males, which becomes larger during the fifth instar [20]. Gonads develop and finally settle at both sides of the gut during embryogenesis in both sexes [21]. The appearance of males allows sexual reproduction to occur [22, 23], when females begin producing haploid eggs requiring fertilization.
Recently, we and others found that male production occurred independently of environmental cues by treatment with exogenous juvenile hormone (JH) or its analogs [24, 25]. Exposure of D. magna to JH analogs at the stage corresponding to the environmentally-sensitive period for sex determination of a cladoceran species of the family Moinidae [26], produced exclusively male broods, suggesting that JH could be a key molecule for understanding mechanisms of ESD [24, 27, 28].
A doublesex (dsx) gene was originally identified in D. melanogaster as a critical and terminal transcription factor in the fly sex determining cascade. Dsx is spatially and temporally transcribed into two sex-specific splice forms conferring sexually dimorphic traits during development [29, 30]. The dsx gene contains two conserved domains: the Dsx/Mab-3 (DM) domain at the N-terminus and the oligomerization domain at the C-terminus [31]. Genes encoding DM-domain (DM-domain genes) were discovered to play a related role in C. elegans[32, 33] and also in vertebrates [34–36]. In contrast, results from numerous studies have shown that other genes in the genetic sex-determination cascade widely diversified among species [1, 2, 37].
To understand the molecular and evolutionary relationships between GSD and ESD, we previously identified and analyzed three DM-domain genes (DMRT11E, DMRT93B and DMRT99B) from D. magna, displaying sexual dimorphic gene expression patterns in adult gonads [38]. However, none of these DM-domain genes exhibited sexually dimorphic expression patterns during embryonic development, suggesting that they are not involved in sex determination [38]. Two additional DM-domain genes were later found in the D. magna expressed sequence tags (ESTs) database [39]. Therefore, we analyzed the function of these two genes from D. magna using gene manipulations that we developed [40]. These experiments revealed that two dsx genes in D. magna were obtained by lineage-specific duplication, and then one of the paralogs, Daphnia magna dsx1 (DapmaDsx1), plays an important role in directing the major sexually dimorphic development of D. magna[41]. In contrast, specific function of Daphnia magna dsx2 (DapmaDsx2) remains unknown. These newly identified dsx genes showed greater sequence similarity at the amino acid sequence level to known insect dsx genes than to the previously identified DM-domain containing genes in D. magna. A genome-wide study of gene functions in D. pulex suggested that lineage-specific duplicated genes are most responsive to varying environmental conditions [42]. In the present study, we investigated the sequence and functional conservation of the two dsx genes in a broader taxonomic sampling of cladocerans by cloning dsx homologs, and determining their sex specific expression in four species representing two families and three genera. We also analyzed the structures of cloned dsx genes of D. magna and D. pulex including their putative regulatory motifs and putative transcription factor binding sites in the 5’ upstream regions of these duplicated dsx genes.
Results and discussion
Molecular cloning of doublesexgenes from cladocerans
To verify whether homologs of dsx genes among daphniids are conserved, we first cloned dsx genes from four cladocerans (D. pulex, D. galeata, C. dubia and M. macrocopa), then characterized them by comparison with dsx genes of D. magna and several insect species [41, 43] (Figure 1). As a result, two dsx homologs were identified from D. pulex, D. galeata and C. dubia, while only one dsx homolog was isolated from M. macrocopa (Figure 1B and Additional file 1). The deduced amino acid sequences of all 9 homologs contained the expected DM- and oligomerization-domains, which are characteristic for all arthropod DSX family members [31, 44] (Figures 2, 3). Phylogenetic analysis with other known DSX of various species revealed that DSX of cladocerans grouped into two distinct monophyletic groups: DSX1 and DSX2 (Figure 4). Because DSX of the giant tiger prawn Penaeus monodon is rooted ancestrally to both DSX1 and DSX2, the gene duplication event likely occurred after the divergence of Branchiopoda and Malacostraca (Figure 4). In the present study, only dsx1, but not dsx2, was identified from M. macrocopa. To test whether another copy might exist, we performed reverse transcription PCR assays using primers corresponding to highly conserved region of dsx1 and dsx2 among Daphniidae. Only a single amplified DNA could be detected from both sexes in M. macrocopa (Additional file 1). These results suggest that the dsx gene duplication occurred prior to the divergence of these cladoceran species, therefore we infer that the M. macrocopa dsx2 gene was secondarily lost.

Nucleotide sequence comparison of dsx1 and dsx2 genes from five cladocerans. (A) Alignment of nucleotide sequence of dsx1 genes from Daphnia magna, D. pulex, D. galeata, Ceriodaphnia dubia and Moina macrocopa. (B) Alignment of nucleotide sequence of dsx2 genes from D. magna, D. pulex, D. galeata and C. dubia.

Schematic diagrams of the DSX1 structures and its sequence comparison of DM- and oligomerization-domains. (A) Domain structures of DSX1 in Daphnia magna, and identity with D. pulex, D. galeata, C. dubia and M. macrocopa. DM- and oligomerization-domains are indicated by black and gray boxes, respectively. (B, C) Alignment of predicted amino acid sequences of DM- and oligomerization-domains of DSX1 from five cladocerans, respectively. Amino acid sequences were aligned using CLUSTAL-X. Dotted boxes highlight the conserved threonine (T) residue in the DM-domain, and arginine (R) residue substituted for glutamine (Q), which is conserved amino acid residues of DSX. Asterisks indicate the zinc chelating residues [43]. Position of non-polar amino acids important in formation of the hydrophobic interface between oligomerization domains in Drosophila DSX are indicated with solid triangles [31, 41].

Schematic diagrams of the DSX2 structures and its sequence comparison of DM- and oligomerization-domains. (A) Domain structures of the DSX2 of Daphnia magna, and identity with D. pulex, D. galeata and C. dubia. DM- and oligomerization-domains are indicated by black and gray boxes, respectively. (B, C) Alignment of predicted amino acid sequences of DM- and oligomerization-domains of DSX2 from four daphniids, respectively. Amino acid sequences were aligned using CLUSTAL-X. Dotted boxes highlight the conserved threonine (T) and glutamine (Q) residues in DSX2. Asterisks indicate the zinc chelating residues. Position of non-polar amino acids important in formation of the hydrophobic interface between oligomerization-domains in Drosophila DSX are indicated with solid triangles [31, 41].

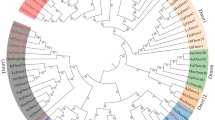
Phylogeny of DM-domain containing genes based on amino-acid sequence conservation. The evolutionary history of DM-domain containing genes was inferred by using the Neighbor-Joining method. The percentage of replicate trees in which the associated genes clustered together in the bootstrap test (1,000 replicates) is shown next to the branches (Bootstrap values below 70 percent are not shown). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. The analysis involved 55 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 62 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 [45]. Red spot indicates duplication period of dsx gene duplication in cladocerans.
By comparing the DapmaDsx1 sequence to orthologs from the four studied species (D. pulex, D. galeata, C. dubia and M. macrocopa), we found that DapmaDsx1 shows 88-58%, 100-95%, 88-48%, 100-78% and 96-61% sequence identities to the A, B (DM-domain), C, D (oligomerization-domain) and E domains, respectively (Figure 2A). Similarly, by comparing the DapmaDsx2 sequence with identified orthologs, we observed that DapmaDsx2 shows comparable relative ratios among each of the domains: 74-66%, 98-97%, 88-67%, 100-78% and 92-69% sequence identities to the A, B, C, D and E domains, respectively (Figure 3A). These results suggest that putative amino acids of both the DM- and oligomerization-domains are highly conserved among the Cladocera. On the other hand, amino acid similarities outside of these domains are lower, and are proportional to the evolutionary distance between each genus; Daphnia, Ceriodaphnia and Moina[46] (Additional files 2 with 3).
The DM-domain contains zinc chelating residues, and among the insects studied to date, two highly conserved amino acid residues, threonine and glutamine (Boxed in Figures 2B, 3B), distinguish the DM-domain of DSX from DM-domains of other insect proteins [43]. Therefore, we searched for similar highly conserved amino acid residues within the DSX DM-domains of cladocerans. Indeed, all zinc chelating residues are found to be conserved in the DM domains of DSX1 and DSX2 among the five cladoceran species (Figures 2B, 3B). Yet, although the threonine and glutamine residues were conserved in DSX2, the glutamine residue in DSX1 was substituted by arginine in all cladoceran species examined (Figures 2B, 3B). These results suggest that DSX1 in cladocerans might have gained a novel function affecting sex determination by amino acid replacement after duplication of dsx in branchiopoda lineage.
Compared to the DM-domains, more amino acid variation is observed in alignments of the oligomerization-domains (Figures 2C, 3C). Dimerization, which enhances specific DNA binding, is mediated by several residues in a non-polar interface that is conserved within oligomerization-domains. Previous study has revealed that, in DSX2 of D. magna, two of three nonpolar amino acids indispensable for formation of the nonpolar interface are substituted with the polar acidic amino acid, aspartic acid [41]. This suggests that the daphniid DSX2 proteins are unable to dimerize and may not be functional or may have a different, unknown functions.
Sex specific expression of dsxgenes in five cladoceran species
We previously reported that both dsx genes in D. magna were transcriptionally up-regulated in males and showed no sex-specific splice isoforms. We and others also reported that exposure to JH analogs reliably produces male daphniids [24, 25]. Moreover, by using gene knock-down (RNAi) and overexpression methods in D. magna[40, 41], we discovered that DapmaDsx1 is necessary and sufficient for sex determination in D. magna, whereas the tandemly duplicated DapmaDsx2 gene does not determine sex, even though its transcription is equally sex-biased [41].
In this study, we confirmed that expression patterns of DapmaDsx1 and DapmaDsx2 homologous genes are conserved in other cladocerans, by studying steady state mRNA levels for dsx transcripts in adult females and males by quantitative PCR. We found that the mRNA levels of these dsx genes range from seven to forty fold greater in males than in females (Figure 5). Our results indicate that the sexual dimorphic mRNA expression patterns of dsx are conserved among daphniids and Moina.

Relative transcriptional expression levels of dsx1 and dsx2 genes in adult males compared with females. The males of D. magna (NIES strain), D. galeata, C. dubia and M. macrocopa were induced by JH analog exposure. D. magna (Belgium strain) and D. pulex males were induced by environmental cues (see Materials and methods). These genes showed higher expression levels in males using both JHs (black) and natural cues (grey) than in females (white). Y-axes indicate relative expression levels normalized by female expression levels. Bars indicate standard errors. Numbers indicate the biological replicates. Asterisks indicate significant differences (P < 0.05, based on Student’s t-test).
Annotation of dsx gene structures in D. Magna and D. Pulex
We previously cloned and described the mRNA transcripts of the DapmaDsx1 and DapmaDsx2 genes [41]. DapmaDsx1 produces two mRNA, DapmaDsx1-α and DapmaDsx1-β, which are expressed in both sexes and differ only in their 5’ UTR, while DapmaDsx2 produces only one mRNA transcript. The D. pulex draft genome sequence was recently published [42], and the D. magna genome sequencing project is currently in progress by the Daphnia Genomics Consortium. The DapmaDsx1-α, DapmaDsx1-β, and DapmaDsx2 mRNA transcripts were aligned to the genome assemblies and used to annotate the dsx1 and dsx2 gene models in the D. magna and D. pulex genomes (Additional file 4).
The D. magna dsx gene cluster is located on scaffold 2190 of the D. magna genome assembly v2.4 with a ~10 Kbp intergenic region between the DapmaDsx1 (~16.1 Kbp length) and DapmaDsx2 (~1.6 Kbp length) genes (Additional file 4A). The second exon of the DapmaDsx1-β mRNA transcript fell within an assembly gap in scaffold 2190, but was located on scaffold 521. We conclude that scaffold 521 (~5.8 Kbp length) characterizes a ~3.1 Kbp gap in scaffold 2190, located in the intragenic region of DapmaDsx1.
The D. pulex dsx gene cluster is located on scaffold 32 of the D. pulex genome assembly v1.1 with a ~9.6 Kbp intergenic region between the DappuDsx1 (~21.5 Kbp length) and DappuDsx2 (~1.6 Kbp length) genes (Additional file 4B). The D. pulex v1.1 gene model predictions did not correctly identify the dsx1 and dsx2 duplicate gene cluster. Our annotations improved the dsx gene models in both D. magna and D. pulex genomes.
Potential transcriptional regulatory elements in the 5’ upstream promoter regions of the dsxgenes
To further annotate putative functional and conserved elements of the dsx genes, we searched and compared transcriptional promoter regions of the genes in D. magna and D. pulex. 1.0 Kbp upstream of the transcription start site (TSS) of dsx1-α, dsx1-β, and dsx2 were extracted as transcriptional regulatory regions. This interval spans the predicted intergenic region between adjacent loci. Promoter sequences are challenging for multiple alignment algorithms, because upstream regulatory regions are not well conserved compared to protein coding regions of genes [47, 48]. We aligned orthologous promoter sequences from D. magna and D. pulex using Pro-Coffee [47], an alignment algorithm specifically designed for homologous promoter regions (Additional files 5, 6, 7). The DapmaDsx1-α and DappuDsx1-α promoter alignment showed 46% sequence identity (Additional file 5), the DapmaDsx1-β and DappuDsx1-β promoter alignment showed 60% sequence identity (Additional file 6), and the DapmaDsx2 and DappuDsx2 promoter alignment showed 62% sequence identity (Additional file 7). The promoter regions of dsx1 and dsx2 have much less conservation than their respective protein coding regions, which have 84% and 83% sequence identity, respectively (92% and 84% sequence identity at synonymous sites on protein coding region, respectively).
We characterized putative known transcription factor binding sites (TFBS) in the dsx upstream promoter regions using transcription factor map (TF-map) alignments [48] between orthologous dsx promoter regions in D. magna and D. pulex, based on matches to position frequency matrices (PFMs) from JASPAR [49] and TRANSFAC [50] TFBS databases. The optimal dsx1-α promoter TF-map alignment contains 20 putative known TFBSs (Figure 6A, Additional file 8). The optimal dsx1-β promoter TF-map alignment contains 31 putative known TFBSs (Figure 6C, Additional file 9). The optimal dsx2 promoter TF-map alignment contains 39 putative known TFBSs (Figure 6E, Additional file 10). The positions of the putative TFBS pairs (between orthologous promoters) are well aligned when annotated onto the promoter sequence Pro-Coffee alignments (Figure 6), suggesting these predicted putative TFBSs are conserved between D. magna and D. pulex.

Annotations mapped onto Pro-Coffee alignment of dsx1-α , dsx1-β and dsx2 5’ upstream promoter regions. (A) Putative known transcription factor binding sites in D. magna and D. pulex dsx1-α promoter regions predicted by TF-map alignment algorithm. (B) dsx1-α promoter locations of conserved regulatory motifs predicted in both D. magna and D. pulex dsx promoter regions. (C) Putative known transcription factor binding sites in D. magna and D. pulex dsx1-β promoter regions predicted by TF-map alignment algorithm. (D) dsx1-β promoter locations of conserved regulatory motifs predicted in all D. magna and D. pulex dsx promoter regions. (E) Putative known transcription factor binding sites in D. magna and D. pulex dsx2 promoter regions predicted by TF-map alignment algorithm. (F) dsx2 promoter locations of conserved regulatory motifs predicted in all D. magna and D. pulex dsx promoter regionArrows underneath denote the conserved regulatory motifs also found D. melanogaster dsx promoter region.
We compared the number of unique predicted transcription factors (TFs) shared amongst the dsx promoter regions (Figure 7A). In total, 32 unique TFs were predicted in the dsx promoters, half (16) are present in at least two of the promoters (Additional file 11). Six unique TFs were predicted in all three dsx promoters; an additional six unique TFs were also shared between dsx1-β and dsx2 promoter regions. Interestingly, 11 unique TFs were predicted in the dsx2 promoter but not in either dsx1 promoters. We previously showed that dsx1-β mRNA expression levels are three times greater than expression levels of dsx1-α during male D. magna development, and that transcription of dsx2 is even greater than both dsx1 mRNAs combined [41]. The shared TF motifs suggest a duplication history involving at least part of the 5’ region upstream of dsx1-β, while numeric differences observed among dsx promoter regions are reflective of these expression level differences. Based on the promoter sequence conservation between D. magna and D. pulex, and the greater number of predicted TFs, the dsx1-β promoter seems to be the more widely used and evolutionarily conserved, while the dsx1-α promoter has experienced more sequence divergence and loss of TFBSs.

Venn diagram of putative transcriptional factors and sequence logos of de novo dsx promoter motifs. (A) Venn diagram showing the number of unique putative transcription factors shared amongst the D. magna and D. pulex dsx1-α, dsx1-β, and dsx2 TF-map alignments. (B-F) Braces under the sequence logos denote the similar regions between the de novo motif and known TFBS. Sequence logos were created with WebLogo [51]. (B) Motif 3 and Mirr TFBS (C) Motif 4 and Vvl TFBS (D) Motif 7 and Gsc TFBS (E) Motif 8 and Pan TFBS (F) Motif 12 and Pan reverse complement TFBS.
Since we previously reported that DapmaDsx1 and DapmaDsx2 are paralogs, and that both DapmaDsx1 and DapmaDsx2 mRNAs are transcriptionally up-regulated in male D. magna[41], we searched for de novo conserved motifs present in all dsx promoter regions in D. magna and D. pulex (DapmaDsx1-α, DappuDsx1-α, DapmaDsx1-β, DappuDsx1-β, DapmaDsx2, and DappuDsx2), without reference to TFBS sequence databases. We identified 14 conserved motifs in the Daphnia dsx promoters (Additional file 12), which can later be functionally investigated as potential TFBSs and/or potential transcriptional promoters of Daphnia dsx. The motifs were labeled M1 through M14 and annotated onto the D. magna/D. pulex dsx promoter alignments (Figures 6B, 6D and 6F). Motifs 1 through 9 were also found in the D. melanogaster dsx upstream promoter region, supporting the conservation and potential regulatory functions of these motifs.
In order to assess whether these de novo conserved motifs are similar to any known TFBSs, we scanned the individual motif sequences for matches to TFBS PFMs and compared each motif consensus sequence to known TFBS consensus sequences. Several of the conserved motifs showed similarity to known TFBSs (Figures 7B-F). Motif 3 matches the TFBS of Mirr, a homeobox transcription factor in the Iroquois complex of transcription factors, which is predicted in all three dsx promoter TF-map alignments (Figure 7B). Motif 4 matches the TFBS of Vvl, a homeobox transcription factor which is predicted in both dsx1-β and dsx2 TF-map alignments (Figure 7C). Motif 7 matches the TFBS of Gsc/Bcd/Oc, three homeobox transcription factors with nearly identical binding sites (Figure 7D), with four of the Motif 7 sequences matching the Gsc/Bcd/Oc consensus TAATC exactly. Gsc was also predicted in the dsx2 TF-map alignment, and Oc was predicted in the dsx1-α TF-map alignment. Motif 8 matches the TFBS of Pan, a high mobility group transcription factor, which is predicted in both dsx1-β and dsx2 TF-map alignments (Figure 7E). Motif 12 also matches the TFBS of Pan, but with the reverse complement of the binding site (Figure 7F). The similarity of these de novo conserved motifs to known TFBSs that were also predicted by the TF-map alignment further supports our results describing the regulatory elements of the Daphnia dsx genes.
Conclusions
In summary, we identified the orthologs of DapmaDsx1 and DapmaDsx2 genes from closely related species belonging to two cladoceran families and three genera: D. pulex, D. galeata, C. dubia (Daphniidae) and M. macrocopa (Moinidae), with highly conserved DM- and oligomerization-domains. All five species examined exhibited sexually dimorphic expression pattern of dsx genes, suggesting that these genes may have similar functions for sex determination in cladocerans. Daphniids are unique animals that exhibit ESD and are; therefore, attractive for understanding the evolution of ESD. We also identified potential regulatory motifs and transcription factor binding sites in the putative promoter regions of these genes in D. magna and D. pulex. This information will facilitate future study of molecular mechanisms underlying sex-determination in cladocerans.
Methods
Daphniastrain and culture conditions
Isoclonal strains of D. magna (NIES and Belgium strains), D. galeata, C. dubia and M. macrocopa were obtained from the National Institute for Environmental Studies (NIES; Tsukuba, Japan) [25, 52]. D. pulex was obtained from Hokkaido University, Sapporo, Japan [53], and maintained as described previously [27]. Briefly, culture medium was prepared using charcoal-filtered tap water and cultures of 20 individuals per liter were incubated at 21 ± 1°C under a 14-h light/10-h dark photoperiod. A 0.01-ml suspension of 4.3 × 108 cells ml−1 Chlorella (Chlorella vulgaris) was added daily to each culture. The water hardness was between 72 and 83 mg L−1, the pH between 7.0 and 7.5, and the dissolved oxygen concentration between 80 and 99%. To obtain natural male embryos, adult D. magna (Belgium clone) was reared in crowded conditions, and D. pulex was incubated at 18°C under a 10-h light/14-h dark photoperiod, and a 0.01-ml suspension of 4.3 × 108 cells ml−1 Chlorella was added every two days. To obtain male embryos of D. magna (NIES clone), D. galeata, C. dubia and M. macrocopa, (in which natural males are rarely seen) adult individuals (about 2 weeks of age) were chemically induced to produce males by treating them with a synthetic JH analog, fenoxycarb (1 μg/L) (technical grade 96.6% pure, Wako Pure Chemical Industries, Ltd., Osaka, Japan) [25]. We confirmed the offspring sexes by the length of the first antenna [19] observed and photographed using a Leica MZ APO dissecting microscope (Leica, Mannheim, Germany).
Cloning of dsxgenes
The nucleotide sequences of the D. magna dsx genes were used for designing primers that amplify dsx genes in four different species. The harvested animals were homogenized using the Micro Smash MS-100R (Tomy, Tokyo, Japan). Total RNA was extracted with ISOGEN reagent according to the manufacturer’s protocol (NIPPON GENE, Tokyo, Japan). Poly (A) + RNA was isolated from purified total RNA using Fast Track (Life Technologies, Carlsbad, CA USA) and converted to cDNA using Superscript III and random primers (Life Technologies) according to the manufacturer’s protocol. cDNAs corresponding to the EST sequences were obtained by PCR amplification, and full-length cDNAs were obtained by RACE (Cap Fishing; SeeGene, Seoul, South Korea) using the oligonucleotide sequences as shown in Additional file 13.
Phylogenetic analysis of the DM-domain genes
A phylogenetic tree of DM-domain genes including newly cloned D. pulex, D. galeata, C. dubia and M. macrocopa dsx genes were constructed using amino acid sequences of DM-domain genes used in the previous study [38] (Additional file 14). A multiple alignment was constructed using Clustal W [45, 54] with the following settings (pairwise alignment parameters: gap opening penalty 15, gap extension penalty 6.66, identity protein weight matrix; multiple alignment parameters: delay divergent cutoff 30%, gap separation distance 4). Phylogenetic reconstruction was performed using the maximum likelihood and the neighbor-joining methods implemented in MEGA version 5 [45].
Quantitative PCR
Two to three weeks old male and female animals of the five cladoceran species were used in quantitative-PCR (q-PCR) assays of gene expression levels. mRNAs were quantified as described previously [38]. Animals were washed briefly and soaked in RNAlater (Life Technologies) for 10 min. Total RNA was purified and cDNA was synthesized as described above except that a random oligonucleotide was used as the primer. PCR was performed in an ABI Prism 7000 (Life Technologies) using the SYBR-Green PCR core reagents kit (Life Technologies), in the presence of appropriate primers. PCR amplifications were performed using the following conditions: 2 min at 50°C and 10 min at 95°C, followed by a total of 40 two-temperature cycles (15 s at 95°C and 1 min at 60°C).
The primers were chosen to amplify short PCR products of 150 bp; the primer sequences are listed in Additional file 15. Ribosomal protein L32 gene was used for normalization purposes [21, 55]. Data acquisition and analysis were performed by ABI Prism 7000 SDS software ver. 1.1 (Life Technologies). The baseline and threshold for the Ct (cycle threshold) were set automatically. Each gene was tested in technical triplicate samples by the relative standard curve method. In the case of D. magna and D. pulex, each experiment was performed in biological triplicate and statistical analyses were applied.
dsxgene annotations
The genomic locations of DapmaDsx1-α, DapmaDsx1-β, and DapmaDsx2 mRNA transcripts were identified using BLASTN sequence similarity searches against a reference blast database of the D. magna genome assembly v2.4 scaffolds, and against a reference blast database of the D. pulex genome assembly v1.1 scaffolds. The best BLAST matches were analyzed and used to map the gene exons onto the D. magna and D. pulex scaffolds. ESTs mapped onto the genome assembly with PASA [56], microarray tiling path expression data, and RNA-Seq data from wFleaBase [57] were used as supporting evidence for exon annotations. The dsx gene annotation figures were created with AnnotationSketch [58] (Figure 6).
Transcription factor map alignments
Matscan [48] was used to search for matches to 125 JASPAR core insect TFBS matrices and 44 TRANSFAC insect TFBS matrices in each dsx promoter region in D. magna and D. pulex. A threshold of 0.85 matrix similarities was used to find TFBS matrix matches in the promoter sequences. For each promoter sequence, the collection of TFBS matrix matches is referred to as its TF-map. The TF-maps for each promoter sequence can be found in the following Additional files: DapmaDsx1-α (Additional file 16), DappuDsx1-α (Additional file 17), DapmaDsx1-β (Additional file 18), DappuDsx1-β (Additional file 19), DapmaDsx2 (Additional file 20), DappuDsx2 (Additional file 21). Meta [48] was then used to find the best meta-alignment of the orthologous promoters TF-maps (with parameters: a = 0.5, l = 0.1, m = 0.1).
de novoconserved promoter motifs
The 5′ 1.0 Kbp upstream region was extracted from all D. pulex gene models and used to create a background frequency model for 8 bp length motifs in Daphnia promoter regions. WeederH [59] was used to search for conserved regulatory motifs of length 8 present in all six dsx promoters in D. magna and D. pulex. The WeederH algorithm measures conservation based on the sequence conservation as well as the motif occurrence’s positions relative to the TSS. WeederH produces a χ2 score assessing how conserved the motif is compared to the rest of the homologous sequences. We used a χ2 score threshold of 3, discarding motifs with a χ2 score less than 3.
References
Marin I, Baker BS: The evolutionary dynamics of sex determination. Science. 1998, 281: 1990-1994.
Zarkower D: Establishing sexual dimorphism: conservation amidst diversity?. Nat Rev Genet. 2001, 2: 175-185.
Bull JJ: Sex determining mechanisms: an evolutionary perspective. Experientia. 1985, 41: 1285-1296. 10.1007/BF01952071.
Organ CL, Janes DE: Evolution of sex chromosomes in Sauropsida. Integr Comp Biol. 2008, 48: 512-519. 10.1093/icb/icn041.
Crews D, Bull JJ: Mode and tempo in environmental sex determination in vertebrates. Semin Cell Dev Biol. 2009, 20: 251-255. 10.1016/j.semcdb.2009.02.004.
Korpelainen H: Sex ratios and conditions required for environmental sex determination in animals. Biol Rev Camb Philos Soc. 1990, 65: 147-184. 10.1111/j.1469-185X.1990.tb01187.x.
Bull JJ: Evolution of environmental sex determination from genotypic sex determination. Heredity. 1981, 47: 173-184. 10.1038/hdy.1981.74.
Bulmer MG, Bull JJ: Models of polygenic sex determination and sex ratio control. Evolution. 1982, 36: 13-26. 10.2307/2407962.
Chandler CH, Phillips PC, Janzen FJ: The evolution of sex-determining mechanisms: lessons from temperature-sensitive mutations in sex determination genes in Caenorhabditis elegans. J Evol Biol. 2009, 22: 192-200. 10.1111/j.1420-9101.2008.01639.x.
Shoemaker CM, Crews D: Analyzing the coordinated gene network underlying temperature-dependent sex determination in reptiles. Semin Cell Dev Biol. 2009, 20: 293-303. 10.1016/j.semcdb.2008.10.010.
Sanchez L: Sex-determining mechanisms in insects. Int J Dev Biol. 2008, 52: 837-856. 10.1387/ijdb.072396ls.
Shukla JN, Nagaraju J: Doublesex: a conserved downstream gene controlled by diverse upstream regulators. J Genet. 2010, 89: 341-356. 10.1007/s12041-010-0046-6.
Gempe T, Beye M: Function and evolution of sex determination mechanisms, genes and pathways in insects. BioEssays. 2011, 33: 52-60. 10.1002/bies.201000043.
Shukla JN, Palli SR: Sex determination in beetles: Production of all male progeny by parental RNAi knockdown of transformer. Sci Rep. 2012, 2: srep00602-
Stenderup JT, Olesen J, Glenner H: Molecular phylogeny of the Branchiopoda (Crustacea)–multiple approaches suggest a ‘diplostracan’ ancestry of the Notostraca. Mol Phylogenet Evol. 2006, 41: 182-194. 10.1016/j.ympev.2006.06.006.
Olesen J: A phylogenetic analysis of the Conchostraca and Cladocera (Crustacea, Branchiopoda, Diplostraca). Zool J Linn Soc. 1998, 122: 491-536. 10.1111/j.1096-3642.1998.tb02161.x.
Taylor DJ, Crease TJ, Brown WM: Phylogenetic evidence for a single long-lived clade of crustacean cyclic parthenogens and its implications for the evolution of sex. Proc R Soc Lond B Biol Sci. 1999, 266: 791-797. 10.1098/rspb.1999.0707.
Kleiven OT, Larsson P, Hobæk A: Sexual reproduction in Daphnia magna requires three stimuli. Oikos. 1992, 65: 197-206. 10.2307/3545010.
Olmstead AW, LeBlanc GA: Effects of endcrine-active chemicals on the development of sex characteristics of Daphnia magna. Environ Toxicol Chem. 2000, 19: 2107-2113.
Mitchell SE: Intersex and male development in Daphnia magna. Hydrobiologia. 2001, 442: 145-156. 10.1023/A:1017564105942.
Sagawa K, Yamagata H, Shiga Y: Exploring embryonic germ line development in the water flea, Daphnia magna, by zinc-finger-containing VASA as a marker. Gene Expr Patterns. 2005, 5: 669-678. 10.1016/j.modgep.2005.02.007.
Stross RG, Hill JC: Diapause induction in Daphnia requires two stimuli. Science. 1965, 150: 1462-1464. 10.1126/science.150.3702.1462.
Hebert PD: Genotypic characteristics of cyclic parthenogens and their obligately asexual derivatives. Experientia Suppl. 1987, 55: 175-195.
Olmstead AW, LeBlanc GA: Juvenoid hormone methyl farnesoate is a sex determinant in the crustacean Daphnia magna. J Exp Zool. 2002, 293: 736-739. 10.1002/jez.10162.
Oda S, Tatarazako N, Watanabe H, Morita M, Iguchib T: Production of male neonates in four cladoceran species exposed to a juvenile hormone analog, fenoxycarb. Chemosphere. 2005, 60: 74-78. 10.1016/j.chemosphere.2004.12.080.
Banta AM, Brown LA: Control of sex in Cladocera. III. Localization of the critical period for control of sex. Proc Natl Acad Sci USA. 1929, 15: 71-81. 10.1073/pnas.15.2.71.
Tatarazako N, Oda S, Watanabe H, Morita M, Iguchi T: Juvenile hormone agonists affect the occurrence of male Daphnia. Chemosphere. 2003, 53: 827-833. 10.1016/S0045-6535(03)00761-6.
Kato Y, Kobayashi K, Oda S, Tatarazako N, Watanabe H, Iguchi T: Sequence divergence and expression of a transformer gene in the branchiopod crustacean, Daphnia magna. Genomics. 2010, 95: 160-165. 10.1016/j.ygeno.2009.12.005.
Robinett CC, Vaughan AG, Knapp J-M, Baker BS: Sex and the single cell. II. There is a time and place for sex. PLoS Biol. 2010, 8: e1000365-10.1371/journal.pbio.1000365.
Burtis KC, Baker BS: Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell. 1989, 56: 997-1010. 10.1016/0092-8674(89)90633-8.
Bayrer JR, Zhang W, Weiss MA: Dimerization of doublesex is mediated by a cryptic ubiquitin-associated domain fold: implications for sex-specific gene regulation. J Biol Chem. 2005, 280: 32989-32996. 10.1074/jbc.M507990200.
Shen MM, Hodgkin J: mab-3, a gene required for sex-specific yolk protein expression and a male-specific lineage in C. elegans. Cell. 1988, 54: 1019-1031. 10.1016/0092-8674(88)90117-1.
Raymond CS, Shamu CE, Shen MM, Seifert KJ, Hirsch B, Hodgkin J, Zarkower D: Evidence for evolutionary conservation of sex-determining genes. Nature. 1998, 391: 691-695. 10.1038/35618.
Raymond CS, Murphy MW, O’Sullivan MG, Bardwell VJ, Zarkower D: Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev. 2000, 14: 2587-2595. 10.1101/gad.834100.
Matson CK, Zarkower D: Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. 2012, 13: 163-174.
Kopp A: Dmrt genes in the development and evolution of sexual dimorphism. Trends Genet. 2012, 28: 175-184. 10.1016/j.tig.2012.02.002.
Williams TM, Carroll SB: Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat Rev Genet. 2009, 10: 797-804.
Kato Y, Kobayashi K, Oda S, Colbourne JK, Tatarazako N, Watanabe H, Iguchi T: Molecular cloning and sexually dimorphic expression of DM-domain genes in Daphnia magna. Genomics. 2008, 91: 94-101. 10.1016/j.ygeno.2007.09.002.
Watanabe H, Tatarazako N, Oda S, Nishide H, Uchiyama I, Morita M, Iguchi T: Analysis of expressed sequence tags of the water flea Daphnia magna. Genome. 2005, 48: 606-609. 10.1139/g05-038.
Kato Y, Shiga Y, Kobayashi K, Tokishita S, Yamagata H, Iguchi T, Watanabe H: Development of an RNA interference method in the cladoceran crustacean Daphnia magna. Dev Genes Evol. 2011, 220: 337-345. 10.1007/s00427-011-0353-9.
Kato Y, Kobayashi K, Watanabe H, Iguchi T: Environmental sex determination in the branchiopod crustacean Daphnia magna: deep conservation of a Doublesex gene in the sex-determining pathway. PLoS Genet. 2011, 7: e1001345-10.1371/journal.pgen.1001345.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, Bauer DJ, Cáceres CE, Carmel L, Casola C, Choi J-H, Detter JC, Dong Q, Dusheyko S, Eads BD, Fröhlich T, Geiler-Samerotte KA, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva EV, Kültz D, Laforsch C, Lindquist E, Lopez J: The ecoresponsive genome of Daphnia pulex. Science. 2011, 331: 555-561. 10.1126/science.1197761.
Oliveira DCSG, Werren JH, Verhuls EC, Giebel JD, Kamping A, Beukeboom LW, Zande L: Identification and characterization of the doublesex gene of Nasonia. Insect Mol Biol. 2009, 18: 315-324. 10.1111/j.1365-2583.2009.00874.x.
Volff J-N, Zarkower D, Bardwell VJ, Schartl M: Evolutionary dynamics of the DM domain gene family in metazoans. J Mol Evol. 2003, 57: 241-249. 10.1007/s00239-003-2470-1.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Swain TD, Taylor DJ: Structural rRNA characters support monophyly of raptorial limbs and paraphyly of limb specialization in water fleas. Proc R Soc Lond B Biol Sci. 2003, 270: 887-896. 10.1098/rspb.2002.2297.
Erb I, Bussotti G, Blanco E, Eyras E, Notredame C, lez-Vallinas JRG: Use of ChIP-Seq data for the design of a multiple promoter-alignment method. Nucleic Acids Res. 2012, 40: e52-10.1093/nar/gkr1292.
Blanco E, Messeguer X, Smith TF, Guigo R: Transcription factor map alignment of promoter regions. PLoS Comp Biol. 2006, 2: e49-10.1371/journal.pcbi.0020049.
Bryne JC, Valen E, Tang M-HE, Marstrand T, Winther O, Piedade I, Krogh A, Lenhard B, Sandelin A: JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008, 36: 102-106. 10.1093/nar/gkn449.
Matys V, Fricke E, Geffers R, Goßling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos D-U, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E: TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31: 374-378. 10.1093/nar/gkg108.
Crooks GE, Hon G, Chandonia J-M, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2002, 14: 1188-1190.
Oda S, Kato Y, Watanabe H, Tatarazako N, Iguchi T: Morphological changes in Daphnia galeata induced by a crustacean terpenoid hormone and its analog. Environ Toxicol Chem. 2011, 30: 232-238. 10.1002/etc.378.
Hiruta C, Nishida C, Tochinai S: Abortive meiosis in the oogenesis of parthenogenetic Daphnia pulex. Chromosome Res. 2010, 18: 833-840. 10.1007/s10577-010-9159-2.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Kato Y, Kobayashi K, Oda S, Tatarazako N, Watanabe H, Iguchi T: Cloning and characterization of the ecdysone receptor and ultraspiracle protein from the water flea Daphnia magna. J Endocrinol. 2007, 193: 183-194. 10.1677/JOE-06-0228.
Haas BJ, Delcher AL, Mount SM, Wortman JR RKS, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, Salzberg SL, White O: Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31: 5654-5666. 10.1093/nar/gkg770.
Colbourne JK, Singan VR, Gilbert DG: wFleaBase: the Daphnia genome database. BMC Bioinformatics. 2005, 6: 45-10.1186/1471-2105-6-45.
Steinbiss S, Gremme G, Scharfer C, Mader M, Kurtz S: AnnotationSketch: a genome annotation drawing library. Bioinformatics. 2009, 25: 533-534. 10.1093/bioinformatics/btn657.
Pavesi G, Zambelli F, Pesole G: WeederH: an algorithm for finding conserved regulatory motifs and regions in homologous sequences. BMC Bioinformatics. 2007, 8: 46-10.1186/1471-2105-8-46.
Acknowledgments
The D. magna and D. pulex sequence data were provided by The Center for Genomics and Bioinformatics at Indiana University, which is supported in part by the METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc., and distributed via wFleaBase in collaboration with Dr. Donald Gilbert and the Daphnia Genomics Consortium https://wiki.cgb.indiana.edu/display/DGC/Home. This work was partly supported by the Japan Society for the Promotion of Science (JSPS) Research Fellowship for Young Scientists. to KT and CH, the Sasakawa Scientific Research Grant from The Japan Science Society to KT, the Saito Ho-on Kai Scientific Research Grant from The Saito Gratitude Foundation to KT, grants from the Ministry of Education, Culture, Sports, Science and Technology (TI), the Ministry of the Environment of Japan (TI), a grant of Long-Range Research Initiative by Japan Chemical Industry Association (TI) and a grant from National Institute for Basic Biology (TI).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KT, YK, HM, HW, NT and TI designed the experiments; KT, YK, MS, NS, SM, HM, SO, YO, CH and TM performed the experiments; KT, HM, JKC, TI, SP and CJ analyzed the data; KT, HM, CJ, JKC and TI wrote the paper. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2012_4947_MOESM1_ESM.doc
Additional file 1: RT-PCR of oligonucleotides corresponding to highly conserved region of dsx1 (A) and dsx2 (B). The amplified cDNAs were analyzed by agarose gel electrophoresis. Lane M: molecular weight marker. Lane 1 to 10: D. magna (female), D. magna (male), D. pulex (female), D. pulex (male), D. galeata (female), D. galeata (male), C. dubia (female), C. dubia (male), M. macrocopa (female), M. macrocopa (male). (DOC 450 KB)
12864_2012_4947_MOESM2_ESM.doc
Additional file 2: Estimation of evolutionary divergence between the DSX1 except for DM- and oligomerization-domain and COI sequences. The number of amino acid differences per site between sequences are shown. The analysis involved 5 amino acid sequences. All positions containing gaps and missing data were eliminated. There were total of 225 and 208 positions in the final dataset of DSX1 and COI, respectively. Evolutionary analyses were conducted in MEGA5 [45]. (DOC 31 KB)
12864_2012_4947_MOESM3_ESM.doc
Additional file 3: Estimation of evolutionary divergence between the DSX2 except for DM- and oligomerization-domain and COI sequences. The number of amino acid differences per site between sequences are shown. The analysis involved 5 amino acid sequences. All positions containing gaps and missing data were eliminated. There were total of 210 and 208 positions in the final dataset of DSX2 and COI, respectively. Evolutionary analyses were conducted in MEGA5 [45]. (DOC 28 KB)
12864_2012_4947_MOESM4_ESM.doc
Additional file 4: Gene model annotations on the D. magna and D. pulex genome assembly. (A) D. magna dsx1 and dsx2 gene model annotations on the D. magna genome assembly. (B) D. pulex dsx1 and dsx2 gene model annotations on the D. pulex genome assembly. Figures were created with AnnotationSketch [58]. (DOC 128 KB)
12864_2012_4947_MOESM5_ESM.doc
Additional file 5: Nucleotide sequence comparison of dsx1-α promoter regions in D. magna and D. pulex. Pro-Coffee alignment of Dsx1-α 1.0 Kbp upstream promoter region from D. magna and D. pulex. (DOC 197 KB)
12864_2012_4947_MOESM6_ESM.doc
Additional file 6: Nucleotide sequence comparison of dsx1-β promoter regions in D. magna and D. pulex.Pro-Coffee alignment of dsx1-β 1.0 Kbp upstream promoter region from D. magna and D. pulex. (DOC 186 KB)
12864_2012_4947_MOESM7_ESM.doc
Additional file 7: Nucleotide sequence comparison of dsx2 promoter regions in D. magna and D. pulex. Pro-Coffee alignment of dsx2 1.0 Kbp upstream promoter region from D. magna and D. pulex. (DOC 184 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Toyota, K., Kato, Y., Sato, M. et al. Molecular cloning of doublesex genes of four cladocera (water flea) species. BMC Genomics 14, 239 (2013). https://doi.org/10.1186/1471-2164-14-239
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-239