
Abstract
Background
Japanese gentians (Gentiana triflora and Gentiana scabra) are amongst the most popular floricultural plants in Japan. However, genomic resources for Japanese gentians have not yet been developed, mainly because of the heterozygous genome structure conserved by outcrossing, the long juvenile period, and limited knowledge about the inheritance of important traits. In this study, we developed a genetic linkage map to improve breeding programs of Japanese gentians.
Results
Enriched simple sequence repeat (SSR) libraries from a G. triflora double haploid line yielded almost 20,000 clones using 454 pyrosequencing technology, 6.7% of which could be used to design SSR markers. To increase the number of molecular markers, we identified three putative long terminal repeat (LTR) sequences using the recently developed inter-primer binding site (iPBS) method. We also developed retrotransposon microsatellite amplified polymorphism (REMAP) markers combining retrotransposon and inter-simple sequence repeat (ISSR) markers. In addition to SSR and REMAP markers, modified amplified fragment length polymorphism (AFLP) and random amplification polymorphic DNA (RAPD) markers were developed. Using 93 BC1 progeny from G. scabra backcrossed with a G. triflora double haploid line, 19 linkage groups were constructed with a total of 263 markers (97 SSR, 97 AFLP, 39 RAPD, and 30 REMAP markers). One phenotypic trait (stem color) and 10 functional markers related to genes controlling flower color, flowering time and cold tolerance were assigned to the linkage map, confirming its utility.
Conclusions
This is the first reported genetic linkage map for Japanese gentians and for any species belonging to the family Gentianaceae. As demonstrated by mapping of functional markers and the stem color trait, our results will help to explain the genetic basis of agronomic important traits, and will be useful for marker-assisted selection in gentian breeding programs. Our map will also be an important resource for further genetic analyses such as mapping of quantitative trait loci and map-based cloning of genes in this species.
Similar content being viewed by others

Background
Gentianaceae (order Gentianales), comprises 87 genera and more than 1,500 species that are distributed worldwide. The genus Gentiana is the largest genus in the Gentianaceae and includes over 400 species [1]. Among them, two major floricultural species, Gentiana triflora and Gentiana scabra, the so-called Japanese gentians, have been cultivated in Japan since the 1960s. In addition to Japanese gentians, the important floricultural plant Eustoma (Eustoma grandiflorum), otherwise known as lisianthus, and a pharmaceutical plant, Swertia (Swertia japonica), also belong to the family Gentianaceae. Japanese gentians are among the most popular floricultural plants in Japan, and have unique vivid blue flowers [2]. The main Japanese gentian is G. triflora, which usually blooms from July to September in Japan. G. scabra blooms later, from September to November, and has some valuable traits, such as an open corolla and resistance to gentian brown leaf spot caused by Mycochaetophora gentianae[3, 4]. The chromosome numbers of G. triflora and G. scabra are 2n=26, whereas those of European pharmaceutical gentians, including Gentiana lutea and Gentiana punctata, are 2n=40. It is difficult to produce interspecific hybrids among members of the genus Gentiana because of the different basal chromosome numbers. The nuclear DNA content in G. triflora is 9.11 to 9.30 pg/2C, and that in G. scabra is 11.35 to 11.75 pg/2C [5]. Thus, these two Japanese gentians have similar genome sizes (approximately 5×109 bp/1C), which are larger than those of some other plants, e.g. Arabidopsis thaliana (1.5×108 bp /1C) [6], Medicago truncatula (3.8×108 bp /1C) [7], rice (4.9×108 bp /1C) [8], and tomato (9.0×108 bp /1C) [9].
There has been extensive molecular and physiological research on flower colors, flowering times, and cold tolerance of winter buds of Japanese gentians [10–14]. Based on these molecular studies, several genetic markers have been developed. For example, we developed genetic markers that discriminate among blue, pink, and white flower color [15, 16]. To protect breeders’ rights, markers based on fragment length polymorphisms [sequence characterized amplified region (SCAR) and SSR markers] were developed to identify Japanese gentian cultivars [17, 18]. The haplotypes of W14/15 alleles, which encode an esterase involved in cold tolerance of winter buds, provided data that clarified phylogenetic relationships in the genus Gentiana, and allowed analysis of the pedigree and breeding history of cultivars derived from those Gentiana spp. [14].
Genetic linkage maps are useful for studies on genomic structure and evolution, and for identification of monogenic traits or Mendelian components of quantitative trait loci (QTLs). Therefore, they are very useful for breeding programs and are the basis for future positional gene cloning [19, 20]. They are also very useful for marker-assisted breeding and introgression of beneficial traits into floricultural plants. Genetic linkage maps have been constructed for several floricultural plants, including rose (Rosa hybrida) [21, 22], carnation (Dianthus caryophyllus) [23, 24], Asiatic hybrid lily (Lilium sp.) [25], and periwinkle (Catharanthus roseus) [26]. However, no genetic linkage maps have been developed for the genus Gentiana or for any member of the family Gentianaceae, including economically important species.
The majority of floricultural plants are allogamous, but most linkage maps for crop plants have been obtained from segregating populations derived from crosses between inbred lines. Therefore, genetic linkage maps for allogamous and vegetatively reproducing species have been constructed using a double pseudo testcross strategy [25, 27]. Conversely, the genetic linkage map for ryegrass (Lolium perenne) was constructed using a backcross population involving a double haploid (DH) tester [28, 29]. Japanese gentians are also highly heterozygous plants that suffer from inbreeding depression. To obtain pure lines, the androgenic double haploid DH line, Aki6PS, was produced recently in G. triflora using anther culture [30].
Here, we report the construction of a genetic linkage map for Japanese gentian, G. scabra, using a BC1 population backcrossed with G. triflora DH line Aki6PS and 308 genetic markers. This is the first reported genetic linkage map for Japanese gentians and the first for any species belonging to the family Gentianaceae. Our results are important for understanding the genetic basis of traits and to apply molecular markers linked to mapped loci for marker-assisted selection in gentian breeding programs.
Results
Simple sequence repeat markers
The enriched SSR libraries from the DH line Aki6PS (G. triflora) yielded 19,416 clones using Roche 454 pyrosequencing. Of these, 8,932 clones (46.0%) contained SSR motifs. Only 599 clones (6.7%) were suitable for designing primer sets for SSR markers; the rest comprised reads that were too short (<200 bp) to design primers from the proximal sequences of the SSR motif. Therefore, in addition to next generation sequencing, 2,539 clones from the enriched SSR libraries were subjected to Sanger DNA sequencing analysis. As a result, 1,048 clones (41.3%) had SSR motifs within the sequence reads, and 630 clones (60.1%) could be used to design primer sets for SSR markers. These results indicated that the conventional strategy using Sanger sequencing was more efficient than next generation sequencing for developing SSR markers, without considering cost, labor, and time.
We screened 1,229 SSR markers using both parents and one F1 individual. Most SSR markers showed no or unstable amplification, or produced amplified fragments with unexpected lengths. Eighty SSR markers detected significant polymorphisms as co-dominant markers. In addition, 21 SSR markers were dominant markers, amplifying PCR fragments from SP6A1 and F1 but not from Aki6PS (Table 1). Ultimately, 101 SSR markers were available to construct the genetic linkage map. Sequence data for the markers have been deposited in GenBank/EMBL/DDBJ nucleotide sequence databases under the accession numbers (AB755678 - AB755777).
Retrotransposon microsatellite amplified polymorphism markers
To increase the number of available genetic markers for Japanese gentian, we developed some retrotransposon microsatellite amplified polymorphism (REMAP markers) using a retrotransposon. A retrotransposable element GsTRIM1 (terminal repeat retrotransposon in miniature) has been reported in G. scabra[31]. Therefore, to isolate novel LTRs of retrotransposons from Japanese gentians, we used iPBS technology as described by Kalendar et al. [32] (Figure 1A). Genomic DNA was amplified using eight iPBS primers (shown in Table 2), and then subjected to sequencing analysis. Three putative LTR sequences, 758 bp (88% identity), 735 bp (86%) and 318 bp (91%), were found in the comparative sequence analyses among 64 independent clones. Although eight primers could be designed from the three putative LTR sequences, a few amplified fragments were obtained using each LTR primer during inter-retrotransposon amplified polymorphism (IRAP) analysis (data not shown). Therefore, each LTR primer was explored in combination with ISSR primers to create REMAP markers (Figure 1B). ISSR primer sets provided from the University of British Columbia (set #9) were pre-screened using Aki6PS genomic DNA. Seventeen primers amplified more than eight stable fragments (data not shown). Seven primer sets combining LTR and ISSR, as shown in Table 3, produced reproducible amplified fragments. These primer combinations provided stable and reproducible results at an annealing temperature of 60°C. REMAP markers were more efficient and reproducible than IRAP and ISSR markers in Japanese gentians.

Schemes of iPBS and REMAP. A) iPBS is an efficient method which isolates long terminal repeat (LTR) sequences of retrotransposons [32]. Most fragments that are amplified between two retrotransposons by iPBS primers annealed at primer binding sites (PBS) contain putative LTR sequences. B) Retrotransposon and microsatellite amplified polymorphism (REMAP) amplified DNA fragments located between LTRs proximal to simple sequence repeats (SSRs).
Amplified fragment length polymorphism markers
Our preliminary experiments showed that conventional amplified fragment length polymorphism (AFLP) amplification using combinations of selective primers with three selective nucleotides resulted in too many faint and overlapping fragments in gentian (data not shown). To solve this problem, we added a fourth selective nucleotide to the Mse I primer, as described by Remington et al. [33] (Table 4). AFLP and RAPD markers are dominantly inherited; therefore, we identified only G. scabra SP6A1-specific fragments among many amplified fragments because Aki6PS-specific fragments were not segregated in the BC1 population backcrossed with G. triflora Aki6PS. Fourteen primer sets, which produced more than 10 SP6A1-specific fragments per primer combination, were selected to generate the genetic linkage map (Table 4).
Long random amplification polymorphic DNA markers
We first screened for reproducible and stable random amplification polymorphic DNA (RAPD) markers using 47 primers, as described by Debener and Mattiesch [21] and by Yamagishi et al. [34]. Among them, twelve 15-mer primers (Table 5) amplified bands reproducibly, and were selected for genetic analysis of Japanese gentians. An average of 10 fragments (< 3 kb in length) with two to seven SP6A1-specific fragments were amplified per primer.
Functional markers
In Japanese gentians, there has been extensive physiological and molecular research on flower pigmentation, flowering time, and cold tolerance of winter buds, and the nucleotide sequences of some important genes have already been deposited in public databases. In addition, our previous study reported significant insertions/deletions (in/dels) in introns and untranslated regions, representing molecular polymorphisms that could be used to discriminate among cultivars and species of Japanese gentians [17]. Therefore, we first identified the intron and proximal sequences of chalcone synthase (CHS) and the flowering locus T1 (FT1) and FT2 genes from both G. triflora Aki6PS and G. scabra SP6A1. Comparative sequencing analysis identified significant in/del polymorphisms with the intron regions of CHS and FT1, and marker primer sets were designed in the proximity of the in/dels of each gene (Table 6). For chalcone isomerase (CHI), flavanone 3-hydroxylase (FHT), flavonoid 3′,5′-hydroxylase (F3′5′H), anthocyanidin synthase (ANS), MYB3, basic helix loop helix 1 (GtbHLH1), terminal flowering 1 (TFL1) and W14/15 genes, we used the primer sets reported in previous studies [11, 12, 14, 16, 17]. No in/dels were detected in the genomic sequences of FT2 and TFL1 genes, but some SNPs were detected using restriction enzymes. Therefore, FT2 and TFL1 markers were distinguished between Aki6PS and SP6A1 as cleaved amplified polymorphic sequence (CAPS) markers, when their amplified fragments were digested by Pst I and Sau 3A I, respectively.
Segregation of stem pigmentation trait
The G. triflora breeding line Aki6PS has a green stem color, while stems of the G. scabra breeding line SP6A1 and F1 are red because of the accumulation of anthocyanins. Among BC1 progeny, 44 and 49 individuals showed green- and red-stem color phenotypes, respectively. Segregation of stem color fitted a 1:1 ratio based on a chi-square test at P = 0.05, suggesting monogenetic inheritance of this trait.
Genetic map construction
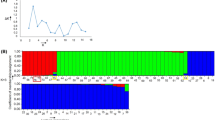
We used a population of 93 BC1 from G. scabra SP6A1 backcrossed with G. triflora Aki6PS DH line (Figure 2). In total, 308 markers, comprising 101 SSR markers, 103 AFLP markers, 54 RAPD markers, 38 REMAP markers and 11 functional markers, and one phenotypic marker were grouped with a LOD score of 9.0. As a result, 19 linkage groups containing at least two markers could be assembled with 273 markers (Figure 3). Thirty-five markers were unmapped and ungrouped in this linkage map, and more RAPD (27.8%) and REMAP (21.0%) markers were excluded from the linkage groups, compared with SSR (3.9%), AFLP (5.8%) and functional markers (9.0%). Of the 10 functional markers, FT1 was closely linked to FT2, an ortholog of FT1. Stem color locus as a phenotypic trait was closely linked to the M03E1-104 AFLP marker in LG9. In total, 19 linkage groups were identified, covering 613.7 cM with an average distance of 2.2 cM between each pair of markers. The size of the linkage groups ranged from 1.1 to 93.6 cM. Twenty-seven, 16, and 38 markers showed distorted segregation at the 5%, 1%, and 0.1% levels, respectively, based on the chi-square test. Five linkage groups (LG3, LG4, LG11, LG14, and LG17) were assembled from markers with distorted segregation at the 0.1% level in the genetic map of Japanese gentian.

Production of BC 1 progeny for genetic linkage analysis. G. scabra breeding line SP6A1 is an unfixed breeding line, because most Japanese gentians exhibit inbreeding depression. Aki6PS is a double haploid line (DH) generated from G. triflora cv. ‘Ashiro-no-Aki’ using anther culture. Crossing the DH line Aki6PS back into one F1 individual yielded 93 BC1 progeny.

Genetic linkage map of G. scabra. Genetic linkage map of G. scabra. based on a BC1 mapping population generated by crossing G. scabra SP6A1 × G. triflora Aki6PS DH line. Map was generated with 307 (101 SSR, 103 AFLP, 54 RAPD, 38 REMAP and 11 functional) polymorphic markers using JoinMap version 4.1 at an LOD value of 9.0, with the Kosambi mapping function. In total, 273 markers (97 SSR, 97 AFLP, 39 RAPD, 30 REMAP and 10 functional) were mapped on 19 linkage groups (LG), which spanned 613.7 cM. Distortions at the 5%, 1% and 0.1% level are indicated as *, ** and ***, respectively. AFLP, RAPD, and REMAP markers are indicated as Mblack circle Eblack circle-, Pblack circle black circle- and REMblack circle-, respectively. Of the 10 functional markers, flowering related genes, FT1, FT2, TFL1 and W14/15 are shown in red, and flower pigmentation related genes CHS, CHI, ANS, F3′5′H1, MYB3 and bHLH1 are shown in blue. Stem pigmentation (Stem Pig), which represents a phenotypic trait, is underlined.
Discussion
We used 454 FLX Titanium pyrosequencing of an SSR-enriched library to develop SSR markers from Japanese gentians. Recently, next generation sequencing has been used successfully to develop several genetic markers, including SSRs [35–37]. In our study, out of approximately 20,000 sequences from the enriched SSR library, only 3.0% could be used to design primers suitable for SSR markers. In contrast, 24.8% of the sequences derived from conventional Sanger DNA sequencing were suitable for designing primers for SSR markers. This is because the reads obtained from pyrosequencing in this study are shorter and less accurate than those obtained from Sanger sequencing. Similarly, in the honey bee (Apis mellira) and zebrafish (Danio rerio), only 1.2% to 5.4% sequences obtained by 454 FLX Titanium pyrosequencing could be used to design primers for SSR markers [37]. In spite of its flaws, next generation sequencing is cheaper and less labor-intensive than conventional Sanger DNA sequencing. As next generation sequencing technology develops further, for example, the long-read protocols based on the GS FLX+ system, this technology will become more efficient for development of SSR markers.
Retrotransposons can be used as molecular markers because their integration creates new junctions between genomic DNA and their conserved ends [38]. IRAP and REMAP are simple methods that do not require restriction enzyme digestion or ligation to generate marker bands [38, 39]. However, the limiting factor for developing genetic markers based on LTR retrotransposons for new plant species is the availability or otherwise of retrotransposon sequences. In Japanese gentian, three dispersed transposable elements, GsTRIM1, Tgt1 (gypsy-Ty3 retrotransposon), and GtMITE1, have been identified [31, 40]; however, there are low copy numbers of Tgt1 and GtMITE1 in the gentian genome [40]. We used iPBS amplification [32], which is based on the universal presence of primer binding sites (PBS) of LTR retrotransposons, to identify new LTR sequences from Japanese gentian. Three putative LTR sequences were predicted from overlapping regions among independent sequences obtained by iPBS amplification. These sequences did not correspond to the terminal direct repeat (TDR) of GsTRIM1 and LTR of Tgt1. We designed primers from these newly identified LTR sequences (Table 3). However, no or few fragments were amplified by the putative LTR primers from any Japanese gentian lines, probably because the LTR retrotransposons were too far apart for efficient amplification. Therefore, these LTR primers were not suitable as IRAP markers. Conversely, REMAP primers yielded more reproducible and stable fragments in Japanese gentians than IRAP and ISSR, and were used to generate the genetic linkage map.
RAPD and AFLP technologies are conventional molecular markers. Their advantages are that they do not require sequence information and have a relatively low start-up cost [41–43]. However, certain procedures must be modified to use RAPD and AFLP markers for Japanese gentians, probably because the large genome size and heterogeneity. RAPDs using10-mer primers and AFLP using three selective nucleotides resulted in too many faint and overlapping fragments (data not shown), whereas long-RAPDs using 15-mer primers amplified reproducible and stable fragments at higher annealing temperatures. The use of 15-mer primers also led to more effective amplification of polymorphic fragments than 10-mer primers in grape [43], rose [21], and Asiatic hybrid lily [34]. The AFLP markers produced reproducible fragments using Mse I-selective primers with four selective nucleotides in Japanese gentians. In conifer, which has a very large genome (2×1010 bp), the use of selective primers with a fourth selective nucleotide also improved amplification of reproducible fragments [33]. Therefore, markers developed by modified RAPD and AFLP were used to generate the gentian genetic linkage map (see below).
A genetic linkage map with 273 markers was constructed using an interspecific BC1 population. Nineteen linkage groups were obtained for the BC1 population at an LOD value of 9.0, and encompassed 613.7 cM (Figure 3). Analysis of published saturated maps for tomato (n = 12, 1,283 cM) [44], melon (n = 12, 1,150 cM) [45], and lettuce (n = 9, 1,505 cM) [46] using the JoinMap program showed that an average map distance of 100 to 150 cM can be expected per chromosome regardless of its physical length. We estimate the map length of Japanese gentians (n =13) to be 1,300 to 1,950 cM. The present genetic linkage map probably covers less than half of the genome; therefore, more genetic markers are required to saturate the linkage map of Japanese gentians.
In the BC1 progeny of Japanese gentians, 30.0% of markers showed segregation distortion. These skewed segregations were also reported in other ornamental plants species, such as the diploid rose [21] and Asiatic hybrid lily [25]. The causes of segregation distortion are not well understood. Segregation distortion has been reported in Rhododendron[47], Asiatic lily [25] and carnation [23, 24], and it was assumed to represent hybrid sterility genes and gametophytic selection genes in interspecific crossing. The BC1 population used in this study was derived from interspecific crossing between G. triflora and G. scabra, and the reciprocal cross showed hybrid weakness. Therefore, hybrid sterility and gametophytic selection genes might be responsible for the segregation distortion in Japanese gentian populations.
Functional markers are those derived from polymorphic sites within genes responsible for phenotype traits [48]. Ten functional markers for genes involved in important traits, such as flower pigmentation (CHS, CHI, F3′5′H, ANS, MYB3 and bHLH1), flowering time (FT1, FT2 and TFL1) and cold tolerance of winter buds (W14/15), were mapped to the 19 linkage groups of Japanese gentian (Figure 3). Our previous research revealed that white-flowered gentians resulted from the functional deficiency of ANS, a structural enzyme, or MYB3, an anthocyanin biosynthetic transcription factor. Genetic analysis showed that the ANS locus was not linked to the MYB3 locus [11, 49]. Consistent with these observations, ANS and MYB3 were assembled into different linkage groups: LG11 and LG4, respectively (Figure 3). Interestingly, two orthologs of the flowering hormone florigen, FT1 and FT2, were closely linked (LG4). These findings will be useful for determining the molecular functions of these genes and to study the genetic regulation of flowering time in gentian, although further research is necessary. The mapping of functional markers corresponding to important agricultural traits will be accelerated by further molecular and physiological studies on Japanese gentians. Our genetic linkage map will be useful for mapping QTLs associated with various traits, and for improving Japanese gentian breeding programs. The map is also applicable to other members of the Gentianaceae family, including several other economically important species.
Conclusions
This study presents the first genetic linkage map for Japanese gentian. The map was constructed using four different types of molecular markers. It was produced from genotypes of 93 BC1 progeny derived from a DH line and included 274 markers (97 SSR, 97 AFLP, 39 RAPD, 30 REMAP, 10 functional markers, and 1 phenotypic trait). The map revealed 19 linkage groups that covered 613.7 cM, with an average intermarker distance of 2.2 cM. This map is a starting point for mapping single or quantitative trait loci affecting agronomically important phenotypes, and will be useful for research on gentian genetics and breeding.
Methods
Plant Materials
In total, 93 BC1 progeny were derived from the interspecific backcross Aki6PS×SP6A1 (Figure 2). Aki6PS is a double haploid (DH) line derived from Gentiana triflora cv. ‘Ashiro-no-Aki’ using anther culture [30]. SP6A1 is a breeding line derived from the G. scabra. F1 was produced by crossing a female Aki6PS with a male SP6A1. BC1 progeny were produced by crossing a female Aki6PS with pollen of a single F1 individual, because the reciprocal hybrid (F1×Aki6PS) showed hybrid weakness and it was difficult to obtain sufficient progeny.
Genomic DNA was isolated from young leaves (500 mg) of each individual using Nucleon PhytoPure (GE Healthcare, Little Chalfont, UK) and stored at −20°C until use.
Simple sequence repeat markers
SSR markers were developed as described by Sato-Ushiku et al. [18]. Genomic DNA was isolated from either G. triflora Aki6PS or G. scabra SP6A1, and then digested by either Alu I, Hae III, Mse I, or Rsa I. The digested genomic DNA was hybridized with biotin-labeled (CA)15 or (GA)15 probes. SSR-enriched libraries were mixed and subjected to sequencing analysis using 454 GS FLX Titanium pyrosequencing (Roche, Basel, Switzerland). The SSR-enriched library was also subcloned into the pGEM-T Easy vector (Promega, Madison, WI, USA), and subjected to sequencing analysis using BigDye terminator version 1.1 cycle sequencing kit and an ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems by Life Technologies, Foster City, CA, USA). SSR markers were predicted and developed using Read2Marker software with the default parameters [50].
PCR amplification was performed in a 20-μL reaction mixture containing 10 ng genomic DNA, 0.5 μM each primer (Table 1), 0.2 mM dNTPs, 1 × Ex buffer, and 0.25 units Ex Taq polymerase (Takara Bio, Otsu, Japan). The PCR conditions were as follows: 94°C for 2 min; 30 cycles at 95°C for 20 s, 60°C for 40 s, and 72°C for 1 min; and a final extension at 72°C for 5 min. The amplified fragments were separated on a high-efficiency genome scanning (HEGS) running system (Nihon Eido, Tokyo, Japan) [51]. In brief, 2 μL of each sample per lane was loaded onto polyacrylamide gels consisting of a stacking gel (5% [w/v] bis-polyacrylamide [29:1] containing 0.5 M Tris–HCl, pH6.8) and running gel (15% [w/v] bis- polyacrylamide [29:1] containing 1.5M Tris–HCl, pH 8.8) and electrophoresed in 1× Tris–glycine buffer (25 mM Tris–HCl, 1.92 M glycine, pH 8.3) at 300 V for 2 h. The sizes of the fragments were estimated based on a 20-bp ladder (Takara Bio). The gels were stained with SYBR Gold nucleic acid gel stain (Molecular Probes by Life Technologies, Helsinki, Finland), and photographed and analyzed using a ImageQuant LAS-4000 luminescent image analyzer (GE Healthcare).
Retrotransposon microsatellite amplified polymorphism markers
The sequences of LTRs of retrotransposons were isolated from gentian genome using the iPBS approach [32]. PCR was performed in a 25-μl reaction mixture containing 1 μg genomic DNA, 1×Ex buffer, 1 μM each primer, 0.2 mM dNTPs, and 1 unit ExTaq DNA polymerase. The PCR program was as follows: 1 cycle at 95°C for 3 min; 30 cycles of 95°C for 15 s, 50–70°C for 1 min, and 72°C for 1 min; and a final extension step of 72°C for 5 min. The sequences and annealing temperatures of iPBS primers are shown in Table 2. The reaction mixtures were purified using a Microspin S-400 HR column (GE Healthcare), and then subcloned into the pCR4-TOPO TA cloning vector (Invitrogen in Life technologies, CA). Sixty-four independent clones were subjected to sequencing analysis, as above. Putative LTRs were found by comparative analysis among sequences obtained by the iPBS approach using stand-alone BLAST [52].
The REMAP markers were created by combining LTR primers with ISSR primers, as shown in Table 3. Each 20-μl reaction mixture comprised 20 ng genomic DNA, 1×Ex buffer, 0.2 mM dNTPs, 0.2 μM each primer, and 1 unit Ex Taq polymerase. The PCR program was as follows: 94°C for 4 min; 30 cycles of 94°C for 40 s, 60°C for 40 s, and 72°C for 2 min; followed by final extension at 72°C for 5 min. The amplified fragments were separated on 1.0 or 1.6% (v/v) agarose gels in TAE buffer, and then photographed and analyzed with an ImageQuant LAS-4000 system after staining with ethidium bromide.
Amplified fragment length polymorphism markers
AFLP analysis was performed as described by Vos et al. [41] with some modifications. One microgram of genomic DNA from each individual was digested with two restriction enzyme, 25 units of Mae I and 50 units of Eco R I, in a reaction volume of 25 μl with restriction enzyme buffer (10 mM Tris–HCl, pH 7.5, 10 mM magnesium acetate, 50 mM potassium acetate, 5 mM dithiothreitol, 0.005%(v/v) bovine serum albumin, pH 7.5) at 37°C for 3 h. The restriction enzyme solution was added to 10 μl adapter ligation solution (1×restriction enzyme buffer, 1mM ATP, 2.5μM Eco R I adapter, 2.5μM Mse I adapter, 50 units T4 DNA ligase), and then incubated at 37°C overnight. To stop the reaction, 500 μl T10E0.1 buffer (10 mM Tris–HCl, 100 mM EDTA, pH 8.0) was added to the ligation solution.
Pre-amplification was conducted in a 25-μl reaction volume containing 2.5 μl diluted adaptor-ligated DNA, 0.4 μM EcoRI+A primer, 1.6μM MseI+C primer, 200 μM dNTPs, 1×ExTaq buffer, and 1.25 units ExTaq DNA polymerase (Takara-bio). The PCR program was as follows: 94°C for 2 min; 20 cycles of 94°C for 30 s, 56°C for 1 min and 72°C for 1 min; followed by final extension at 72°C for 10 min. The PCR products were diluted 100-fold in T10E0.1 buffer. Four labeled-Eco R I selective primers with three selective nucleotides (NED-AAC, JOE-AAG, FAM-ACA and JOE-AGG) and 10 Mse I selective primers with four selective nucleotides (CACA, CACT, CACC, CATA, CATT, CATC, CATG, CTTA, CTAC and CTAG) were used for selective amplification. We screened 14 primer combinations because they provided the highest number of heterozygous bands (Table 4). For the selective PCR reaction, the 10-μl reaction mixture consisted of 5 μl pre-selective PCR product, 1×ExTaq buffer, 200 μM dNTPs, 0.5 μM labeled-Eco R I-ANN primer, 0.5 μM Mse I-ANNN primer, and 0.5 units ExTaq DNA polymerase. The touchdown PCR profile was as follows: 1 cycle at 94°C for 30 s, 68°C for 30 s, and 72°C for 1 min; 17 cycles with the annealing temperature reduced by 0.7°C/cycle; 23 cycles with an annealing temperature of 56°C; and a final extension at 72°C for 10 min. PCR products were separated and detected using an ABI 3130 genetic analyzer. The sizes of the amplified products were calculated based on internal standard DNA (GeneScan-500ROX size standard, Applied Biosystems) using GeneMapper software (Applied Biosystems).
Long random amplified polymorphic DNA markers
The long RAPD markers were developed as described by Debener and Mattiesch [21] and Yamagishi et al. [34]. After pre-screening, twelve 15-mer primers (Table 5) were selected to generate the genetic linkage map. The 20-μl reaction mixture comprised 20 ng genomic DNA, 1×Ex buffer, 0.2 mM dNTPs, 0.4 μM 15-mer primer, and 1 unit Ex Taq polymerase. The PCR program consisted of 94°C for 90 s; 40 cycles of 95°C for 30 s, 53°C for 1 min 30 s, and 72°C for 2 min; followed by final extension at 72°C for 5 min. The amplified fragments were separated on 1% (v/v) agarose gels in TAE buffer, and then photographed and analyzed as described in the REMAP markers section.
Functional markers
Our previous research revealed that the intron lengths of several flavonoid biosynthetic genes, including CHS, CHI, FHT, F3′5′H, ANS, and the transcriptional factors GtMYB3 and GtbHLH1 exhibited significant polymorphisms among Japanese gentian cultivars [10, 16, 17]. Primers for GtFT1, GtFT2, and GtTFL1, which are regulator genes of flowering time [12], were also designed from the difference in the genomic sequences between Aki6PS and SP6A1. The W14/15 esterase is related to cold tolerance of gentian winter buds, and the W14/15 alleles of Aki6PS and SP6A1 are known as 15a′/15a′ and 14b1/14b1′, respectively [14]. The primer sets for the functional markers are shown in Table 6. The genomic sequences of GtFT2 and GtTFL1 have no large insertions or deletions between Aki6PS and SP6A1. Therefore, CAPS markers were used for GtFT2 and GtTFL1 by digesting the amplified fragments with Hae III and Sau 3A I, respectively.
PCR amplification was performed in a 20-μL reaction mixture containing 20 ng genomic DNA, 0.5 μM each primer (as shown in Table 6), 0.2 mM dNTPs, 1× Ex buffer, and 0.25 units Ex Taq polymerase. The PCR conditions were as follows: 94°C for 2 min; 30 cycles at 95°C for 20 s, 60°C for 40 s, and 72°C for 1 min; and a final extension at 72°C for 5 min. The amplified fragments were separated on the HEGS running system or 2% agarose gels in TBE buffer, and then photographed and analyzed as described above.
Evaluation of stem pigmentation
Stem colors were investigated three times in the mapping population. Green and red stem colors were scored as G. triflora (A) and G. triflora × G. scabra hybrid (H) genotypes, respectively.
Linkage analysis
We used 308 markers, including 101 SSRs, 38 REMAPs, 103 AFLPs, 54 long RAPDs, 11 functional markers, and one phenotypic trait to construct a genetic linkage map using JoinMap ver. 4.1 (Kyazma, Wageningen, Netherlands). The Kosambi function was used to convert recombination units into genetic distances. The mapping analysis was conducted by using a minimum LOD score of 9.0. Distorted markers analyzed by the chi-square test were used in the construction of the linkage maps.
Abbreviations
- AFLP:
-
Amplified fragment length polymorphism
- ANS:
-
Anthocyanidin synthase
- bHLH:
-
Basic helix-loop-helix
- CAPS:
-
Cleaved amplified polymorphic sequence
- CHS:
-
Chalcone synthase
- CHI:
-
Chalcone isomerase
- DH:
-
Doubled haploid
- F3′5′H:
-
Flavonoid 3′,5′-hydroxylase
- FHT:
-
Flavanone 3-hydroxylase
- FT:
-
Flowering locus T
- ISSR:
-
Inter-simple sequence repeat
- LTR:
-
Long terminal repeat
- MITE:
-
Miniature inverted-repeat in transposable element
- PBS:
-
Primer binding site
- QTL:
-
Quantitative trait loci
- RAPD:
-
Random amplification polymorphic DNA
- REMAP:
-
Retrotransposon microsatellite amplified polymorphism
- SCAR:
-
Sequence characterized amplified region
- SSR:
-
Simple sequence repeat
- TDR:
-
Terminal direct repeat
- TFL1:
-
Terminal flowering 1
- TRIM:
-
Terminal repeat retrotransposon in miniature.
References
Kohlein: Gentians. 1991, London: Christopher Helm
Nishihara M, Nakatsuka T, Mizutani-Fukuchi M, Tanaka Y, Yamamura S: Gentians: From gene cloning to molecular breeding. Floricultural and Ornamental Biotechnology V. 2008, UK: Global Science Books, 57-67.
Nekoduka S, Tanaka K, Harada Y, Sano T: Phylogenetic affinity of Mycochaetophora gentianae, the causal fungus of brown leaf spot on gentian (Gentiana triflora), to Pseudocercosporella-like hyphomycetes in Helotiales. Mycoscience. 2010, 51: 123-133.
Nekoduka S, Tanaka K, Sano T: Pathogenicity of Mycochaetophora gentianae, causal fungus of gentian brown leaf spot, as affected by host species, inoculum density, temperature, leaf wetness duration, and leaf position. J Gen Plant Pathol. 2010, 76: 370-376.
Mishiba K, Yamane K, Nakatsuka T, Nakano Y, Yamamura S, Abe J, Kawamura H, Takahata Y, Nishihara M: Genetic relationships in the genus Gentiana based on chloroplast DNA sequence data and nuclear DNA content. Breed Sci. 2009, 59: 119-127.
Arabidopsis-genome-initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408: 796-815.
Young ND, Debelle F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, et al: The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2012, 480: 520-524.
International-Rice-Genome-Sequencing-Project: The map-based sequence of the rice genome. Nature. 2005, 436: 793-800.
The-tomato-genome-consortium: The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012, 485: 635-641.
Nakatsuka T, Nishihara M, Mishiba K, Yamamura S: Temporal expression of flavonoid biosynthesis-related genes regulates flower pigmentation in gentian plants. Plant Science. 2005, 168: 1309-1318.
Nakatsuka T, Haruta KS, Pitaksutheepong C, Abe Y, Kakizaki Y, Yamamoto K, Shimada N, Yamamura S, Nishihara M: Identification and characterization of R2R3-MYB and bHLH transcription factors regulating anthocyanin biosynthesis in gentian flowers. Plant & cell physiology. 2008, 49: 1818-1829.
Imamura T, Nakatsuka T, Higuchi A, Nishihara M, Takahashi H: The gentian orthologs of the FT/TFL1 gene family control floral initiation in Gentiana. Plant & cell physiology. 2011, 52: 1031-1041.
Hikage T, Saitoh Y, Tanaka-Saito C, Hagami H, Satou F, Shimotai Y, Nakano Y, Takahashi M, Takahata Y, Tsutsumi K: Structure and allele-specific expression variation of novel alpha/beta hydrolase fold proteins in gentian plants. Mol Genet Genomics. 2007, 278: 95-104.
Hikage T, Kogusuri K, Tanaka-Saito C, Watanabe S, Chiba S, Kume K, Doi H, Saitoh Y, Takahata Y, Tsutsumi K: W14/15 esterase gene haplotype can be a genetic landmark of cultivars and species of the genus Gentiana L. Mol Gen Genomics. 2011, 285: 47-56.
Kakizaki Y, Nakatsuka T, Kawamura H, Abe J, Abe Y, Yamamura S, Nishihara M: Development of codominant DNA marker distinguishing pink from blue flowers in Gentiana scabra. Breed Res. 2009, 11: 9-14. (Japanese)
Nakatsuka T, Saito M, Sato-Ushiku Y, Yamada E, Nakasato T, Hoshi N, Fujiwara K, Hikage T, Nishihara M: Development of DNA markers that discriminate between white- and blue-flowers in Japanese gentian plants. Euphytica. 2012, 184: 335-344.
Shimada N, Nakatsuka T, Nakano Y, Kakizaki Y, Abe Y, Hikage T, Nishihara M: Identification of gentian cultivars using SCAR markers based on intron-length polymorphisms of flavonoid biosynthetic genes. Sci Hort. 2009, 119: 292-296.
Sato-Ushiku Y, Shimada N, Saito M, Yamada E, Hikage T, Nakatsuka T, Nishihara M: Development of simple sequence repeat markers for identification of Japanese gentian cultivars. J Japan Soc Hort Sci. 2011, 80: 475-485.
Ribaut JM, Hoisington D: Marker-assisted selection: new tools and strategies. Trends Plant Sci. 1998, 3: 236-239.
Jones N, Ougham H, Thomas H, Pasakinskiene I: Markers and mapping revisited: finding your gene. New Phytol. 2009, 183: 935-966.
Debener T, Mattiesch L:: Construction of a genetic linkage map for roses using RAPD and AFLP markers. Theor Appl Genet. 1999, 99: 891-899.
Yan Z, Denneboom C, Hattendorf A, Dolstra O, Debener T, Stam P, Visser PB: Construction of an integrated map of rose with AFLP, SSR, PK, RGA, RFLP, SCAR and morphological markers. Theor Appl Genet. 2005, 110: 766-777.
Yagi M, Onozaki T, Taneya M, Watanabe H, Yoshimura T, Yoshinari T, Ochiai Y, Sibata M: Construction of genetic linkage map for the carnation by using RAPD and SSR marker and mapping quantitative trait loci (QTL) for resistance to bacterial wilt caused by Burkholderia caryophylli. J Japan Soc Hort Sci. 2006, 75: 166-172.
Yagi M, Kimura T, Yamamoto T, Isobe S, Tabata S, Onozaki T: QTL analysis for resistance to bacterial wilt (Burkholderia caryophylli) in carnation (Dianthus caryophyllus) using an SSR-based genetic linkage map. Mol Breeding. 2012, 30: 495-509.
Abe H, Nakano M, Nakatsuka A, Nakayama M, Koshioka M, Yamagishi M: Genetic analysis of floral anthocyanin pigmentation traits in Asiatic hybrid lily using molecular linkage maps. Theor Appl Genet. 2002, 105: 1175-1182.
Shokeen B, Choudhary S, Sethy NK, Bhatia S: Development of SSR and gene-targeted markers for construction of a framework linkage map of Catharanthus roseus. Ann Bot. 2011, 108: 321-336.
Grattapaglia D, Sederoff R: Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics. 1994, 137: 1121-1137.
Bert PF, Charmet G, Sourdille P, Hayward MD, Balfourier F: A high-density molecular map for ryegrass (Lolium perenne) using AFLP markers. Theor Appl Genet. 1999, 99: 445-452.
Jones S, Dupal P, Dumsday L, Hughes J, Forster W: An SSR-based genetic linkage map for perennial ryegrass ( Lolium perenne L.). Theor Appl Genet. 2002, 105: 577-584.
Doi H, Takahashi R, Hikage T, Takahata Y: Embryogenesis and doubled haploid production from anther culture in gentian (Gentiana triflora). Plant Cell Tissue Organ Cult. 2011, 102: 27-33.
Nakatsuka T, Nishihara M, Mishiba K, Hirano H, Yamamura S: Two different transposable elements inserted in flavonoid 3',5'-hydroxylase gene contribute to pink flower coloration in Gentiana scabra. Mol Gen Genomics. 2006, 275: 231-241.
Kalendar R, Antonius K, Smykal P, Schulman AH: iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor Appl Genet. 2010, 121: 1419-1430.
Remington DL, Whetten RW, Liu BH, O'Malley DM: Construction of an AFLP genetic map with nearly complete genome coverage in Pinus taeda. Theor Appl Genet. 1999, 98: 1279-1292.
Yamagishi M, Abe H, Nakano M, Nakatsuka A: PCR-based molecular markers in Asiatic hybrid lily. Sci Hort. 2002, 96: 225-234.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML: Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature reviews. 2011, 12: 499-510.
Santana Q, Coetzee M, Steenkamp E, Mlonyeni O, Hammond G, Wingfield M, Wingfield B: Microsatellite discovery by deep sequencing of enriched genomic libraries. Biotechniques. 2009, 46: 217-223.
Martin JF, Pech N, Meglecz E, Ferreira S, Costedoat C, Dubut V, Malausa T, Gilles A: Representativeness of microsatellite distributions in genomes, as revealed by 454 GS-FLX titanium pyrosequencing. BMC genomics. 2010, 11: 560-
Kalendar R, Schulman AH: IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat Protoc. 2006, 1: 2478-2484.
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A: IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet. 1999, 98: 704-711.
Nishihara M, Hikage T, Yamada E, Nakatsuka T: A single-base substitution suppresses flower color mutation caused by a novel miniature inverted-repeat transposable element in gentian. Mol Genet Genomics. 2011, 286: 371-382.
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, et al: AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23: 4407-4414.
Vuylsteke M, Peleman JD, van Eijk MJ: AFLP technology for DNA fingerprinting. Nat Protoc. 2007, 2: 1387-1398.
Ye GN, Hemmat M, Lodhi MA, Weeden NF, Reisch BI: Long primers for RAPD mapping and fingerprinting of grape and pear. Biotechniques. 1995, 20: 368-371.
Haanstra JP, Lauge R, Meijer-Dekens F, Bonnema G, de Wit PJ, Lindhout P: An integrated high-density RFLP-AFLP map of tomato based on two Lycopersicon esculentum × L. pennellii F2 populations. Theor Appl Genet. 1999, 99: 254-271.
Diaz A, Fergany M, Formisano G, Ziarsolo P, Blanca J, Fei Z, Staub JE, Zalapa JE, Cuevas HE, Dace G, et al: A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon (Cucumis melo L.). BMC plant biology. 2011, 11: 111-
Truco MJ, Antonise R, Lavelle D, Ochoa O, Kozik A, Witsenboer H, Fort SB, Jeuken MJ, Kesseli RV, Lindhout P, et al: A high-density, integrated genetic linkage map of lettuce (Lactuca spp.). Theor Appl Genet. 2007, 115: 735-746.
Dunemann F, Kahnau R, Stange I: Analysis of complex leaf and flower characters in Rhododendron using a molecular linkage map. Theor Appl Genet. 1999, 98: 1146-1155.
Andersen JR, Lubberstedt T: Functional markers in plants. Trends Plant Sci. 2003, 8: 554-560.
Nakatsuka T, Nishihara M, Mishiba K, Yamamura S: Two different mutations are involved in the formation of white-flowered gentian plants. Plant Science. 2005, 169: 949-958.
Fukuoka H, Nunome N, Minamiyama Y, Kono I, Namiki N, Kojima A: read2Marker: a data processing tool for microsatellite marker development from a large data set. Biotechniques. 2005, 39: 472-476.
Kawasaki S, Murakami Y: Genome analysis of Lotus japonicus. J Plant Res. 2000, 113: 497-506.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402.
Acknowledgements
The authors are especially grateful to Drs. Toshiya Yamamoto and Miyuki Kunihisa, National Institute of Fruit Tree Science (NIFTS), and Drs. Masafumi Yagi and Takashi Onozaki, National Institute of Floricultural Science (NIFS), for helpful discussions and technical advice. We thank Dr. Masumi Yamagishi, Hokkaido University, for providing long RAPD primers and technical advice. We thank Drs. Tomohiro Imamura and Hideyuki Takahashi, Iwate Biotechnology Research Center, for providing sequence information for flowering time genes and for helpful discussions. The authors thank Drs. Hisako Doi and Yoshihito Takahata, Iwate University, for providing the double haploid gentian line Aki6PS. We thank Dr. Hiroyuki Fukuoka, National Institute of Vegetable and Tea Science (NIVTS), for technical advice on developing SSR markers. The authors thank Mses. Chiharu Yoshida and Yoshiko Abe, members of our laboratory, for technical support. This study was supported by a ‘Research Project for Utilizing Advanced Technologies in Agriculture, Forestry and Fisheries’ from the Ministry of Agriculture, Forestry and Fisheries of Japan (MAFF), and by the Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TN participated in the design of the experiments, and developed and genotyped the SSR, AFLP and functional markers, performed statistical analysis, data interpretation and wrote the paper. EY developed and genotyped the RAPD and REMAP markers. TH participated in the design of the experiments and produced populations for genetic analysis. MS developed and genotyped the SSR and AFLP markers. YU developed the SSR markers. MN participated in the design of the experiments and wrote the paper. All the authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nakatsuka, T., Yamada, E., Saito, M. et al. Construction of the first genetic linkage map of Japanese gentian (Gentianaceae). BMC Genomics 13, 672 (2012). https://doi.org/10.1186/1471-2164-13-672
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-672