
Abstract
Background
Xanthomonas albilineans causes leaf scald, a lethal disease of sugarcane. X. albilineans exhibits distinctive pathogenic mechanisms, ecology and taxonomy compared to other species of Xanthomonas. For example, this species produces a potent DNA gyrase inhibitor called albicidin that is largely responsible for inducing disease symptoms; its habitat is limited to xylem; and the species exhibits large variability. A first manuscript on the complete genome sequence of the highly pathogenic X. albilineans strain GPE PC73 focused exclusively on distinctive genomic features shared with Xylella fastidiosa—another xylem-limited Xanthomonadaceae. The present manuscript on the same genome sequence aims to describe all other pathogenicity-related genomic features of X. albilineans, and to compare, using suppression subtractive hybridization (SSH), genomic features of two strains differing in pathogenicity.
Results
Comparative genomic analyses showed that most of the known pathogenicity factors from other Xanthomonas species are conserved in X. albilineans, with the notable absence of two major determinants of the “artillery” of other plant pathogenic species of Xanthomonas: the xanthan gum biosynthesis gene cluster, and the type III secretion system Hrp (hypersensitive response and pathogenicity). Genomic features specific to X. albilineans that may contribute to specific adaptation of this pathogen to sugarcane xylem vessels were also revealed. SSH experiments led to the identification of 20 genes common to three highly pathogenic strains but missing in a less pathogenic strain. These 20 genes, which include four ABC transporter genes, a methyl-accepting chemotaxis protein gene and an oxidoreductase gene, could play a key role in pathogenicity. With the exception of hypothetical proteins revealed by our comparative genomic analyses and SSH experiments, no genes potentially involved in any offensive or counter-defensive mechanism specific to X. albilineans were identified, supposing that X. albilineans has a reduced artillery compared to other pathogenic Xanthomonas species. Particular attention has therefore been given to genomic features specific to X. albilineans making it more capable of evading sugarcane surveillance systems or resisting sugarcane defense systems.
Conclusions
This study confirms that X. albilineans is a highly distinctive species within the genus Xanthomonas, and opens new perpectives towards a greater understanding of the pathogenicity of this destructive sugarcane pathogen.
Similar content being viewed by others

Background
Members of the Xanthomonas genus are Gram-negative bacteria belonging to the γ-subdivision of the Proteobacteriae and to the Xanthomonadaceae family. They are exclusively plant-associated bacteria. The plant pathogenic xanthomonads target over 120 monocotyledonous species (including rice, sugarcane, banana, etc.) and over 260 dicotyledonous species (e.g. citrus, cauliflower, cabbage, bean, pepper, etc.), causing dramatic economic losses worldwide. A recently published comprehensive list of plant pathogenic bacteria divided the members of the Xanthomonas genus into 27 species and, at the infra-species level, into over 120 pathovars [1]. Over the past few decades, many attempts have been made to classify members of the Xanthomonas genus using a large variety of biochemical, pathogenicity-based or genomic methods [2–5]. A recent extensive multilocus sequence analysis (MLSA) performed with 119 strains spanning the whole Xanthomonas genus clearly showed that the strains are distributed into two uneven groups, with group 2 containing all but five species, namely X. albilineans, X. sacchari, X. theicola, X. hyacinthi and X. translucens, which were clustered into group 1 [5]. This distribution is consistent both with that based on 16S rDNA sequences [6] and with a distribution based on intergenic sequences between 16S rDNA and 23S rDNA [7]. To date, four strains belonging to group 1 were sequenced, i.e. X. albilineans strain GPE PC73 [8] and Xanthomonas spp. strains NCPPB4393, NCPPB1131 and NCPPB1132 [9]. Xanthomonas spp. strain NCPPB4393 was isolated from an insect collected on a diseased banana plant, but ended by being described as X. sacchari which was also isolated on sugarcane and milled rice [10, 11]. However, no disease caused by this species on any plants has been described to date. Xanthomonas spp. strains NCPPB1131 and NCPPB1132 were both isolated from Musa and belong to unidentified species, although they are phylogenetically related to X. albilineans and to X. sacchari, respectively [9].
Xanthomonas albilineans is the causal agent of leaf scald—one of the major diseases of sugarcane (Saccharum spp.) that occurs in at least 66 countries worldwide [12]. Host plants for this species are restricted to some Poaceae, of which sugarcane is the main known target. Disease symptoms vary from a single, white, narrow, sharply defined stripe to complete wilting and necrosis of infected sugarcane leaves, leading to plant death and large yield losses in the field. X. albilineans produces albicidin, a secreted antibiotic with phytotoxic properties [13]. Albicidin is a potent DNA gyrase inhibitor that targets chloroplastic DNA gyrase A, inhibits chloroplast DNA replication and blocks chloroplast differentiation, resulting in the white foliar stripe symptom [13, 14]. Dissemination of X. albilineans occurs mainly mechanically through use of contaminated harvesting tools and by distribution and planting of infected cuttings. However, aerial transmission and potential for epiphytic survival have also been reported for this pathogen [15–17]. The sugarcane leaf scald outbreaks that occurred in the late 1980s and early 1990s, especially in Guadeloupe [18], Cuba [19], Florida [20], Taiwan [21], Louisiana [22] and Texas [23], were associated with strains of X. albilineans belonging to a specific genetic sub-group known as pulsed-field gel electrophoresis group B (PFGE-B) [24]. The complete genome sequence of strain GPE PC73 (= CFBP 7063) of X. albilineans from Guadeloupe, which belongs to PFGE-B, was published recently (NCBI reference sequence: NC_013722.1). Previous pathogenicity studies identified this strain as highly pathogenic in sugarcane [25]. The genetic information of X. albilineans is encoded on a 3.8-Mb circular chromosome, much smaller than those described to date in other sequenced Xanthomonas (approximately 5 Mb), and on three plasmids of 25, 27 and 32 kb, respectively. The G+C content is 63%, close to values from other sequenced Xanthomonas genomes, and 3115 protein-coding sequences have been annotated manually [8].
Within its sugarcane host, the habitat of X. albilineans is limited to xylem [13, 26]. Our earlier study of the genome sequence of X. albilineans strain GPE PC73 [8] focused on the description of genomic features shared with an evolutionary distant Xanthomonadaceae that is also xylem-limited: Xylella fastidiosa—the causal agent of numerous lethal plant diseases [27]. This first paper, however, did not describe pathogenicity-related features shared by X. albilineans and other species of Xanthomonas. This initial study by Pieretti et al. showed that the two xylem-limited Xanthomonadaceae experienced convergent reductive genome evolution during their descent from a common ancestral parent and possess similar cellulases, which are adapted to the use of plant cell breakdown products as carbon source. These genomic features are not shared with any of the other sequenced species of Xanthomonas. Another genomic feature common to X. albilineans and X. fastidiosa is the absence of a type III secretion system (T3SS) of the Hrp1 and Hrp2 (hypersensitive response and pathogenicity-1 and 2, respectively) injectisome families that are used by most Gram negative phytopathogenic bacteria to deliver bacterial effector proteins or virulence factors into the host plant cell. This latter genomic feature is shared with other sequenced species of group 1 (X. sacchari and the two Xanthomonas spp. isolated on Musa), but not with sequenced species of group 2. The genomic features shared between X. albilineans and X. fastidiosa were proposed to be associated with the cloistered environmental niche of xylem vessels and to adaptation to nutrient-poor xylem elements [8]. However, X. albilineans and X. fastidiosa also differ in several important genomic features: (i) genome erosion was less important in X. albilineans than in X. fastidiosa (890 genes lost by X. fastidiosa were conserved in X. albilineans); (ii) X. albilineans lost xanthan gum biosynthesis genes that were partially conserved by X. fastidiosa; and (iii) only 11 genes are specific to the two xylem-limited Xanthomonadaceae[8]. X. albilineans, while sharing important genomic features with X. fastidiosa, remains closer to xanthomonads than to this distant cousin species from another genus.
The only pathogenicity factor of X. albilineans that has been studied extensively to date is albicidin. Some albicidin-deficient mutants are unable to produce disease symptoms, indicating that this toxin potentially plays an important role in disease expression [13]. However, other albicidin-deficient mutants are still able to produce severe leaf symptoms, indicating that albicidin alone cannot fully explain the pathogenicity mechanism(s) of X. albilineans[28]. Albicidin production in vitro also varies according to the strain of the pathogen [29]. However, no relationship was found between the amount of albicidin produced in vitro and the pathotypes or genetic diversity of the pathogen [29].
The present study of the genome sequence of X. albilineans strain GPE PC73 aimed to identify other virulence factors of X. albilineans in order to gain further insights into the pathogenicity of this sugarcane pathogen. To this end, we listed and described all predicted pathogenicity-related genomic features and compared, by suppression subtractive hybridization (SSH), two strains of X. albilineans that differ in pathogenicity. The genome sequence of X. albilineans strain GPE PC73 was compared to eight available genome sequences of Xanthomonas X. axonopodis pv. vesicatoria strain 85–10 [30], X. axonopodis pv. citri strain 306 [31], X. campestris pv. campestris strain ATCC 33913 [31], X. campestris pv. vasculorum NCPPB702 [32], X. campestris pv. musacearum NCPPB4381 [32], X. oryzae pv. oryzae MAFF 311018 [33], X. oryzae pv. oryzicola strain BLS256 [34] and X. sacchari[9]]. Genomic features shared between these eight genomes and X. albilineans strain GPE PC73 were surveyed in order to better understand the strategies developed by X. albilineans to compensate for the absence of the xanthan gum biosynthesis genes and the type III secretion system Hrp. In the rather cloistered environmental niche inside xylem vessels, X. albilineans may have largely avoided surveillance by general and specific plant defense systems. The absence of T3SS Hrp may be partially palliated due to the xylem-limited lifestyle of X. albilineans, as this pathogen lives and multiplies essentially in a dead-cell environment. However, like other bacterial vascular pathogens, X. albilineans may interact with living xylem parenchyma cells through pit membranes [35]. Genomic features of X. albilineans strain GPE PC73 were also analyzed to better understand the adaptation of X. albilineans to the nutrient-poor xylem elements within xylem vessels.
Results and discussion
Secretion systems
To date, six different types of secretion systems in Gram-negative bacteria have been described, differing both in their structures and in the molecules transferred, which can be proteins, including both peptides and enzymes, and other small and large molecules, including DNA [36]. The secreted molecules can play an important role in cellular homeostasis and even in bacterial lifestyles: some are literally injected into host cells in which they modify host physiology to promote colonization.
In order to establish themselves successfully in their hosts, Xanthomonas species rely on the presence of various types of secretion systems [37, 38]. The type I secretion system (T1SS) is required for secretion of a variety of degradative enzymes and offensive molecules, including antibiotics and molecules involved in plant or animal pathogenicity. These include a variety of hydrolases (proteases, phosphatases, esterases, nucleases and glucanases) and proteic toxins (hemolysins or bacteriocins) [39, 40]. T1SS utilises an outer membrane protein factor, TolC, which traverses both the periplasm and the outer membrane [40–43]. TolC is also essential for the action of multidrug resistance (MDR) efflux pumps, and was shown to be required for pathogenicity of X. fastidiosa[44] and for secretion of an elicitor in X. oryzae pv. oryzae[45]. The gene encoding TolC in X. albilineans (XALc_0747) shares 32% amino acid identity with the tolC gene of Escherichia coli strain K12, and more than 61% amino acid identity with the TolC protein of X. fastidiosa or other xanthomonads. Like other xanthomonads, and unlike X. fastidiosa, X. albilineans carries other outer membrane factor genes sharing some similarities with tolC. These are located in the same genomic regions as genes encoding MDR efflux pumps. They share less than 20% amino acid identity with the tolC gene of E. coli. Six of these genes are conserved in other Xanthomonas strains (XALc_0344, XALc_1555, XALc_2009, XALc_2167, XALc_2468 and XALc_3155) whereas a seventh is specific to X. albilineans (XALc_1770).
The type II secretion system (T2SS) secretes cell wall-degrading enzymes like cellulases and xylanases. Two independent T2SS gene clusters, Xps and Xcs, are apparently dedicated to distinct substrates and roles in the genus Xanthomonas[31, 46]. Both are present in X. axonopodis pv. vesicatoria, X. campestris pv. campestris, X. axonopodis pv. citri, X. fuscans subsp. aurantifoli, X. gardneri and X. perforans. Xcs is missing in the X. oryzae pv. oryzae and X. oryzae pv. oryzicola genomes, whereas only two genes of the cluster (xcsN and xcsM, respectively) are conserved in X. campestris pv. vasculorum and X. campestris pv. musacearum. The chromosome of X. albilineans strain GPE PC73, like the chromosome of X. sacchari, possesses only the xps gene cluster, which contains 11 genes annotated as xpsD to xpsN (XALc_2654 to XALc_2664). The Xps T2SS of X. axonopodis pv. vesicatoria has been shown to be involved in pathogenicity [46]. The contribution to virulence of X. albilineans of the Xps T2SS is so far unknown, but 364 genes are found in the genome that encode proteins predicted to be T2SS-secreted using SignalP 4.0 server http://www.cbs.dtu.dk/services/SignalP/ (see Additional file 1). They include 159 hypothetical proteins as well as many additional proteins known to be secreted by a T2SS.
Type III secretion systems (T3SS), also called injectisomes, are molecular syringes that deliver into the cytoplasm of host cells effector proteins capable of targeting and disrupting key functions of the host. Seven T3SS families have been described [47], but phytopathogenic bacteria usually rely on T3SS from the Hrp families to fulfil their pathogenicity. The remarkable absence of a T3SS-Hrp in the genome of X. albilineans implies that secretion of effectors interacting with sugarcane cells relies on other secretion systems. Interestingly, a T3SS gene cluster belonging to the SPI-1 (for Salmonella Pathogenicity Island-1) injectisome family, found mainly in animal pathogens and insect symbionts, is present in the genome of X. albilineans strain GPE PC73 (XALc_1472 to XALc_1511) near the termination site of chromosome replication (Figure 1). Genomic and evolutionary features of the T3SS SPI-1 of X. albilineans were described recently [48]. This system, which is partially conserved in X. axonopodis pv. phaseoli, shares only low similarity with other available T3SS SPI-1 sequences but encodes all components required for secretion [48]. However, functional analysis with knockout insertional mutants showed that the T3SS SPI-1 is not required by X. albilineans to spread within the xylem and to cause disease. Involvement of a T3SS SPI-1 in adherence to plant surfaces was proposed for the strictly epiphytic Erwinia tasmaniensis strain Et1/99 [49] suggesting that the T3SS SPI-1 of X. albilineans may be involved in leaf surface colonization. Alternatively, T3SS SPI-1 may play an important role in association with an animal host, although insect vectors of X. albilineans have not yet been reported.

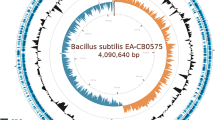
Circular representation of the chromosome of strain GPE PC73 of X. albilineans . The scale is shown in kbp in the center and around the periphery. Boxes: Albicidin biosynthesis gene cluster (yellow), other large NRPS loci (orange), T3SS SPI-1 gene cluster (red) and flagellum gene cluster (blue). Arrows: 12 copies of the 5-kb gene cluster present at 12 different locations of the chromosome (purple), locus CRISPR-1 (green), locus CRISPR-2 (sky blue) and short NRPS loci containing only a short NRPS gene (orange). The grey circle shows the GC skew (G-C)/(G+C) using a 100-base window. The black circle shows the G+C content using a 100-base window
The type IV secretion system (T4SS) has been described as an important bacterial factor helping bacterial adaptation to new hosts [50]. A diversity of structures and functions has been reported as well as the versatile nature of this system, which can be used to mediate horizontal gene transfer or to secrete virulence factors [51]. In Agrobacterium and Helicobacter, VirB2 and VirB5 form an extracellular pilus that was proposed to be an adhesion-like protein susceptible to interact with specific host-cell receptors [52]. A recent study demonstrated that the T4SS of Bartonella mediates host-specific adhesion to erythrocytes [53]. The chromosome of X. albilineans strain GPE PC73 possesses a T4SS, which may correspond to an ancestral gene cluster because genes encoding this system share 53–83% amino acid identity with genes located at the same position in other xanthomonads, i.e. downstream of the same uvrB gene (XALc_1870) and the same valine tRNA (XALc_1869). However, this T4SS seems to be complete only in X. axonopodis pv. citri, in which the protein encoded by XAC2622 was characterized structurally recently by NMR and X-ray crystallography as an outer membrane transport protein, confirming that this protein corresponds to VirB7 [54]. The T4SS gene cluster of X. albilineans strain GPE PC73 possesses the same virB7 gene (XALc_1843) and contains a phage-related sequence (XALc_1844 to XALc_1866) but lacks both the virB5 gene and the virD4 gene that encodes an NTPase required to power the conjugation/secretion apparatus. Nonetheless, four putative virD4-like genes (XALc_2330, XALc_2379, XALc_2612 and XALc_3055) are present on the chromosome in a repeated region (see below and Figure 2) and an additional putative virD4-like gene (XALr_3251) is present on one of the three plasmids, suggesting that trans-complementation of this gene might occur. Still, no functional T4SS involving a VirD4 encoded elsewhere in the genome (i.e. not encoded in the T4SS gene cluster) has been described in any bacterium to date. The missing virB5 gene may also be complemented by virB5-like genes present elsewhere in the genome. Each of the three plasmids present in X. albilineans strain GPE PC73 harbors an incomplete conjugal transfer system similar to a T4SS, which includes a VirB5-like protein (XALp_3186, XALq_3225 and XALr_3258). Another putative virB5- like gene (XALc_2643) is present in a phage-related region of the chromosome.

Representation of the 12 copies of the 5-kb gene cluster present at 12 different locations within the chromosome of strain GPE PC73 of X. albilineans. The light-coloured boxes correspond to the ORFs of the 5-kb gene cluster. They are bordered by unframed plain-coloured boxes assigned to phage, IS transposases, virD4, Rhs or other specific sequences. Similar ORFs (orthologs) are represented by the same coloured squares. The dotted line illustrates two non-adjacent ORFs. The blue and the red stars above the green framed boxes show positions of the serine from the triade Ser-His-Asp and of the Poly-S locations, respectively. MTase: DNA methyltransferase. MTase-XamI: XamI DNA methyltransferase-restriction endonuclease system
Type V secretion systems (T5SS) form a heterogeneous family of transporters for non-fimbrial adhesins. Three subtypes have been described for this family: autotransporter, trimeric autotransporter and two-partner system (TPS) [37, 55, 56]. Since we have identified genes encoding non-fimbrial adhesins in the genome of X. albilineans GPE PC73 (see below), one might expect the occurrence of at least one T5SS.
In contrast to most of the xanthomonad species, X. albilineans strain GPE PC73 does not possess a type VI secretion system (T6SS). The genome of X. oryzae pv. oryzae strain MAFF 311018 contains two T6SS gene clusters, while the genome of X. axonopodis pv. vesicatoria strain 85–10 harbours only one copy split over two loci [57]. Despite T6SSs being found in pathogenic and nonpathogenic bacteria, this system has been shown to be essential for pathogenicity in many animal pathogens [58, 59].
In X. albilineans, four secretion systems are potentially involved in secretion of pathogenicity effectors, namely T1SS, T2SS, T5SS and T3SS SPI-1. This latter system may be involved in interactions with an animal host rather than with sugarcane. In X. oryzae pv. oryzae, the elicitor Ax21, formerly known as AvrXa21, has been described as being secreted by a T1SS [60, 61]. As with Ax21, further functional analysis and biochemical experimentation will be necessary to identify such effectors in X. albilineans. Small molecules, which are secreted by specific transporters usually encoded within their biosynthesis gene clusters, are also good candidates for secretion into the xylem by X. albilineans. These could then diffuse into targeted adjacent living parenchymal cells and act as pathogenicity effectors, or may enter plant cells by hijacking nutrient transporters.
Nonribosomal peptide synthetase genes
Bacteria use nonribosomal peptide synthetases (NRPSs) to produce peptides or small molecules of broad structural and biological activity that can contribute to virulence, adaptation to unfavorable environments or competition with rival microorganisms in their natural habitat (for reviews, [62, 63]). Small molecules synthesized by NRPS may play an important role in the pathogenicity of X. albilineans. This assumption is based on the presence in the genome of X. albilineans strain GPE PC73 of 12 genes encoding NRPSs comprising 4% of the chromosome (Figure 1). Three of these NRPS genes belong to the albicidin biosynthesis gene cluster XALB1, which was sequenced previously from X. albilineans strain Xa23R1 [64]. Albicidin, which is secreted by X. albilineans using a specific transporter encoded by XALB1, can enter sugarcane chloroplasts using nucleoside transporters similar to the nucleoside transporter Tsx, which mediates albicidin uptake in E. coli[65]. The structure and function of small molecules synthesized by the nine other NRPS genes identified in the genome of X. albilineans strain GPE PC73 are currently unknown. In silico features of these nine new NRPS genes were analyzed and seven of these genes are predicted to share features with NRPS genes identified in the genome of other Xanthomonas spp. strains also associated with monocotyledonous species (X. oryzae pv. oryzae, X. oryzae pv. orizycola and X. translucens) (Royer et al., unpublished data). This suggests that unknown small molecules synthesized by these NRPS gene clusters are involved in specific interaction with these plants (Royer et al., unpublished data). Furthermore, four of these new NRPS genes are grouped in a gene cluster encoding a specific transporter, suggesting that at least one small molecule synthesized by this gene cluster is secreted and may interact with sugarcane cells. Small molecules synthesized by NRPS that act as surfactants have been described for Pseudomonas syringae, which is found in various environments and can survive on leaf surfaces as an epiphyte by producing several surfactants synthesized by NRPSs [66, 67]. NRPSs have also been described as being involved in the biosynthesis of siderophores [68]. These iron-chelating structures are used widely by phytopathogenic bacteria to achieve survival in iron-depleted environments or to out-compete other bacterial strains in the same environment [69]. To date, NRPS genes are the only genes identified in the genome of X. albilineans strain GPE PC73 that can be considered as good candidates to produce the small molecules required for interactions with sugarcane cells.
Cell wall degrading enzymes
The genome of X. albilineans strain GPE PC73 exhibits 19 genes encoding putative cell wall degrading enzymes (e.g. cellulases, polygalacturonases, rhamnogalacturonases, beta-glucosidases and xylanases), all of which being predicted to be secreted by the Xps-T2SS (see Additional file 1). As previously described, X. albilineans and X. fastidiosa possess similar enzymes that help adapt these organisms to use plant cell breakdown products as carbon sources [8]. Such enzymes include endoglucanase EngXCA and 1,4-beta cellobiosidase CbhA which, in both of the xylem-limited Xanthomonadaceae, but not in other Xanthomonadaceae, have a cellulose binding domain (CBD) at their C-terminal extremity, and a long linker region consisting of simple or repetitive sequences rich in proline, threonine, serine, or glycine. Such long linker regions are known to enhance substrate accessibility [70, 71], indicating that these enzymes are particularly well adapted to the degradation of carbohydrate substrates.
X. albilineans possesses one copy of the 1,4-beta cellobiosidase cbhA gene (XALc_0484). Interestingly, the 1,4-beta cellobiosidase cbhA gene is missing in the non vascular Xanthomonas species (X. oryzae pv. oryzicola, X. axonopodis pv. citri, X. axonopodis pv. vesicatoria and other sequenced xanthomonads including X. sacchari) but is conserved in the xylem-invading Xanthomonas species (X. oryzae pv. oryzae, X. campestris pv. campestris, X. campestris pv. vasculorum and X. campestris pv. musacearum). However, in the latter species, CbhA has neither a linker region nor a CBD (Figure 3). The cbhA gene was shown to contribute to virulence of the xylem-invading pathogen Ralstonia solanacearum[72]. The presence of a cbhA gene in the xylemic xanthomonads X. fastidiosa and R. solanacearum suggests that this enzyme is absolutely required for spread within xylem vessels. A specific feature of these vessels is that they are interconnected by channels, called bordered pits, which allow the passage of xylem sap but block the passage of larger objects due to the presence of a pit membrane. This pit membrane constitutes the primary cell wall barrier that separates adjacent xylem water conduits, limiting in particular the colonization of the plant by a pathogen [73]. The movement of X. fastidiosa within the plant is an active process and appears to depend on its ability to disrupt pit membranes (for a review, see [27]). The pit membrane is composed of pectin and cellulose microfibrils [74], and may require several enzymes for complete dissolution. The presence of a cbhA gene in the xylemic xanthomonads, X. fastidiosa and R. solanacearum, might indicate that this enzyme is required for degradation of the pit membrane. X. albilineans possesses two polygalacturonase genes (XALc_0811 and XALc_1916) that are conserved in all sequenced Xanthomonas and in R. solanacearum and potentially required for degradation of pectin. XALc_0811 is not conserved in X. fastidiosa. XALc_1916 is not conserved in strain 9a5c of X. fastidiosa (isolated from citrus), but is conserved in strain Temecula1 of X. fastidiosa (isolated from grapevine) where it was shown to be required for colonization and pathogenicity of this strain in grapevine [75].

Comparison of four cell wall degrading enzymes encoded by Xanthomonadaceae species. Sacch: X. sacchari strain NCPPB4393; XOC: X. oryzae pv. oryzicola strain BLS256; XCC: X. campestris pv. campestris strain ATCC 33913; XCV: X. campestris pv. vesicatoria strain 85–10; XAC: X. axonopodis pv. citri strain 306; XOO: X. oryzae pv. oryzae strain MAFF 311018; Xva: X. campestris pv. vasculorum strain NCPPB702; Xmu: X. campestris pv. musacearum strain NCPPB4381; XAL: X. albilineans strain GPE PC73; Xyl 9a5c: X. fastidiosa strain 9a5c; XYL Temecula 1: X. fastidiosa strain Temecula1. Blue boxes: linker regions consisting of simple or repetitive sequences rich in proline, threonine, serine, or glycine; box length is proportional to the length of linker region (number of amino acids of each linker region is indicated inside each box). Orange boxes: cellulose binding domains (CBD). Aa = amino acids
The genome of X. albilineans strain GPE PC73 contains two copies of the endoglucanase engXCA gene (XALc_2969 and XALc_2967). Endoglucanase EngXCA is conserved in all other Xanthomonas and also possesses a linker region and a CBD, but the linker region in these species is much smaller than that in X. albilineans and X. fastidiosa (Figure 2). Interestingly, the genome of X. albilineans strain GPE PC73 encodes two additional enzymes harboring a long linker region and a CBD at their C-termini, namely cellulase CelS (XALc_0865) and a putative secreted protein (XALc_0874), which may be considered as putative cell wall degrading enzymes because of the presence of a CBD. The cellulase gene celS has a frameshift mutation in X. axonopodis pv. citri, is not present in X. fastidiosa and is conserved in seven other sequenced genomes of Xanthomonas, but it does not possess any linker region or any CBD in these species. The putative cell wall degrading enzyme XALc_0874 is absent in both X. campestris pv. campestris and X. fastidiosa. It is, however, present in the seven other sequenced genomes of Xanthomonas, but does not possess any linker region or any CBD in these species (Figure 3). The presence of five genes that encode enzymes harboring a long linker region and a CBD at their C-termini indicates that X. albilineans is adapted to the utilization of cell breakdown products as a carbon source. Xylem is a water transport network of vessels composed of dead, lignified cells. Xylem sap is therefore rich in cell-wall breakdown products. X. albilineans strain GPE PC73 encodes putative sugar transporter systems, further supporting the concept that cell-wall derived sugars are consumed by this pathogen.
TonB-dependent transporters
Outer membrane TonB-dependent transporters (TBDT) are involved in the active transport of plant nutrients, more precisely iron-siderophore complexes, vitamin B12, nickel or other large molecules such as plant carbohydrates [76, 77]. A large proportion of TBDT genes were described as related to carbohydrate utilization in X. campestris pv. campestris[76]. There is a large variation in the number of TBDT genes among xanthomonads, even at the infra-species level. This number does not seem to be linked to genome size but rather to the ecological niche and lifestyle of the species considered [77]. Seventy-two TBDT genes were identified in the genome of X. campestris pv. campestris, of which only nine were assigned to iron uptake; several others were associated with plant carbohydrate utilization and the pathogenicity of this species [76]. Thirty-five putative TBDT genes were found in the chromosome of X. albilineans strain GPE PC73, including 26 canonical TBDTs and five TBDT/Oar-like genes. Orthologous or paralogous genes were found in other sequenced genomes of Xanthomonas, except for gene XALc_1949 which encodes a non-canonical TBDT/Oar-like protein specific to X. albilineans and for gene XALc_2962 which encodes a canonical TBDT specific to X. albilineans and X. sacchari. The presence of 35 TBDT genes in X. albilineans indicates that this species, like other species of Xanthomonas, is adapted to plant scavenging and to living in nutrient-poor environments. A recent transposon mutagenesis study of strain XaFL07-1 of X. albilineans revealed the presence of two pathogenicity-related TBDT loci (XALc_0643 and XALc_0723) involved in disease severity and extent of stalk colonization, although orthologous genes of these two TBDTs have not yet been reported as pathogenicity factors in other xanthomonads [28]. Further studies will be necessary to characterize the nature of the nutrients transported by these two TBDTs. XALc_0643 is present in a putative operon upstream from another TBDT (XALc_0646) and a putative nucleoside hydrolase. XALc_0723 is present just downstream of a putative HpcH/HpaI aldolase involved in the (d)-glucarate/galactarate catabolic pathway. These three TBDTs (XALc_0643, XALc_0646 and XALc_0723) may be adapted to the transport of carbohydrate products provided by the five cellulases specific to X. albilineans.
Lipopolysaccharides
Lipopolysaccharides (LPS) are essential for protecting the cell from hostile environments. They can also play a direct role in interactions between bacteria and eukaryotic host cells. In animal pathogenic bacteria, lps loci involved in LPS biosynthesis are under host selection, with large variations in the lps gene cluster being found [78]. Similarly, the lps loci of plant pathogenic bacteria are also under selection, especially to escape host defense responses. Bacterial LPSs have been found to act as elicitors of plant innate immunity. In Xanthomonas axonopodis pv. citri, the O-antigen moiety of the LPS has been shown recently to act as a PAMP (Pathogen-Associated Molecular Pattern) and therefore to activate the basal response of the attacked plant, in particular by inducing the expression of defense-related genes and promoting callose deposition, the latter being accompanied by an oxidative burst [79]. Comparison of the LPS biosynthetic gene clusters of Xanthomonas species shows a high variability in the number and identity of genes [80, 81]. Multiple horizontal gene transfer events are considered to be responsible for the high variation in LPS biosynthetic gene clusters between various xanthomonads [82]. LPS clusters are bordered by the highly conserved etfA and metB genes in all xanthomonads sequenced to date. In X. albilineans strain GPE PC73, the LPS gene cluster is also bordered by etfA (XALc_2699) and metB (XALc_2712), and comprises 12 genes (XALc_2700 to XALc_2711), five of which are specific to X. albilineans (XALc_2700 to XALc_2704). Seven genes (XALc_2705, XALc_2706 (gmd), XALc_2707 (rmd), XALc_2708, XALc_2709, XALc_2710 (xzm) and XALc_2711) show the highest similarity with genes from the vascular sugarcane pathogen X. campestris pv. vasculorum (see Additional file 2). The relatedness of LPS-encoding genes between X. albilineans strain GPE PC73 and X. campestris pv. vasculorum is incongruent with their phylogenetic relationship, which separates the two pathogens into two distinct and distant clades [5]. In the draft genome sequence of X. campestris pv. vasculorum, the LPS cluster is found at the borders of two contigs (Additional file 2). One contig contains seven genes sharing the highest similarity with genes from X. albilineans; this contig additionally contains an insertion sequence (IS), which probably explains why the complete LPS cluster could not be assembled in one contig. The occurrence of this IS suggests recent horizontal transfer of LPS genes from X. albilineans to X. campestris pv. vasculorum, both bacterial species spreading in the xylem of sugarcane. Interestingly, the seven LPS-encoding genes exhibiting the highest similarity with X. albilineans are involved in sugar metabolism or transport and thus, are thought to be involved in O-antigen biosynthesis. Genes shared by X. campestris pv. vasculorum and X. albilineans may therefore be required for biosynthesis of specific LPSs that are adapted to interactions with sugarcane and are possibly not recognized as PAMPs in this plant. Interestingly, the complete LPS gene cluster is highly conserved between X. sacchari and X. campestris pv. vasculorum. Recently, it was shown that Tn5-mutants of X. albilineans in XALc_2705 or XALc_2707 were affected in production of disease symptoms and in their capacity to spread within the sugarcane stalk xylem [28].
Exopolysaccarides
Extracellular polysaccharides (EPS) contribute to the virulence of xanthomonads, in particular by the formation of biofilm [83, 84]. The major EPS produced by Xanthomonas spp. is called xanthan gum, the production of which is acomplished by a cluster of 12 genes (annotated as gumB through gumM). X. fastidiosa contains nine out of the 12 gum genes [85] and the complete cluster is present in X. sacchari genome. However, the genome of X. albilineans strain GPE PC73 harbors no gum gene at all [8]. Furthermore, occurrence of biofilm in the xylem of sugarcane infected by X. albilineans has not been reported to date.
X. albilineans is the only species of Xanthomonas to lack the xanthan gum gene cluster, although this pathogen harbors genes involved in nucleotide sugar biosynthesis using "raw material" during the biosynthesis of carbohydrates found on the bacterial cell surface. Among these genes, some, such as the two key genes xanA, encoding a phosphoglucomutase (XALc_2692), and xanB encoding a phosphomannose isomerase (XALc_2693), are involved in biosynthesis of xanthan. Other EPS genes, such as pgi, galU and ugd, which encode a glucose-6-phosphate isomerase, an UTP-glucose-1-phosphate uridylyltransferase and an UDP-glucose 6-dehydrogenase, respectively, are also present on the chromosome of X. albilineans strain GPE PC73. The role of these genes in biosynthesis of EPS and pathogenicity of X. albilineans remains to be investigated. However, it has already been shown that mutation of xanB results in an incapacity to produce symptoms and to colonize the sugarcane stalk [28].
Recently, the polysaccharide biosynthesis operon XagABC from X. campestris pv. campestris was found to be involved in protection against oxidative damage [86]. The xag gene cluster may be involved in biofilm formation during the first stage of infection to favor bacterial survival and systemic infection in planta. Through quorum sensing during bacterial growth, this cluster is expected to be down-regulated and relayed by up-regulation of manA (xanB in X. albilineans)—a gene that is also involved in biofilm formation in the later systemic stage of infection [86]. Interestingly, the XagABC operon, which occurs in other xanthomonads, was not identified in the genomes of X. albilineans and X. fastidiosa.
Despite the absence of gum genes, the production in planta of a xanthan-like polysaccharide by X. albilineans strain NCPPB887 has been reported. This polysaccharide, which was purified from diseased sugarcane plants, is formed by repeated tetrasaccharide motifs, each formed by two molecules of glucose, one of mannose, and one of glucuronic acid [87]. The occurrence of glucuronic acid in this xanthan-like polysaccharide was thought to result from the activity of a protease-sensitive UDP-glucose-dehydrogenase. This enzyme, whose N-terminal protein sequence starts with IQPYNH, was purified from sugarcane plants infected by X. albilineans strain NCPPB887 [87]. It was hypothesized that X. albilineans produces proteases that inhibit this UDP-glucose-dehydrogenase, thus preventing glucuronic acid synthesis and production of the xanthan-like polysaccharide, as observed in vitro[87, 88]. To explain the production in planta of this polysaccharide, it was suggested that glycoproteins produced by sugarcane in response to infection by X. albilineans act as powerful inhibitors of proteases synthesized by the pathogen, thus preventing degradation of the UDP-glucose-dehydrogenase and permitting the production of the xanthan-like polysaccharide. As a consequence, this polysaccharide can be produced only in sugarcane plants infected by X. albilineans. The genome of X. albilineans strain GPE PC73 contains two UDP-glucose-dehydrogenase genes that are conserved in all sequenced Xanthomonadaceae (XALc_1655 and XALc_1695), but no gene with an N-terminal or internal IQPYNH sequence could be identified. Further studies are therefore needed to decipher the mechanisms of biosynthesis of this polysaccharide and to confirm its bacterial origin in sugarcane plants infected by the leaf scald pathogen. Indeed, this polysaccharide may be produced by sugarcane to limit spread of X. albilineans.
Lack of production of xanthan gum may be a crucial advantage for X. albilineans, allowing it to spread within xylem vessels without obstructing them. Complete obstruction of xylem may lead to rapid death of the infected sugarcane and may be unfavorable for X. albilineans, which is transmitted mainly by infected cuttings.
Flagellum and chemotaxis
X. albilineans cells, like other Xanthomonas spp., carry a single polar flagellum [13]. This flagellum is involved in swimming motility and constitutes a motor organelle that propulses a bacterium through its environment to find optimal conditions via chemotaxis (allowing the bacterium to find nutrients or avoid toxic molecules). Chemoreceptors, also called Methyl-accepting Chemotaxis Proteins (MCPs), are associated with the bacterial membrane and are sensitive to various chemical signals to modulate the rotational direction of the flagellum [89, 90]. Two sets of genes encoding flagellar assembly (Figure 1) and chemotaxis-related proteins are contiguous in the chromosome of X. albilineans strain GPE PC73. The first set of genes, encoding the polar flagellum, shows high homology with the corresponding genes in the other Xanthomonas spp. Interestingly, the FliD protein from X. albilineans strain GPE PC73 (XALc_1416) harbors a polyserine linker (PSL) that is absent in the corresponding protein encoded by other Xanthomonas spp. including X. sacchari and also strains NCPPB1131 and NCPPB1132. FliD is the filament-capping protein that forms a cap at the tip of the growing filament structure, which helps the folding process and promotes flagellin subunit insertion and polymerization during helical filament growth [91–93]. FliD is implicated in the virulence of many pathogenic bacteria [94]. In some species, the structural diversity of the cap protein has been reported to be a mean of escaping the host immune system [95]. Indeed, among Pseudomonas aeruginosa strains, two distinct FliD proteins were observed, with differences in their primary amino acid sequences resulting in distinct conformations of the flagellum. These two types of FliD proteins differ immunologically. They may play a role in escaping host defenses and, more likely, be responsible for differential binding of strains to respiratory mucins, suggesting adaptability during infections ranging from acute to chronic respiratory infection of cystic fibrosis patients [95]. In X. albilineans strain GPE PC73, the presence of a PSL of 12 serine residues in the FliD protein may modify the tertiary structure of the flagellar cap protein when compared with other Xanthomonas and, as a consequence, may modify its detection by the sugarcane defense system or be related to a specific interaction with its host.
The second set of genes, encoding chemotaxis-related proteins (from XALc_1353 to XALc_1378), contains five tandemly-repeated genes encoding MCPs, 12 other chemotaxis proteins (Che-like proteins) and six hypothetical proteins (see Additional file 3). In other Xanthomonas spp., the number of MCPs is higher, especially in X. axonopodis pv. vesicatoria (14 tandemly-repeated MCP genes) [30] (Additional file 3). Although the majority of genes encoding chemotaxis receptors proteins are found within a genomic cluster dedicated to chemotaxis and mobility [30, 90], some MCP-encoding genes are found scattered on the chromosome. In X. albilineans strain GPE PC73, nine additional MCPs were found dispersed on the chromosome (XALc_2151 to 2153, XALc_0649, XALc_0760, XALc_1440, XALc_1926, XALc_2239 and XALc_3131, respectively).
Bacterial pili or fimbriae
Bacterial pili or fimbriae are proteinaceous multi-subunits structures forming filamentous cell-surface appendages involved in various bacterial virulence processes, including adhesion, biofilm formation, twitching motility, cellular invasion or protein and DNA transport across membranes (DNA uptake during transformation, phage transduction) [96–98]. Like other Xanthomonas sequenced to date, the complete genome sequence of X. albilineans strain GPE PC73 revealed the presence of a Chaperone-Usher (CU) pilus. Genes involved in fimbriae assembled by the CU-dependent pathway are clustered in an operon together with a periplasmic chaperone (XALc_2025), a predicted outer membrane protein corresponding to the assembly platform called “usher” (XALc_2022) and two candidate structural fimbrial subunits (annotated as hypothetical proteins XALc_2023 and XALc_2021, although they do contain a spore coat U domain also found in pili proteins). These genes share at most 47% overall amino acid identity with their counterparts from other Xanthomonas species. Furthermore, like all other sequenced Xanthomonas, the genome of X. albilineans strain GPE PC73 contains genes able to encode a type IV pilus. Biogenesis of a type IV pilus involves a large number of proteins that are highly conserved within the Xanthomonas genus, with the notable exception of the PilA pilin and the PilV-W-X-Y-E operon. The PilA protein is the main structural protein of the type IV pilus. The sequence variability of the pilA gene observed among the sequenced Xanthomonas could be correlated with host specificity [99]. In X. fuscans subsp. fuscans strain CFBP4834-R, PilA is involved in adhesion and transmission to seeds, and mutation of pilA resulted in reduced pathogenicity on bean [100]. Regarding the PilV-W-X-Y-E operon, a low identity is observed among the whole pool of sequenced xanthomonads. PilQ protein has a kind of intermediate status: it shares over 94% of amino acid sequence identity between all sequenced Xanthomonas, with the remarkable exception of X. albilineans, the PilQ protein of which showing less than 77% identity. PilQ in X. oryzae pv. oryzae plays a critical role in virulence, twitching motility and biofilm formation [101], but has no effect on leaf attachment or entry into the host plant [98]. Type IV pilus proteins specific to X. albilineans (the PilA pilin and the PilV-W-X-Y-E operon) may be adapted specifically to the xylem of sugarcane. Interestingly, X. fastidiosa, which does not possess any flagellum, has both type I and type IV pili at the same pole. Type I (also called Chaperone-Usher), corresponding to the shorter pilus, is involved in adhesion and biofilm formation, whereas the longer type IV pilus is also involved in upstream migration of X. fastidiosa against the xylem flow rate [102]. However, the Chaperone-Usher pilus from X. fastidiosa seems to differ from that found in xanthomonads. Low sequence conservation and a different gene syntheny suggest that they may play different roles [103].
Non-fimbrial adhesins
Non-fimbrial (or afimbrial) adhesins are single proteins located on the bacterial cell surface, which are secreted via one of three subtypes of the type V secretion system (T5SS) (monomeric autotransporter, trimeric autotransporter and two-partner system [55, 104, 105]). Non-fimbrial adhesins play a role in attachment and infection processes and, more widely, promote the virulence of phytopathogenic bacteria. Several type V-secreted adhesins are duplicated, frameshifted or truncated in the various sequenced Xanthomonas genomes. As a consequence, this genetic variability can lead to misannotated genomes and many predicted adhesins are probably not functional [103, 106]. Among all the afimbrial adhesin genes identified in the sequenced Xanthomonas, only the following orthologs have been found in the genome of X. albilineans strain GPE PC73: XALc_2666 encoding XadA, XALc_2291 encoding an hemolysin protein close to FhaC and XALc_2290 encoding an hemagglutinin/hemolysin protein close to FhaB (followed by XALc_2288 encoding a truncated FhaB form). Moreover, unlike some species of Xanthomonas in which a few paralogs of this locus exist that could play specific roles during infection and dissemination [107], X. albilineans possesses only one XadA locus. This is illustrated by XadA1 and XadA2 from X. axonopodis pv. phaseoli in which only paralog XadA2 is involved in vascular transmission to bean seeds [100]. Furthermore, although the genome sequence of X. albilineans strain GPE PC73 does not possess a gene encoding the filamentous hemagglutinin YapH that is found in other Xanthomonas spp., it encodes two specific non-fimbrial adhesins (XALc_1305 and XALc_1884) matching proteins encoded by Agrobacterium radiobacter strain K84 with 54 and 42 percent of amino acid identities, respectively. These non-fimbrial adhesins of X. albilineans may play a role in epiphytic survival or in xylem colonization of sugarcane, thus corroborating the adaptation of this pathogen to its specific host and lifestyle. Indeed, a recent study showed that adhesins play a role in the adaptation of xanthomonads to their host plant [107].
Quorum sensing genes
The rpf (for regulation of pathogenicity factors) gene cluster is involved in cell-cell signaling and control of various cellular processes [108]. The rpf cluster, first characterized in X. campestris pv. campestris by [109], comprises nine genes (annotated as rpfA through rpfI) involved in biosynthesis and detection of DSF (for Diffusible Signal Factor). DSF is a signaling molecule that plays a main role in regulation of the expression of genes required for production of extracellular polysaccharides, biofilm formation and colonization in planta[110]. DSF biosynthesis is governed mainly by rpfF—a gene encoding an enoyl-CoA hydratase—whereas both rpfC (encoding a hybrid two-component DSF sensor) and rpfG (encoding a two-component regulator) are implicated in DSF perception and signal transduction, respectively. The rpf genes are unique and specific to the xanthomonads X. fastidiosa and S. maltophilia[90, 111], although a Burkholderia cepacia gene showing 37% identity at the peptide level with rpfF from X. campestris pv. campestris has been shown recently to be involved in biosynthesis of a DSF functional analog called BDSF [111]. In X. albilineans and X. fastidiosa, rpfD, rpfH and rpfI are missing. Similarly, X. axonopodis pv. citri does not possess rpfH and rpfI[31]. The RpfH protein is related structurally to the sensory input domain of RpfC [112]. The rpfD gene encodes a transcriptional regulator and rpfI encodes a protein that positively regulates the biosynthesis of proteases, endoglucanases and EPS in X. campestris pv. campestris[113]. Even if the rpf gene cluster appears to be incomplete, it plays a key role in pathogenicity and insect transmission of X. fastidiosa[27, 114]. DSF is also produced by X. albilineans[115], and the importance of this molecule in the pathogenicity of this species is currently under investigation. Studying the regulatory Rpf/DSF pathway by comparing gene expression in the wild type strain versus an rpf mutant may be a promising approach with which to identify pathogenicity-related genes of X. albilineans.
Two-component signal transduction systems
The genomes of Xanthomonas spp. encode numerous two-component signal transduction systems (TCSTSs). These molecular sense-response mechanisms are able to act to regulate the expression of genes involved in cell motility, virulence, biofilm formation, chemotaxis, quorum-sensing and many others cellular processes (e.g. [116, 117]). It has been suggested that the number of these signaling proteins occurring in the genome reflects the adaptation of xanthomonads to stochastic environmental changes and heterogeneous ecological niches [116, 118]. We identified 98 two-component system proteins in the chromosome of X. albilineans strain GPE PC73 (Additional file 4). These proteins include 35 two-component system sensor proteins, 45 two-component system regulatory proteins and 18 two-component system sensor-response regulator hybrid proteins. The number of TCSTSs identified in X. albilineans is lower than that found in X. axonopodis pv. vesicatoria strain 85–10 (121 genes) but higher than the number found in X. oryzae pv. oryzae strain KACC10331 (92 genes) [116].
Transposases
Transposases catalyze the transposition mechanisms of mobile genetic elements. They are involved in DNA rearrangement and play a role in both genome evolution and cellular function [119]. The chromosome of X. albilineans strain GPE PC73 harbors 86 transposases or transposase fragments (Additional file 5). This number is similar to those reported for other Xanthomonas spp. with the notable exception of X. oryzae pv. oryzae and X. oryzae pv. oryzicola strains, which possess a very high number of insertion sequences (IS) covering 20% of the genome [120]. Twenty-two transposases of X. albilineans are unique to this pathogen, while the others are shared or partially shared by many Xanthomonas species. However, the open reading frames XALc_2324, XALc_2365, XALc_2622 and XALc_2931 match only with transposase sequences from the vascular rice pathogen X. oryzae pv. oryzae. Interestingly, transposases encoded by XALc_1244 and XALc_2604 were found only in two sugarcane pathogens (X. albilineans and X. campestris pv. vasculorum) and in a banana pathogen (X. campestris pv. musacearum) that is closely related to one of these sugarcane pathogens [32].
A 5-kb gene cluster present in 12 different locations of the chromosome
An intriguing genomic feature of X. albilineans strain GPE PC73 is the presence of 12 similar, but not identical, copies of a 5-kb gene cluster in 12 locations of the chromosome of strain GPE PC73 (Figures 1 and 2). Most of these genes are not conserved in all 12 copies, but some genes conserved in several copies share high percentages of overall amino acid identity (Figure 2), suggesting that they are involved in similar molecular processes. Proteins encoded by these 5-kb gene clusters, annotated as hypothetical proteins, do not contain any conserved functional domain and match only partially with hypothetical proteins from X. campestris pv. musacearum, X. campestris pv. vasculorum or X. oryzae pv. oryzae. Although these proteins are not found in the genome of X. sacchari strain NCPPB4393, two of them (XALc_1969 and XALc_1970) are partially conserved in a single copy in the genome of Xanthomonas spp. NCPPB1131.
The 12 copies of the gene cluster each encode a protein harboring the triade Ser-His-Asp (SHD) characteristic of some members of the α/β-hydrolase fold enzyme superfamily. This very large group of hydrolytic enzymes exhibits a catalytic site involving a nucleophilic amino acid (serine or cysteine) within a Gx(S/C)xG signature sequence, a histidine and an acidic amino acid (aspartic acid or glutamic acid) [121]. These three catalytic residues are dispersed in the primary sequence, but the 3D-structure of the enzyme brings them together to form the catalytic site. In X. albilineans, the signature sequence is GxSxG, and all the catalytic triades are from the SHD form found in thioester hydrolases, serine carboxypeptidases, haloperoxydases, hydroxynitrile lyases, C-C bond hydrolases and lipid hydrolases. Moreover, all SHD triades found in the X. albilineans genome are organized in almost the same way as lipases from Pseudomonas fluorescens[122].
Additionally, nine proteins harboring the triade SHD also contain a polyserine linker (PSL) that separates two different putative functional domains of unknown functions. Similar PSLs have been described as flexible spacer regions that enhance substrate accessibility, thus facilitating the enzymatic activities of proteins [70]. The presence of a PSL and/or the SHD triade, may indicate that the 12 copies of the 5-kb gene cluster are involved in molecular processes required for the use of substrates specifically present in sugarcane sap. Five copies of this gene cluster contain a virD4-like gene encoding a NTPase which may be required to power such specific molecular processes. The 12 proteins containing a PSL and/or the triade SHD, each present in one copy of the cluster, share some amino acid similarity with each other, but they can be clustered in three distinct groups according to their amino acid identity. XALc_1969, XALc_2380, XALc_1070, XALc_3014, XALc_2331, XALc_2613 and XALc_2074-75 share 59–88% amino acid identity. XALc_3056 and XALc_0685 share 93% amino acid identity. XALc_1798, XALc_1863 and XALc_1627 share at least 84% amino acid identity. The three groups share together less than 45% amino acid identity, indicating that they may be involved in three distinct molecular processes. Nine copies of this 5-kb gene cluster are flanked by phage and/or recombination hot spot (Rhs) sequences, indicating that the 12 copies of this 5-kb gene cluster were probably acquired by lateral gene transfer.
Clustered regularly interspaced short palindromic repeats systems (CRISPRs)
CRISPRs are repetitive structures in bacteria and Archaea composed of exact 24- to 48-bp repeated sequences separated by unique spacers of similar length. Over 40 gene families, which are found nowhere except near these repeats, have been designated collectively as CRISPR-associated (cas) genes [123, 124]. CRISPR/cas systems belong to different classes, with different repeat patterns or sets of genes, and are distributed widely in a large range of species. Recent data showed that CRISPR/cas systems participate in an antiviral response, probably by an RNA interference-like mechanism [125]. CRISPR interference may occur when the spacer sequences within CRISPR/cas systems match with corresponding bacteriophage or plasmid sequences. We identified two different CRISPR/cas systems in the genome of strain GPE PC73 of X. albilineans (Figure 1). The first system, CRISPR-1, is similar to that found in X. oryzae pv. oryzae, X. axonopodis pv. citri, X. campestris pv. vasculorum and X. campestris pv. musacearum. This system is associated with seven cas genes: cas3 (XALc_2885), cas5d (XALc_2887), csd1 (XALc_2888), csd2 (XALc_2889), cas4 (XALc_2890), cas1 (XALc_2891), cas2 (XALc_2892), and contains thirty-four 31-base pair repeats and thirty-three 33- to 38-base pair spacers. The number of spacers in CRISPR-1 in X. albilineans strain GPE PC73 is lower than that in X. oryzae pv. oryzae (37–77 spacers depending on sequenced strains [126]). The second system, CRISPR-2, is associated with six cas genes: cas1 (XALc_3048), cas3 (XALc_3049), csy1 (XALc_3050), csy2 (XALc_3051), csy3 (XALc_3052), csy4 (XALc_3053), and contains twenty-four repeats of 28 base pairs and twenty-three spacers of 32 base pairs. There is only one other Xanthomonas pathovar that is known to contain a similar CRISPR-2 system, namely X. campestris pv. raphani[34]. Interestingly, spacers of CRISPR-1 and CRISPR-2 of X. albilineans strain GPE PC73 are identical to the phage-related DNA sequences present only in the chromosome of this strain (prophage; Additional file 6), indicating the existence of an hitherto unknown world of X. albilineans-specific bacteriophages. Additionally, orthologs of some ORFs encoded by one of these phage-related sequences are present in X. fastidiosa strain 9a5C (XALc_0178 = XF0935, XALc_0206 = XF0508, XALc_0209 = XF0510 and XALc_1544 = XF1859). Only one CRISPR spacer matches a non phage-related gene (XALc_0969 encoding a dCTP deaminase, Additional file 6). The presence of two CRISPR systems may indicate that X. albilineans is adapted to live in environments containing phages. Many endophytic and pathogenic bacteria have been isolated from sugarcane xylem [127]. Resistance to phages may confer a competitive advantage to X. albilineans cohabiting in sugarcane xylem vessels with rival bacteria that might produce phages. Resistance to phages may also be required to survive in environments other than xylem vessels (rain water, leaf surfaces…) during aerial spread of X. albilineans[16].
Putative pathogenicity-related genes identified by comparison by suppression subtractive hybridization (SSH) of two strains of X. albilineans differing in pathogenicity
A large diversity exists among strains of X. albilineans; ten genetic groups (PFGE-A to PFGE-J) have been identified by PFGE analyses [24] Rott et al., personal communication). Additionally, disease severity and capacity to colonize sugarcane stalks also varies between strains of X. albilineans[16, 25, 128, 129]. As a first step towards establishing the phylogenetic relationship between the different PFGE-groups, we performed a multi-locus sequence analysis (MLSA) using a set of seven housekeeping genes fragments (gyrB, groEL, atpD, dnaK, efp, glnA and recA) with 16 strains spanning the diversity of X. albilineans (Table 1). The dendrogram obtained using the maximum likelihood method proposed a phylogeny in which PFGE groups are clustered in two main distant clades called MLSA-1 and MLSA-2 (Figure 4). The robustness of the tree topology clustering PFGE groups in these two main clades was confirmed by high bootstrap values (86% and 100%). MLSA-1 contains four PFGE groups and MLSA-2 contains six PFGE groups. Interestingly, all highly pathogenic strains are grouped in clade MLSA-2. To further survey genes required for pathogenicity of X. albilineans strains, we performed suppression subtractive hybridization (SSH) to compare the genome of a highly pathogenic strain (XaFL07-1) belonging to clade MLSA-2 with a less pathogenic strain (Xa23R1) belonging to clade MLSA-1. Both strains originate from Florida. Their respective genome sequences are not available to date. Strain XaFL07-1 was recently used to identify new putative pathogenicity genes by transposon mutagenesis [28], and strain Xa23R1 was used in a previous study to clone the albicidin biosynthesis gene cluster [64]. The 42-kb genomic region of strain Xa23R1 encoding the T3SS SPI-1 was also sequenced recently [48]. Interestingly, strains XaFL07-1 and Xa23R1 belong to PFGE groups B and A, respectively. Davis et al. (1997) [24] previously showed in a greenhouse experiment that strains of X. albilineans belonging to genetic group B are able to infect a higher number of stalks than strains of group A after inoculation of sugarcane plants by the decapitation technique. Additionally, strains of group B are spread aerially whereas strains of group A are not.

Phylogenetic analyses of 16 strains of X. albilineans spanning the genetic diversity of this species and belonging to different PFGE groups: A: MLSA dendrogram obtained with 16 strains of X. albilineans from diverse geographical origins and rooted with X. albilineans strain XaS3 (outgroup). Strain XaS3 was isolated in Guadeloupe from the surface of a sugarcane leaf and failed to induce symptoms in sugarcane after plant inoculation [16]. The tree was constructed with concatenated sequence of the PCR fragments of seven housekeeping genes (gyrB, groEL, atpD, dnaK, efp, glnA and recA) using the maximum likelihood method and GTR as substitution model. Strains on a grey background correspond to strains Xa23R1, HVO082, GPE PC73 and XaFL07-1 used for SSH comparative in silico analysis. B: PFGE groups assigned to each X. albilineans strain, data from Davis et al. (1997) [24] and Rott P. (unpublished data). PFGE groups on a grey background are groups corresponding to strains Xa23R1, HVO082, GPE PC73 and XaFL07-1 used for SSH comparative in silico analysis. C: Pathogenicity in sugarcane rated as disease severity and extent of stalk colonisation (data from [25, 130]; Rott P, unpublished data): H = highly aggressive group, L = low or nonaggressive group and nd = no data. Groups on a grey background are groups corresponding to strains Xa23R1, HVO082, GPE PC73 and XaFL07-1 used for SSH comparative in silico analysis
We enriched a library of unique DNA sequences from strain XaFL07-1 (tester strain) using Xa23R1 DNA as the driver strain. A total of 143 XaFL07-1-specific clones were generated and sequenced. Sequences were compared with the genome sequences of two highly pathogenic X. albilineans strains, i.e. strain GPE PC73 which belongs to the same PFGE-B group as strain XaFL07-1, and strain HVO082, which belongs to group PFGE-C. Strains XaFL07, GPE PC73 and HVO082 all belong to clade MLSA-2 (Figure 4). We used the complete genome sequence of strain GPE PC73 available in Genbank and an unpublished draft genome sequence of strain HVO082 (Rott P. et al., unpublished data). Within the library of 143 clones, 50 clones were absent in the genome of strains GPE PC73 and HVO082, and were consequently considered as specific to strain XaFL07-1. Twenty-five clones targeting the same ORF as another clone were considered as duplicates. The occurrence of these duplicates reflected the good coverage of the genome and reliability of the method used to construct the SSH library. The remaining 68 SSH fragments were present in strain GPE PC73, but only 18 of them occurred also in strain HVO082. These 18 SSH fragments were therefore considered specific to the three highly pathogenic strains (XaFL07-1, GPE PC73 and HVO082).
The 18 SSH fragments correspond to 20 annotated ORFs and to one large intergenic sequence (2,117 bp) of strain GPE PC73 (Table 2). This large intergenic sequence, specific to the three highly pathogenic strains, is bordered in strain GPE PC73 by one tRNA (XALc_0254) and an entericidin gene conserved in all other sequenced xanthomonads (XALc_0255). Among the 20 ORFs that were absent in the less pathogenic strain (Xa23R1) and present in three highly pathogenic strains, five of them are in the same genomic region: they encode an ATP-binding cassette (ABC) transporter (XALc_1232 and XALc_1231) and a hypothetical protein (XALc_1229) which are conserved in X. oryzae pv. oryzicola BLS256, and two hypothetical proteins (XALc_1226 and XALc_1228) specific to X. albilineans (Table 2). Two other ORFs specific to highly pathogenic strains encode an ABC transporter specific to X. albilineans (XALc_0703 and XALc_0704). One ORF, which encodes a hypothetical protein, and which is specific to highly pathogenic strains, is present in strain GPE PC73 in a region encoding ABC transporters (XALc_2561), suggesting that it may also be involved in such a function. ABC transporters specific to highly pathogenic strains could contribute to pathogenicity by secretion of small molecules or proteins or, alternatively, could confer resistance against toxins produced by the sugarcane plant. Two additional ORFs with an assigned function that are specific to the three highly pathogenic strains are also candidate pathogenicity genes based on their predicted function. One encodes a methyl-accepting protein (XALc_1361) belonging to the flagellum gene cluster, which may be required in response to temporal changes in the chemical environment during colonization of the sugarcane xylem vessels. The other encodes an oxidoreductase (XALc_2283) that could protect the pathogen from the oxidative burst resulting from recognition of X. albilineans by the sugarcane plant. Interestingly, the reciprocal best BLAST hit in GenBank for this ORF belongs to X. campestris pv. vasculorum. This ORF is also conserved in strain ATCC35937 of X. axonopodis pv. vesicatoria, but is not conserved in any other sequenced xanthomonads.
Finally, one ORF, which encodes a hypothetical protein containing an integral membrane sensory domain, and which is specific to highly pathogenic strains (XALc_1622), is present in two copies in the genome of strain GPE PC73. The remaining ORFs are not considered as putative pathogenicity genes. The smallest ORF (XALc_2284) is probably a pseudogene deriving from an ancestral copy of XALc_2292 (RNA methylase). The recombinase-resolvase (XALc_2384) probably belongs to a phage sequence. The remaining hypothetical protein genes specific to highly pathogenic strains are bordered by phage-related sequences and probably also belong to a phage sequence (XALc_2627, XALc_2628, XALc_1545, XALc_1546, XALc_1631 and XALc_1803). The absence in strain Xa23R1 of the locus CRISPR-1 was confirmed by PCR using several pairs of primers specific of this locus. This result suggests that strain Xa23R1 may be less adapted to life in phages-containing environments than highly pathogenic strains.
Conclusions
In this study, we present a comparative genomic analysis of the complete genome sequence of the highly pathogenic strain GPE PC73 of X. albilineans. This in silico analysis revealed genomic features specific to X. albilineans that may play key roles in strategies used by this pathogen to spread within sugarcane xylem vessels with a reduced artillery compared to other pathogenic Xanthomonas species.
The most important strength of X. albilineans is that it is very well equipped for spread in a nutrient-poor environment. In this respect, the 35 TBDT genes found on the chromosome of X. albilineans strain GPE PC73 could play a key role in the active transport of plant nutrients. Interestingly, two TBDT genes (XALc_0643 and XALc_0723) were previously identified to contribute to disease severity and extent of stalk colonization [28]. The five cellulases exhibiting a CBD and a long linker region (XALc_2967, XALc_2969, XALc_0484, XALc_0865 and XALc_0874) may be adapted specifically to use cell breakdown products as carbon sources. The presence of 12 copies of a 5-kb gene cluster encoding hypothetical proteins, which are predicted to be involved in the degradation of nutrients because they harbor a PSL or a lipase-specific triade, may also be linked to the adaptation to the poor nutrient conditions prevailing in the xylem of sugarcane. Additionally, life in the iron-poor xylem sap may require unknown siderophores that are possibly synthesized by the new NRPS genes identified in the genome of strain GPE PC73 of X. albilineans.
In the rather cloistered environmental niche inside xylem vessels, X. albilineans most likely uses strategies to avoid detection by plant defense systems. The presence of a PSL in the FliD protein might help mask the bacterium from the sugarcane's defense surveillance. LPS genes shared with X. campestris pv. vasculorum, another sugarcane-interacting species, might be involved in biosynthesis of specific LPS that are poorly recognized as PAMP by sugarcane surveillance systems. Similarly, the reductive genome evolution experienced by X. albilineans during its descent from the common ancestor of Xanthomonas may have favored an adaptation to sugarcane xylem vessels by allowing the loss of genes encoding PAMPs recognized by sugarcane surveillance systems.
As shown in other plant-microbe interactions, X. albilineans most likely uses various strategies to protect itself against sugarcane defense systems. ABC transporters specific to highly pathogenic strains may confer resistance against antibacterial toxins produced by sugarcane. The gene XALc_1762, which encodes the membrane fusion of an MDR efflux pump, may be involved in a similar resistance process because it was shown previously to confer resistance to novobiocin and to be required for efficient stalk colonization of sugarcane by X. albilineans[28]. Interestingly, another ABC transporter (XALc_0028) was shown previously to be required for spreading within sugarcane [28]. Additionally, three ABC transporter proteins from X. albilineans GPE PC73 (XALc_941, XALc_942 and XALc_944) have also been identified in the genome of Leifsonia xyli subsp. xyli, Acidovorax avenae subsp. avenae and X. sacchari, respectively, which all are able to spread within xylem vessels of sugarcane, making these ABC transporter proteins also good candidates for future studies (orthoMCL analysis; Pieretti I., personal communication). Additionally, the oxidoreductase specific to highly pathogenic X. albilineans strains may play a role by protecting the bacteria from the oxidative burst resulting from the recognition of X. albilineans by the sugarcane plant. Another oxidoreductase (XALc_1972) that has been shown as shared only by X. albilineans strain GPE PC73, X. sacchari and L. xyli subsp. xyli may have a similar function. TBDTs could also play a key role in the detoxification of antibacterial toxins produced by sugarcane that target external components of X. albilineans by allowing their active and fast uptake inside the bacterium.
Additionally, X. albilineans may use specific and unknown strategies to suppress or weaken sugarcane plant defenses. Because neither a T3SS-Hrp nor the corresponding effectors have been identified in the genome of X. albilineans, the pathogen might rely on other secretion systems identified in the genome of X. albilineans strain GPE PC73. Secretion systems T1SS and T2SS might possibly be involved in the secretion of yet-to-be identified effectors that could be secreted in the xylem and diffuse to the parenchyma target cells. Potentially, such effectors may be synthesized by the new NRPS genes identified in the genome of strain GPE PC73 of X. albilineans. Some of the 216 hypothetical protein genes specific to X. albilineans may be required to suppress or weaken sugarcane plant defenses. However, screening of a random mutant library of strain XaFL07-1 of X. albilineans failed to identify any pathogenicity gene predicted to be involved in an offensive or counter-defensive mechanism [28].
To conclude, neither comparative genomic analyses nor SSH experiments have revealed the existence of genes involved in any offensive or counter-defensive mechanism specific to X. albilineans, at least unless the role of hypothetical proteins can be determined, suggesting that this pathogen is able to spread within sugarcane xylem vessels with only a reduced artillery. This is even more surprising with respect to preliminary confocal microscopy data, which revealed recently that highly pathogenic strains XaFL07-1 and GPE PC73 of X. albilineans are able to spread efficiently in the xylem and also in other leaf and stalk tissues (Mensi I., personal communication).
Methods
Bacterial strains and media
The origin and main characteristics of the bacterial strains studied in this paper are summarized in Table 1. X. albilineans strains Xa23R1 and XaFL07-1 stored at −80°C were retrieved by plating on Wilbrink’s agar medium (peptone 5 g, yeast extract 5 g, sucrose 10 g, sodium sulfite (Na2SO3, 7 H2O) 0.05 g, magnesium sulfate (MgSO4, 7 H2O) 0.25 g, dipotassium phosphate (K2HPO4) 0.5 g, Benomyl® 12.5 mg, distilled water 1 l, pH 6.9-7.0).
Genome sequence of X. albilineans strain GPE PC73
X. albilineans strain GPE PC73 is referred to as CFBP 7063 in the French Collection of Plant Pathogenic Bacteria (http://www.angers.inra.fr/cfbp/). The complete annotated genetic map, search tools (BLAST), annotation and classification process are available at [http://iant.toulouse.inra.fr/X.albilineans].
Draft genome sequence of X. albilineans strain HVO082
X. albilineans strain HVO082 from the Bacterial Collection of UMR BGPI at Cirad in Montpellier, France, was sequenced recently by Genoscope (Evry, France) (Rott P., unpublished sequence).
Extraction of bacterial genomic DNA
Bacterial genomic DNA was extracted from 5 ml of culture grown in liquid Wilbrink’s medium for 48 h at 28°C with the Illustra GenomicPrep Bacteria Mini Spin Kit (GE Healthcare, Buckinghamshire, UK), following the manufacturer's instructions.
Construction of suppression subtractive hybridization librairies
Genomic DNA was extracted from X. albilineans strains GPE PC73 and XaFL07-1 as described above. The subtracted DNA library was generated following the instructions with the PCR-Select Bacterial Genome Subtraction Kit (Clontech Laboratories, Inc., Mountain View, CA, USA). Approximately 2 μg of both XaFL07-1 and Xa23R1 genomic DNA were digested with Hae III for 5 h at 37°C with 25 units of enzyme. Hae III-digested DNA from XaFL07-1 was used as the tester and Xa23R1 was used as the driver for XaFL07-1 library construction. Two different adaptors (1 and 2R) were ligated to the 5’ end of two aliquots of the tester DNA fragments. Two hybridizations were performed. In the first, the driver DNA fragments were added to each adaptor-ligated tester DNA (ratio 2:1). Then, the DNA fragments were denatured for 90 s at 98°C and allowed to anneal at 63°C for 1.5 h. The two samples (one with adaptor 1 and the other with adaptor 2R) were mixed together and freshly denatured driver DNA was added before the second hybridization. The second hybridization was carried out for 16 h at 63°C. Two sequential nested PCR reactions were carried out. The subtraction efficiency was measured by PCR amplification of the rpfB gene, present in these two strains and which does not contain any Hae III restriction site. The rpfB gene was amplified using rpfB-forward (CTCTGGTGGAAGCCTACGGAC) and rpfB-reverse (CAATACGCCTGGCATCATCGC) primers, under the PCR conditions of the supplier. Five μl of amplimers were removed after 18, 21, 24 and 27 PCR cycles, respectively, and examined by agarose gel electrophoresis. The difference in the number of cycles required for equal amplification of the corresponding PCR product in the subtracted and unsubtracted samples indicated that the efficiency of our subtraction was six. According to the supplier’s instructions, six cycles corresponds roughly to a 12-fold enrichment.
The PCR products were cloned into the pCR®8/GW/TOPO® TA Cloning® vector system (Invitrogen, Carlsbad, CA, USA). The SSH library was constructed by transforming the cloned vector into One Shot® TOP10 Chemically Competent E. coli cells according to the supplier’s instructions (Invitrogen, Carlsbad, CA, USA). For each colony, bacterial suspension samples after dilution were amplified by PCR using T7 and M13 forward primers to confirm the presence and size of the different inserts by agarose gel electrophoresis.
Dot blot screening
Each PCR clone was subjected to differential dot blotting to identify those that hybridized preferentially to the tester XaFL07-1 DNA. Briefly, 2 μl of each denatured PCR sample were dotted in duplicate on positively-charged nylon membranes (Roche, Mannheim, Germany). DNA fixation was carried out by irradiation under a UV transilluminator for 2 min. A digoxigenin-11-2'-dUTP DNA labeling and detection kit (Roche, Mannheim, Germany) was used to prepare the probes. The genomic DNA of both strains, Xa23R1 and XaFL07-1, were used independently as a probe for each of the duplicate membranes. Prehybridization and hybridization were carried out according to the manufacturer's instructions (Roche, Mannheim, Germany).
DNA sequencing and analysis
Specific inserts were sequenced using T7 and M13 forward sequencing primers. Sequencing reactions were performed using the Big Dye kit (Applied Biosystems, Foster City, CA, USA) and were analysed on an Applied Biosystems 3100 automated DNA sequencer by CGRC/ICBR (University of Florida, Gainesville, FL, USA). Sequence assembly and further editing were performed with Bioedit Analysis software (Tom Hall, Carlsbad, CA, USA). Blastx sequence homology analyses were performed using the Basic Local Aligment Search Tool (BLAST) [131] network service of the National Center for Biotechnology Information (NCBI) and using iANT (integrated ANnotation Tool) [132].
Genomic comparison
For comparative analyses, the annotated genome sequences of X. albilineans strain GPE PC73, GenBank accession no. NC_013722.1 [8] was compared to the unpublished draft sequence of strain HVO082 of X. albilineans (Rott P., unpublished data) and to seven published genome sequences of Xanthomonas: X. axonopodis pv. vesicatoria strain 85–10, GenBank accession no. NC_007508.1 [30], X. axonopodis pv. citri strain 306, GenBank accession no. NC_003919.1 [31], X. campestris pv. campestris strain ATCC 33913, GenBank accession no. NC_003902.1 [31], X. campestris pv. vasculorum NCPPB702, GenBank accession no. NZ_ACHS00000000.1 [32], X. campestris pv. musacearum NCPPB4381, GenBank accession no. NZ_ACHT00000000.1 [32], X. oryzae pv. oryzae MAFF 311018 [33] and X. oryzae pv. oryzicola strain BLS256, GenBank accession no. CP003057 [34]. Homology searches were conducted at the nucleotide and amino acid sequence levels using the BLAST server [131].
OrthoMCL analysis
OrthoMCL clustering analyses [133] were performed using the following parameters: P-value Cut-off = 1 × 10-5; Percent Identity Cut-off = 0; Percent Match Cut-off = 80; MCL Inflation = 1.5; Maximum Weight = 316. We modified OrthoMCL analysis by inactivating the filter query sequence during the BLASTP pre-process. Best BLASTP hit analyses were performed with database UniProt by excluding all accessions from the xanthomonads using expectation value lower than 1 × 10-5.
Phylogenetic analysis
The phylogenetic MLSA tree was reconstructed using the maximum likelihood method implemented in the PhyML program. The GTR substitution model was selected assuming an estimated proportion of invariant sites (of 0.01) and four gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data (α= 0.010). Five hundred bootstrap replicates were performed with the PhyML program. The seven analyzed loci (gyrB, groEL, recA, dnaK, efp, atpD, and glnA) are typical housekeeping genes in the chromosome of X. albilineans GPE PC73, located 0.004, 0.348, 1.369, 1.983, 2.245, 3.442, and 3.655 Mb, respectively, from the origin of replication. The total length of the partial CDS nucleotide sequences concatenated for each taxon was 4,016 bp. Multiple alignments of the nucleotide sequences of the seven housekeeping fragment genes and for all taxa were performed using ClustalW.
Abbreviations
- ABC:
-
ATP-binding cassette
- BLAST:
-
Basic Local Alignment Search Tool
- CBD:
-
Cellulose Binding Domain
- CRISPRs:
-
Clustered Regularly Interspaced Short Palindromic Repeats systems
- CU:
-
Chaperone-Usher
- DSF:
-
Diffusible Signal Factor
- Hrp:
-
Hypersensitive Response and Pathogenicity
- IS:
-
Insertion Sequence
- LPS:
-
Lipopolysaccharides
- MCP:
-
Methyl-accepting Chemotaxis Proteins
- MDR:
-
Multidrug resistance
- MLSA:
-
Multilocus sequence analysis
- NCBI:
-
National Center for Biotechnology Information
- NRPS:
-
Nonribosomal peptide synthetases
- PAMP:
-
Pathogen-Associated Molecular Pattern
- PFGE:
-
Pulsed-Field Gel Electrophoresis
- PSL:
-
Polyserine linker
- Rhs:
-
Recombination hot spot
- rpf:
-
Regulation of pathogenicity factors
- SPI-1:
-
Salmonella Pathogenicity Island-1
- SSH:
-
Suppression Subtractive Hybridization
- T1SS:
-
Type I Secretion System
- T2SS:
-
Type II Secretion System
- T3SS:
-
Type III Secretion System
- T4SS:
-
Type IV Secretion System
- T5SS:
-
Type V Secretion System
- T6SS:
-
Type VI Secretion System
- TBDT:
-
TonB-Dependent Transporters
- TCSTS:
-
Two-Component Signal Transduction Systems
- TPS:
-
Two-Partner System.
References
Bull CT, De Boer SH, Denny TP, Firrao G, Fischer-Le Saux M, Saddler GS, Scortichini M, Stead DE, Takikawa Y: Comprehensive list of names of plant pathogenic bacteria, 1980–2007. J Plant Pathol. 2010, 92: 551-592.
Van Den Mooter M, Swings J: Numerical analysis of 295 phenotypic features of 266 Xanthomonas strains and related strains and an improved taxonomy of the genus. Int J Syst Bacteriol. 1990, 40: 348-369. 10.1099/00207713-40-4-348.
Vauterin L, Rademaker J, Swings J: Synopsis on the taxonomy of the genus Xanthomonas. Phytopathology. 2000, 90: 677-682. 10.1094/PHYTO.2000.90.7.677.
Rademaker J, Louws F, Schultz M, Rossbach U, Vauterin L, Swings J, de Bruijn F: A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology. 2005, 95: 1098-1111. 10.1094/PHYTO-95-1098.
Young JM, Park DC, Shearman HM, Fargier E: A multilocus sequence analysis of the genus Xanthomonas. Syst Appl Microbiol. 2008, 31: 366-377. 10.1016/j.syapm.2008.06.004.
Hauben L, Vauterin L, Swings J, Moore ERB: Comparison of 16S ribosomal DNA sequences of all Xanthomonas species. Int J Syst Bacteriol. 1997, 47: 328-335. 10.1099/00207713-47-2-328.
Goncalvez ER, Rosato YB: Phylogenetic analysis of Xanthomonas species based upon 16S-23S rDNA intergenic spacer sequences. Int J Syst Evol Microbiol. 2002, 52: 355-361.
Pieretti I, Royer M, Barbe V, Carrere S, Koebnik R, Cociancich S, Couloux A, Darrasse A, Gouzy J, Jacques M-A, et al: The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics. 2009, 10: 616-10.1186/1471-2164-10-616.
Studholme D, Wasukira A, Paszkiewicz K, Aritua V, Thwaites R, Smith J, Grant M: Draft genome sequences of Xanthomonas sacchariand two banana-associated xanthomonads reveal insights into theXanthomonasgroup 1 clade. Genes. 2011, 2: 1050-1065. 10.3390/genes2041050.
Vauterin L, Hoste B, Kersters K, Swings J: Reclassification of Xanthomonas. Int J Syst Bacteriol. 1995, 45: 472-489. 10.1099/00207713-45-3-472.
Ikeda S, Fujji S, Sato T, Furuya H, Naito H, Ytow N, Ezura H, Minamisawa K, Fujimura T: Microbial diversity in milled rice as revealed by ribosomal intergenic spacer analysis. Microb Environ. 2007, 22: 165-174. 10.1264/jsme2.22.165.
Rott P, Davis M: Leaf scald. A guide to sugarcane diseases. Edited by: Rott P, Bailey R, Comstock J, Croft B, Saumtally A. 2000, Montpellier: CIRAD-ISSCT, 339-
Birch RG: Xanthomonas albilineans and the antipathogenesis approach to disease control. Mol Plant Pathol. 2001, 2: 1-11. 10.1046/j.1364-3703.2001.00046.x.
Hashimi SM, Wall MK, Smith AB, Maxwell A, Birch RG: The phytotoxin albicidin is a novel inhibitor of DNA gyrase. Antimicrob Agents Chemother. 2007, 51: 181-187. 10.1128/AAC.00918-06.
Autrey L, Saumtally S, Dookun A, Sullivan S, Dhayan S: Aerial transmission of the leaf scald pathogen,Xanthomonas albilineans. Proc Int Soc Sugar Cane Technol. 1995, 21: 508-526.
Daugrois JH, Dumont V, Champoiseau P, Costet L, Boisne-Noc R, Rott P: Aerial contamination of sugarcane in Guadeloupe by two strains of Xanthomonas albilineans. Eur J Plant Pathol. 2003, 109: 445-458. 10.1023/A:1024259606468.
Champoiseau P, Rott , Daugrois JH: Epiphytic populations of Xanthomonas albilineans and subsequent sugarcane stalk infection are linked to rainfall in Guadeloupe. Plant Dis. 2009, 93: 339-346. 10.1094/PDIS-93-4-0339.
Rott P, Abel M, Soupa D, Feldman P, Letourmy P: Population dynamics of Xanthomonas albilineans in sugarcane plants as determined with an antibiotic-resistant mutant. Plant Dis. 1994, 78: 241-247. 10.1094/PD-78-0241.
Diaz M, Peralta EL, Iglesia A, Pazos V, Carvajal O, Perez MP, Giglioti EA, Gagliardi PR, Wendland A, Camargo LEA: Xanthomonas albilineans haplotype B responsible for a recent sugarcane leaf scald disease outbreak in Cuba. Plant Dis. 2001, 85: 334-
Comstock JC, Shine JM: Outbreaks of leaf scald of sugarcane, caused by Xanthomonas albilineans, in Florida. Plant Dis. 1992, 76: 426-
Chen CT, Lin CP, Liang YG: Leaf scald of sugarcane in Taiwan. Taiwan Sugar. 1993, 40: 8-16.
Grisham MP, Legendre BL, Comstock JC: First report of leaf scald, caused by Xanthomonas albilineans, of sugarcane in Louisiana. Plant Dis. 1993, 77: 537-
Isakeit T, Irvine JE: First report of leaf scald, caused by Xanthomonas albilineans, of sugarcane in Texas. Plant Dis. 1995, 79: 860-
Davis M, Rott P, Warmuth C, Chatenet M, Baudin P: Intraspecific genomic variation within Xanthomonas albilineans, the sugarcane leaf scald pathogen. Phytopathology. 1997, 87: 316-324. 10.1094/PHYTO.1997.87.3.316.
Champoiseau P, Daugrois JH, Girard JC, Royer M, Rott PC: Variation in albicidin biosynthesis genes and in pathogenicity of Xanthomonas albilineans, the sugarcane leaf scald pathogen. Phytopathology. 2006, 96: 33-45. 10.1094/PHYTO-96-0033.
Birch R, Patil S: The relation of blocked chloroplast differenciation to sugarcane leaf scald disease. Phytopathology. 1983, 73: 1368-1374. 10.1094/Phyto-73-1368.
Chatterjee S, Almeida RPP, Lindow S: Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa. Annu Rev Phytopathol. 2008, 46: 243-271. 10.1146/annurev.phyto.45.062806.094342.
Rott P, Fleites L, Marlow G, Royer M, Gabriel DW: Identification of new candidate pathogenicity factors in the xylem-invading pathogen Xanthomonas albilineans by transposon mutagenesis. Mol Plant Microbe Interact. 2011, 24: 594-605. 10.1094/MPMI-07-10-0156.
Renier A, Vivien E, Cociancich S, Letourmy P, Perrier X, Rott PC, Royer M: Substrate specificity-conferring regions of the nonribosomal peptide synthetase adenylation domains involved in albicidin pathotoxin biosynthesis are highly conserved within the species Xanthomonas albilineans. Appl Environ Microbiol. 2007, 73: 5523-5530. 10.1128/AEM.00577-07.
Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Buttner D, Caldana C, Gaigalat L, Goesmann A, Kay S, et al: Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J Bacteriol. 2005, 187: 7254-7266. 10.1128/JB.187.21.7254-7266.2005.
da Silva A, Ferro J, Reinach F, Farah C, Furlan L, Quaggio R, Monteiro-Vitorello C, Van Sluys M, Almeida N, Alves L, et al: Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002, 417: 459-463. 10.1038/417459a.
Studholme DJ, Kemen E, MacLean D, Schornack S, Aritua V, Thwaites R, Grant M, Smith J, Jones JDG: Genome-wide sequencing data reveals virulence factors implicated in banana Xanthomonas wilt. FEMS Microbiol Lett. 2010, 310: 182-192. 10.1111/j.1574-6968.2010.02065.x.
Ochiai H, Horino O, Miyajima K, Kaku H: Genetic diversity of Xanthomonas oryzae pv. oryzae strains from Sri Lanka. Phytopathology. 2000, 90: 415-421. 10.1094/PHYTO.2000.90.4.415.
Bogdanove AJ, Koebnik R, Lu H, Furutani A, Angiuoli SV, Patil PB, Van Sluys M-A, Ryan RP, Meyer DF, Han S-W, et al: Two new complete genome sequences offer insight into host and tissue specificity of plant pathogenic Xanthomonas spp. J Bacteriol. 2011, 193: 5450-5464. 10.1128/JB.05262-11.
Hilaire E, Young SA, Willard LH, McGee JD, Sweat T, Chittoor JM, Guikema JA, Leach JE: Vascular defense responses in rice: peroxidase accumulation in xylem parenchyma cells and xylem wall thickening. Mol Plant Microbe Interact. 2001, 14: 1411-1419. 10.1094/MPMI.2001.14.12.1411.
Büttner D, Bonas U: Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev. 2010, 34: 107-133. 10.1111/j.1574-6976.2009.00192.x.
Gerlach RG, Hensel M: Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int J Med Microbiol. 2007, 297: 401-415. 10.1016/j.ijmm.2007.03.017.
Desvaux M, Hébraud M, Talon R, Henderson IR: Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 2009, 17: 139-145. 10.1016/j.tim.2009.01.004.
Wandersman C, Delepelaire P: TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc Natl Acad Sci USA. 1990, 87: 4776-4780. 10.1073/pnas.87.12.4776.
Koronakis V, Eswaran J, Hughes C: Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu Rev Biochem. 2004, 73: 467-489. 10.1146/annurev.biochem.73.011303.074104.
Nikaido H: Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996, 178: 5853-5859.
Paulsen IT: Multidrug efflux pumps and resistance: regulation and evolution. Curr Opin Microbiol. 2003, 6: 446-451. 10.1016/j.mib.2003.08.005.
Zgurskaya HI, Nikaido H: Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol. 2000, 37: 219-225. 10.1046/j.1365-2958.2000.01926.x.
Reddy JD, Reddy SL, Hopkins DL, Gabriel DW: TolC is required for pathogenicity of Xylella fastidiosa in vitis vinifera grapevines. Mol Plant Microbe Interact. 2007, 20: 403-410. 10.1094/MPMI-20-4-0403.
da Silva FG, Shen Y, Dardick C, Burdman S, Yadav RC, de Leon AL, Ronald PC: Bacterial genes involved in type I secretion and sulfation are required to elicit the rice Xa21-mediated innate immune response. Mol Plant Microbe Interact. 2004, 17: 593-601. 10.1094/MPMI.2004.17.6.593.
Szczesny R, Jordan M, Schramm C, Schulz S, Cogez V, Bonas U, Büttner D: Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv vesicatoria. New Phytol. 2010, 187: 983-1002. 10.1111/j.1469-8137.2010.03312.x.
Cornelis GR: The type III secretion injectisome. Nat Rev Micro. 2006, 4: 811-825. 10.1038/nrmicro1526.
Marguerettaz M, Pieretti I, Gayral P, Puig J, Brin C, Cociancich S, Poussier S, Rott P, Royer M: Genomic and evolutionary features of the SPI-1 type III secretion system that is present in Xanthomonas albilineans but is not essential for xylem colonization and symptom development of sugarcane leaf scald. Mol Plant Microbe Interact. 2011, 24: 246-259. 10.1094/MPMI-08-10-0188.
Kube M, Migdoll AM, Müller I, Kuhl H, Beck A, Reinhardt R, Geider K: The genome of Erwinia tasmaniensis strain Et1/99, a non-pathogenic bacterium in the genus Erwinia. Environ Microbiol. 2008, 10: 2211-2222. 10.1111/j.1462-2920.2008.01639.x.
Saenz HL, Engel P, Stoeckli MC, Lanz C, Raddatz G, Vayssier-Taussat M, Birtles R, Schuster SC, Dehio C: Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat Genet. 2007, 39: 1469-1476. 10.1038/ng.2007.38.
Rêgo AT, Chandran V, Waksman G: Two-step and one-step secretion mechanisms in Gram-negative bacteria: contrasting the type IV secretion system and the chaperone-usher pathway of pilus biogenesis. Biochem J. 2010, 425: 475-488. 10.1042/BJ20091518.
Backert S, Fronzes R, Waksman G: VirB2 and VirB5 proteins: specialized adhesins in bacterial type-IV secretion systems?. Trends Microbiol. 2008, 16: 409-413. 10.1016/j.tim.2008.07.001.
Vayssier-Taussat M, Le Rhun D, Deng HK, Biville F, Cescau S, Danchin A, Marignac G, Lenaour E, Boulouis HJ, Mavris M, et al: The Trw type IV secretion system of Bartonella mediates host-specific adhesion to erythrocytes. PLoS Pathog. 2010, 6: e1000946-10.1371/journal.ppat.1000946.
Souza DP, Andrade MO, Alvarez-Martinez CE, Arantes GM, Farah CS, Salinas RK: A component of the Xanthomonadaceae type IV secretion system combines a VirB7 motif with a N0 domain found in outer membrane transport proteins. PLoS Pathog. 2011, 7: e1002031-10.1371/journal.ppat.1002031.
Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala’Aldeen D: Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev. 2004, 68: 692-744. 10.1128/MMBR.68.4.692-744.2004.
Tseng T-T, Tyler B, Setubal J: Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 2009, 9 (Suppl 1): S2-10.1186/1471-2180-9-S1-S2.
Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I: Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources?. BMC Genomics. 2009, 10: 104-10.1186/1471-2164-10-104.
Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, et al: A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006, 312: 1526-1530. 10.1126/science.1128393.
Schell MA, Ulrich RL, Ribot WJ, Brueggemann EE, Hines HB, Chen D, Lipscomb L, Kim HS, Mrázek J, Nierman WC, et al: Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol Microbiol. 2007, 64: 1466-1485. 10.1111/j.1365-2958.2007.05734.x.
Lee SW, Jeong KS, Han SW, Lee SE, Phee BK, Hahn TR, Ronald P: The Xanthomonas oryzae pv. oryzae PhoPQ two-component system is required for AvrXA21 activity, hrpG expression, and virulence. J Bacteriol. 2008, 190: 2183-2197. 10.1128/JB.01406-07.
Lee SW, Han SW, Sririyanum M, Park CJ, Seo YS, Ronald PC: A type I secreted, sulfated peptide triggers XA21-mediated innate immunity. Science. 2009, 326: 850-853. 10.1126/science.1173438.
Donadio S, Monciardini P, Sosio M: Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat Prod Rep. 2007, 24: 1073-1109. 10.1039/b514050c.
Gross H, Loper JE: Genomics of secondary metabolite production by Pseudomonas spp. Nat Prod Rep. 2009, 26: 1408-1446. 10.1039/b817075b.
Royer M, Costet L, Vivien E, Bes M, Cousin A, Damais A, Pieretti I, Savin A, Megessier S, Viard M, et al: Albicidin pathotoxin produced by Xanthomonas albilineans is encoded by three large PKS and NRPS genes present in a gene cluster also containing several putative modifying, regulatory, and resistance genes. Mol Plant Microbe Interact. 2004, 17: 414-427. 10.1094/MPMI.2004.17.4.414.
Birch RG, Pemberton JM, Basnayake WVS: Stable albicidin resistance in Escherichia coli involves an altered outer-membrane nucleoside uptake system. J Gen Microbiol. 1990, 1: 51-58.
Morikawa M, Daido H, Takao T, Murata S, Shimonishi Y, Imanaka T: A new lipopeptide biosurfactant produced by Arthrobacter sp. strain MIS38. J Bacteriol. 1993, 175: 6459-6466.
Berti AD, Greve NJ, Christensen QH, Thomas MG: Identification of a biosynthetic gene cluster and the six associated lipopeptides involved in swarming motility of Pseudomonas syringae pv. tomato DC3000. J Bacteriol. 2007, 189: 6312-6323. 10.1128/JB.00725-07.
Barry SM, Challis GL: Recent advances in siderophore biosynthesis. Curr Opin Chem Biol. 2009, 13: 205-215. 10.1016/j.cbpa.2009.03.008.
Leeman M, Ouden E, Pelt J, Dirkx F, Steijl H, Bakker P, Schippers B: Iron availability affects induction of systemic resistance to fusarium wilt of radish by Pseudomonas fluorescens. Phytopathol. 1996, 86: 149-155. 10.1094/Phyto-86-149.
Howard H, Otto K, Gent D: Relation of temperature and rainfall to development of Xanthomonas and Pantoea leaf blights of onion in Colorado. Plant Dis. 2003, 87: 11-14. 10.1094/PDIS.2003.87.1.11.
Boraston AB, Bolam DN, Gilbert HJ, Davies GJ: Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004, 382: 769-781. 10.1042/BJ20040892.
Liu H, Zhang S, Schell MA, Denny TP: Pyramiding unmarked deletions in Ralstonia solanacearum shows that secreted proteins in addition to plant cell-wall degrading enzymes contribute to virulence. Mol Plant Microbe Interact. 2005, 18: 1296-1305. 10.1094/MPMI-18-1296.
Pérez-Donoso AG, Sun Q, Roper MC, Greve LC, Kirkpatrick B, Labavitch JM: Cell wall-degrading enzymes enlarge the pore size of intervessel pit membranes in healthy and Xylella fastidiosa-infected grapevines. Plant Physiol. 2010, 152: 1748-1759. 10.1104/pp.109.148791.
Choat B, Cobb AR, Jansen S: Structure and function of bordered pits: new discoveries and impacts on whole-plant hydraulic function. New Phytol. 2008, 177: 608-625. 10.1111/j.1469-8137.2007.02317.x.
Roper MC, Greve LC, Warren JG, Labavitch JM, Kirkpatrick BC: Xylella fastidiosa requires polygalacturonase for colonization and pathogenicity in vitis vinifera grapevines. Mol Plant Microbe Interact. 2007, 20: 411-419. 10.1094/MPMI-20-4-0411.
Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, Vasse J, Lauber E, Arlat M: Plant carbohydrate scavenging through TonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One. 2007, 2: e224-10.1371/journal.pone.0000224.
Schauer K, Rodionov DA, de Reuse H: New substrates for TonB-dependent transport: do we only see the 'tip of the iceberg'?. Trends Biochem Sci. 2008, 33: 330-338. 10.1016/j.tibs.2008.04.012.
Reeves PP, Wang L: Genomic organization of LPS-specific loci. Curr Top Microbiol Immunol. 2002, 264: 109-135. 10.1007/978-3-642-56031-6_7.
Casabuono A, Petrocelli S, Ottado J, Orellano EG, Couto AS: Structural analysis and involvement in plant innate immunity of Xanthomonas axonopodis pv. citri lipopolysaccharide. J Biol Chem. 2011, 286: 25628-25643. 10.1074/jbc.M110.186049.
Patil P, Bogdanove A, Sonti R: The role of horizontal transfer in the evolution of a highly variable lipopolysaccharide biosynthesis locus in xanthomonads that infect rice, citrus and crucifers. BMC Evol Biol. 2007, 7: 243-10.1186/1471-2148-7-243.
Lu H, Patil P, Van Sluys M-A, White FF, Ryan RP, Dow JM, Rabinowicz P, Salzberg SL, Leach JE, Sonti R, et al: Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in Xanthomonas. PLoS One. 2008, 3: e3828-10.1371/journal.pone.0003828.
Patil P, Sonti R: Variation suggestive of horizontal gene transfer at a lipopolysaccharide (lps) biosynthetic locus in Xanthomonas oryzae pv. oryzae, the bacterial leaf blight pathogen of rice. BMC Microbiol. 2004, 4: 40-10.1186/1471-2180-4-40.
Aslam SN, Newman M-A, Erbs G, Morrissey KL, Chinchilla D, Boller T, Jensen TT, De Castro C, Ierano T, Molinaro A, et al: Bacterial polysaccharides suppress induced innate immunity by calcium chelation. Curr Biol. 2008, 18: 1078-1083. 10.1016/j.cub.2008.06.061.
Li J, Wang N: Genome-wide mutagenesis of Xanthomonas axonopodis pv. citri reveals novel genetic determinants and regulation mechanisms of biofilm formation. PLoS One. 2011, 6: e21804-10.1371/journal.pone.0021804.
da Silva FR, Vettore AL, Kemper EL, Leite A, Arruda P: Fastidian gum: the Xylella fastidiosa exopolysaccharide possibly involved in bacterial pathogenicity. FEMS Microbiol Lett. 2001, 203: 165-171.
Tao F, Swarup S, Zhang L-H: Quorum sensing modulation of a putative glycosyltransferase gene cluster essential for Xanthomonas campestris biofilm formation. Environ Microbiol. 2010, 12: 3159-3170. 10.1111/j.1462-2920.2010.02288.x.
Blanch M, Legaz M-E, Vicente C: Xanthan production by Xanthomonas albilineans infecting sugarcane stalks. J Plant Physiol. 2008, 165: 366-374. 10.1016/j.jplph.2007.03.008.
Blanch M, Legaz M-E, Vicente C: Purification and properties of an unusual UDP-glucose dehydrogenase, NADPH-dependent, from Xanthomonas albilineans. Microbiol Res. 2008, 163: 362-371. 10.1016/j.micres.2006.07.011.
Macnab RM: The bacterial flagellum: reversible rotary propellor and type III export apparatus. J Bacteriol. 1999, 181: 7149-7153.
Moreira LM, de Souza RF, Almeida NF, Setubal JC, Oliveira JCF, Furlan LR, Ferro JA, da Silva ACR: Comparative genomics analyses of citrus-associated bacteria. Annu Rev Phytopathol. 2004, 42: 163-184. 10.1146/annurev.phyto.42.040803.140310.
Ikeda T, Oosawa K, Hotani H: Self-assembly of the filament capping protein, FliD, of bacterial flagella into an annular structure. J Mol Biol. 1996, 259 (4): 679-686. 10.1006/jmbi.1996.0349.
Yonekura K, Maki S, Morgan DG, DeRosier DJ, Vonderviszt F, Imada K, Namba K: The bacterial flagellar cap as the rotary promoter of flagellin self-assembly. Science. 2000, 290: 2148-2152. 10.1126/science.290.5499.2148.
Macnab RM: Type III flagellar protein export and flagellar assembly. Biochim Biophys Acta, Mol Cell Res. 2004, 1694: 207-217. 10.1016/j.bbamcr.2004.04.005.
Ichinose Y, Shimizu R, Ikeda Y, Taguchi F, Marutani M, Mukaihara T, Inagaki Y, Toyoda K, Shiraishi T: Need for flagella for complete virulence of Pseudomonas syringae pv. tabaci: genetic analysis with flagella-defective mutants fliC and fliD in host tobacco plants. J Gen Plant Pathol. 2003, 69: 244-249. 10.1007/s10327-003-0045-z.
Arora SK, Dasgupta N, Lory S, Ramphal R: Identification of two distinct types of flagellar cap proteins, FliD, in Pseudomonas aeruginosa. Infect Immun. 2000, 68: 1474-1479. 10.1128/IAI.68.3.1474-1479.2000.
Scott JR, Zähner D: Pili with strong attachments: gram-positive bacteria do it differently. Mol Microbiol. 2006, 62: 320-330. 10.1111/j.1365-2958.2006.05279.x.
Fronzes R, Remaut H, Waksman G: Architectures and biogenesis of non-flagellar protein appendages in Gram-negative bacteria. EMBO J. 2008, 27: 2271-2280. 10.1038/emboj.2008.155.
Das A, Rangaraj N, Sonti RV: Multiple adhesin-like functions of Xanthomonas oryzae pv. oryzae are involved in promoting leaf attachment, entry, and virulence on rice. Mol Plant Microbe Interact. 2009, 22: 73-85. 10.1094/MPMI-22-1-0073.
Van Sluys MA, Monteiro-Vitorello CB, Camargo LEA, Menck CFM, da Silva ACR, Ferro JA, Oliveira MC, Setubal JC, Kitajima JP, Simpson AJ: Comparative genomic analysis of plant-associated bacteria. Annu Rev Phytopathol. 2002, 40: 169-189. 10.1146/annurev.phyto.40.030402.090559.
Darsonval A, Darrasse A, Durand K, Bureau C, Cesbron S, Jacques MA: Adhesion and fitness in the bean phyllosphere and transmission to seed of Xanthomonas fuscans subsp. fuscans. Mol Plant Microbe Interact. 2009, 22: 747-757. 10.1094/MPMI-22-6-0747.
Lim S-H, So B-H, Wang J-C, Song E-S, Park Y-J, Lee B-M, Kang H-W: Functional analysis of pilQ gene in Xanthomanas oryzae pv. oryzae, bacterial blight pathogen of rice. J Microbiol. 2008, 46: 214-220. 10.1007/s12275-007-0173-9.
Meng Y, Li Y, Galvani CD, Hao G, Turner JN, Burr TJ, Hoch HC: Upstream migration of Xylella fastidiosa via pilus-driven twitching motility. J Bacteriol. 2005, 187: 5560-5567. 10.1128/JB.187.16.5560-5567.2005.
Mhedbi-Hajri N, Jacques M-A, Koebnik R: Adhesion mechanisms of plant-pathogenic Xanthomonadaceae. Bacterial Adhesion. Edited by: Linke D, Goldman A. 2011, Netherlands: Springer, 71-89. 715
Jacob-Dubuisson F, Locht C, Antoine R: Two-partner secretion in Gram-negative bacteria: a thrifty, specific pathway for large virulence proteins. Mol Microbiol. 2001, 40: 306-313. 10.1046/j.1365-2958.2001.02278.x.
Jacob-Dubuisson F, Fernandez R, Coutte L: Protein secretion through autotransporter and two-partner pathways. Biochim Biophys Acta, Mol Cell Res. 2004, 1694: 235-257. 10.1016/j.bbamcr.2004.03.008.
Van Sluys MA, de Oliveira MC, Monteiro-Vitorello CB, Miyaki CY, Furlan LR, Camargo LEA, da Silva ACR, Moon DH, Takita MA, Lemos EGM, et al: Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J Bacteriol. 2003, 185: 1018-1026. 10.1128/JB.185.3.1018-1026.2003.
Mhedbi-Hajri N, Darrasse A, Pigne S, Durand K, Fouteau S, Barbe V, Manceau C, Lemaire C, Jacques M-A: Sensing and adhesion are adaptive functions in the plant pathogenic xanthomonads. BMC Evol Biol. 2011, 11: 67-10.1186/1471-2148-11-67.
Ryan RP, Dow JM: Communication with a growing family: diffusible signal factor (DSF) signaling in bacteria. Trends Microbiol. 2011, 19: 145-152. 10.1016/j.tim.2010.12.003.
Tang JL, Liu YN, Barber CE, Dow JM, Wootton JC, Daniels MJ: Genetic and molecular analysis of a cluster of rpf genes involved in positive regulation of synthesis of extracellular enzymes and polysaccharide in Xanthomonas campestris pathovar campestris. Mol Gen Genet. 1991, 226: 409-417.
Vojnov A, Slater H, Newman M-A, Daniels M, Dow M: Regulation of the synthesis of cyclic glucan in Xanthomonas campestris by a diffusible signal molecule. Arch Microbiol. 2001, 176: 415-420. 10.1007/s002030100341.
Deng Y, Wu J, Tao F, Zhang L-H: Listening to a new language: DSF-based quorum sensing in Gram-negative bacteria. Chem Rev. 2011, 111: 160-173. 10.1021/cr100354f.
Slater H, Alvarez-Morales A, Barber CE, Daniels MJ, Dow JM: A two-component system involving an HD-GYP domain protein links cell–cell signalling to pathogenicity gene expression in Xanthomonas campestris. Mol Microbiol. 2000, 38: 986-1003.
Dow JM, Feng J-X, Barber CE, Tang J-L, Daniels MJ: Novel genes involved in the regulation of pathogenicity factor production within the rpf gene cluster of Xanthomonas campestris. Microbiology. 2000, 146: 885-891.
Dow M: Diversification of the function of cell-to-cell signaling in regulation of virulence within plant pathogenic Xanthomonads. Sci Signaling. 2008, 1: pe23-10.1126/stke.121pe23.
Wang L-H, He Y, Gao Y, Wu JE, Dong Y-H, He C, Wang SX, Weng L-X, Xu J-L, Tay L, et al: A bacterial cell–cell communication signal with cross-kingdom structural analogues. Mol Microbiol. 2004, 51: 903-912.
Qian W, Han Z-J, He C: Two-component signal transduction systems of Xanthomonas spp.: a lesson from genomics. Mol Plant Microbe Interact. 2008, 21: 151-161. 10.1094/MPMI-21-2-0151.
Wang X, Wang Q, Yang M, Xiao J, Liu Q, Wu H, Zhang Y: QseBC controls flagellar motility, fimbrial hemagglutination and intracellular virulence in fish pathogen Edwardsiella tarda. Fish Shellfish Immunol. 2011, 30: 944-953. 10.1016/j.fsi.2011.01.019.
Galperin M: A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 2005, 5: 35-10.1186/1471-2180-5-35.
Hickman AB, Chandler M, Dyda F: Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol. 2010, 45: 50-69. 10.3109/10409230903505596.
Salzberg S, Sommer D, Schatz M, Phillippy A, Rabinowicz P, Tsuge S, Furutani A, Ochiai H, Delcher A, Kelley D, et al: Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics. 2008, 9: 204-10.1186/1471-2164-9-204.
Holmquist M: Alpha/Beta-hydrolase fold enzymes: structures, functions and mechanisms. Curr Protein Pept Sci. 2000, 1: 209-235. 10.2174/1389203003381405.
Angkawidjaja C, Kanaya S: Family I.3 lipase: bacterial lipases secreted by the type I secretion system. Cell Mol Life Sci. 2006, 63: 2804-2817. 10.1007/s00018-006-6172-x.
Grissa I, Vergnaud G, Pourcel C: CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35 (suppl 2): W52-W57.
Kunin V, Sorek R, Hugenholtz P: Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8: R61-10.1186/gb-2007-8-4-r61.
Al-Attar S, Westra E, van der Oost J, Brouns S: Clustered regularly interspaced short palindromic repeats (CRISPRs): the hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol Chem. 2011, 392: 277-289.
Semenova E, Nagornykh M, Pyatnitskiy M, Artamonova II, Severinov K: Analysis of CRISPR system function in plant pathogen Xanthomonas oryzae. FEMS Microbiol Lett. 2009, 296: 110-116. 10.1111/j.1574-6968.2009.01626.x.
Blanco Y, Blanch M, Pinon D, Legaz M-E, Vicente C: Antagonism of Gluconacetobacter diazotrophicus (a sugarcane endosymbiot) against Xanthomonas albilineans (pathogen) studied in alginate-immobilized sugarcane stalk tissues. J Biosci Bioeng. 2005, 99: 366-371. 10.1263/jbb.99.366.
Huerta-Lara M, Rojas-Martinez RI, Bautista-Calles J, Reyes-Lopez D, Becerril-Herrera M, Romero-Arenas O, Franco-Mora O, Jimenez-Garcia D, Aragon-Garcia A, Simon-Baez A, et al: Genetic and pathogenic diversity of Xanthomonas albilineans (Ashby) Dowson, in Mexico. Res J Biol Sci. 2009, 4: 312-319.
Mohamed IS, Rott P, Davis MJ, Chatenet M: Differentiation of Xanthomonas albilineans strains based on multiplication of the pathogen in sugarcane varieties. Proc Int Soc Sugar-Cane Technol. 1996, 22: 486-492.
Champoiseau P, Daugrois JH, Pieretti I, Cociancich S, Royer M, Rott P: High variation in pathogenicity of genetically closely related strains of Xanthomonas albilineans, the sugarcane leaf scald pathogen, in Guadeloupe. Phytopathology. 2006, 96: 1081-1091. 10.1094/PHYTO-96-1081.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Thébaut P, Servant F, Schiex T, Kahn D, Gouzy J: JOBIM Conf Proc: 2000. 2000, Montpellier: ENSA, LIRM, 361-365.
Li L, Stoeckert CJ, Roos D: OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13: 2178-2189. 10.1101/gr.1224503.
Acknowledgements
IP was supported by an outgoing Masters Fellowship awarded by the Fondation Agropolis, France (AO2007 07 012) to work for three months at the University of Florida to perform SSH experiments. PR was supported by a Marie Curie Outgoing International Fellowship from the European Commission. We thank Dr Helen Rothnie for English editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IP and MR contributed to manual annotation of the genome, analysed the data, drafted part of the manuscript and coordinated the annotation project. IP also performed SSH experiments. VB, AC, SM, BS (Segurens) performed sequencing of the genome. SC (Carrere) and JG performed automatic annotation of the genome and OrthoMCL analysis. RK contributed to manual annotation of the genome and drafted part of the manuscript. CM, VV and MA conceived the study and revised the manuscript. AD, M-A J, EL, SP, BS (Szurek) contributed to manual annotation of the genome and revised the manuscript. DWG conceived and supervised the SSH study. PR and SC (Cociancich) conceived the study, contributed to manual annotation of the genome and drafted part of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4578_MOESM1_ESM.xls
Additional file 1: List of proteins from X. albilineans strain GPE PC73 putatively T2SS-secreted based on SignalP 4.0 prediction [http://www.cbs.dtu.dk/services/SignalP/].(XLS 60 KB)
12864_2012_4578_MOESM2_ESM.pdf
Additional file 2: LPS gene cluster comparison between X. albilineans strain GPE PC73, X. campestris pv. campestris strain B100 and X. campestris pv. vasculorum strain NCPPB702. Orthologs shared by at least two species are represented by arrows with identical colours. Specific genes for each species are shown by specific and neutral colour: grey for X. campestris pv. campestris, sky blue for X. albilineans and beige for X. campestris pv. vasculorum. HP = hypothetical protein. IS = insertion sequence. (PDF 173 KB)
12864_2012_4578_MOESM3_ESM.pdf
Additional file 3: Chemotaxis gene cluster comparison between Xanthomonas species. Orthologs shared by at least two species are represented by arrows with identical colours; the length of each arrow is proportional to the length in amino acids of each ORF. Specific proteins are shown by specific colours: grey for hypothetical protein, black for IS element or transposases, all other colours except pink for Methyl-accepting proteins and pink for other chemotaxis proteins. (PDF 244 KB)
12864_2012_4578_MOESM4_ESM.xls
Additional file 4: List of two-component signal transduction system identified in the genome of strain GPE PC73 of X. albilineans . Two-component system regulatory proteins (I), two-component system sensor proteins (II) and two-component system sensor-response regulator hybrid proteins (III) are listed. (XLS 34 KB)
12864_2012_4578_MOESM5_ESM.xls
Additional file 5: List of transposases found in the chromosome of the strain GPE PC73 of X. albilineans . The ORFs named “ISXal” are specific to X. albilineans. However, the recent publication of the complete genome sequence of X. oryzae pv. oryzicola revealed that XALc_2324, XALc_2365, XALc_2622 and XALc_2931, corresponding to ISXal5, ISXal7 and ISXal9 in X. albilineans strain GPE PC73, showed similarities with transposases found in the strain BLS256 of X. oryzae pv. oryzicola.(XLS 28 KB)
12864_2012_4578_MOESM6_ESM.pdf
Additional file 6: CRISPR-1 and CRISPR-2 spacer distribution in X. albilineans strain GPE PC73. Each box represents a CRISPR spacer, with the spacer positions numbered inside each box from the trailer end spacer to the leader end spacer. Pink boxes = CRISPR-1 spacers. Green boxes = CRISPR-2 spacers. Spacers showing nucleic acid identity with sequences of strain GPE PC73 of X. albilineans are listed in three separated tables according to the origin of these sequences (a same prophage region located between XALc_0170 to XALc_0242, phage and plasmid sequences and a housekeeping gene, respectively). Some spacers are 100% identical to corresponding sequences of strain GPE PC73 of X. albilineans. Percentage value is indicated in brackets only for spacers for which the percentage identity is less than 100%. (PDF 126 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pieretti, I., Royer, M., Barbe, V. et al. Genomic insights into strategies used by Xanthomonas albilineans with its reduced artillery to spread within sugarcane xylem vessels. BMC Genomics 13, 658 (2012). https://doi.org/10.1186/1471-2164-13-658
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-658