
Abstract
Background
HacA/Xbp1 is a conserved bZIP transcription factor in eukaryotic cells which regulates gene expression in response to various forms of secretion stress and as part of secretory cell differentiation. In the present study, we replaced the endogenous hacA gene of an Aspergillus niger strain with a gene encoding a constitutively active form of the HacA transcription factor (HacACA). The impact of constitutive HacA activity during exponential growth was explored in bioreactor controlled cultures using transcriptomic analysis to identify affected genes and processes.
Results
Transcription profiles for the wild-type strain (HacAWT) and the HacACA strain were obtained using Affymetrix GeneChip analysis of three replicate batch cultures of each strain. In addition to the well known HacA targets such as the ER resident foldases and chaperones, GO enrichment analysis revealed up-regulation of genes involved in protein glycosylation, phospholipid biosynthesis, intracellular protein transport, exocytosis and protein complex assembly in the HacACA mutant. Biological processes over-represented in the down-regulated genes include those belonging to central metabolic pathways, translation and transcription. A remarkable transcriptional response in the HacACA strain was the down-regulation of the AmyR transcription factor and its target genes.
Conclusions
The results indicate that the constitutive activation of the HacA leads to a coordinated regulation of the folding and secretion capacity of the cell, but with consequences on growth and fungal physiology to reduce secretion stress.
Similar content being viewed by others


Background
The secretion of extracellular proteins is very important to the natural saprophytic lifestyle of Aspergillus niger. The inherent ability of efficient protein secretion, found among several Aspergillus species such as A. niger and A. oryzae, has led to their biotechnological exploitation as hosts for homologous and heterologous protein production [1–5]. As protein yields for heterologous proteins are often reported as low, efforts have been made in order to describe and understand the processes that limit their secretion [6, 7], as well as efforts to prevent proteolytic activity outside the cell [4, 8, 9].
Secretory proteins begin their journey by entering the endoplasmic reticulum (ER) where they are assembled, folded and modified. Then, they are packed into COPII coated vesicles and transported into the Golgi-like structures where further modifications take place. Proteins destined for secretion are packed into secretory vesicles to be transported to the tip of the growing hyphae, where the proteins are released extracellularly [6, 10, 11]. Among the factors that disturb efficient secretion of heterologous proteins is the mis-folding of these proteins in the ER and the consequence that those proteins are recognized as mis-folded by the Quality Control system present in the ER [12, 13]. The presence or accumulation of aberrant proteins in the ER may become fatal to the cell and to deal with the presence of mis-folded proteins in the ER, eukaryotic cells react with the expression of several genes related to protein folding and degradation, a response termed the Unfolded Protein Response (UPR) [14]. The basic sensing pathway to detect ER stress or an increase in folding load is highly conserved from yeast to man. In Saccharomyces cerevisiae, the sensor protein is Ire1p which is an ER-resident trans-membrane protein that contains a luminal domain that functions as the sensor of the folded state of the proteins, and has a site-specific endoribonuclease (RNase) domain at the cytoplasmic C-terminus [15, 16]. The accumulation of unfolded proteins is sensed through a dynamic interaction between Ire1p and the chaperone Kar2p (also known as Bi nding P rotein - BiP) [17, 18] or by direct sensing by Ire1p [19]. As BiP/Kar2p is recruited to help with the folding of the ER accumulating proteins, its release from Ire1p leads to the oligomerization of Ire1p proteins. In turn, the formed Ire1p oligomer is activated by autophosphorylation and the RNase domain is responsible for the splicing of a 252 nt intron present in mRNA of the bZIP transcription factor Hac1p (HacA in filamentous fungi and XBP-1 in the mammalian system), a process well characterized in fungi [20–22] and higher eukaryotes [23–26]. Alternatively, from the known structures of the yeast and human lumenal and cytoplasmic domains of Ire1p, a model for direct binding of Ire1p to unfolded proteins has been postulated that leads to structural changes in Ire1p, oligomerization and activation of the kinase and endoribonuclease domains [16, 18, 27, 28]. In A. niger , the hacA mRNA splicing event results in the excision of a 20 nt intron [29], releasing it from a translational block [30]. Although it has not yet been shown in the S. cerevisiae or mammalian homologues, in addition to the intron splicing, the hacA mRNA of A. niger Aspergillus nidulans and Trichoderma reesei is truncated at the 5’-end during UPR induction [31, 32]. However, Mulder and Nikolaev [30] showed that in A. niger truncation of hacA is not a requirement for induction of the pathway. Once translated, HacA migrates into the nucleus where it binds to palindromic UPR elements at the promoter regions of UPR targets [32]. Transcriptome analysis under UPR inducing conditions in both fungi and mammalian cells has revealed the induced expression of subsets of genes involved in folding, secretion, phospholipid biosynthesis and protein degradation [14, 33–35]. Most of the UPR studies performed have induced this pathway through the presence of harsh chemicals (DTT or tunicamycin), which by itself may impose collateral responses that might provoke ER stress, and by expressing heterologous proteins such as tPA and chymosin [35–37]. However, a recent study has illustrated that the induction of UPR-target genes may not be a stress response only induced by the presence of mis-folded proteins, but may represent a more physiologically natural mechanism required and induced under conditions where there is a demand for an increased secretion capacity [38].
Manipulation of the UPR pathway and its components, like BiP1 and PDI [39–41], has been a common approach to improve the secreted production of heterologous proteins. Valkonen et al. [42] have shown, in S. cerevisiae, that controlling Hac1p expression has effects on native and foreign protein production; hac1 deletion led to a decrease of heterologous α-amylase and endoglucanase production whereas overexpression of this transcription factor resulted in an increase in the production of these proteins when compared to the respective parental strains. Similar results have been demonstrated in A. niger var awamori, where a constitutive induction of the UPR pathway enhanced the production of heterologous laccase and of bovine preprochymosin [43]. The UPR is activated to alleviate the stress caused by the accumulation of mis-folded protein in the ER lumen by improving protein folding, degrading unwanted proteins [14, 37] and reducing the entry of secretory proteins into the ER, a mechanism known as REpression under Secretion Stress (RESS) [44]. Studies have shown that there is a selective down-regulation of genes coding extracellular enzymes in the presence of chemicals which inhibit protein folding [44–46].
In this study, we present a genome-wide overview of the HacA responsive genes by comparing the transcriptomic profiles of two genetically engineered A. niger strains expressing either the wild-type hacA gene or the active form of the HacA transcription factor. The comparison suggests HacA as a master regulator, coordinating several processes within the secretory pathway such as the induction of protein folding, protein glycosylation and intracellular transport. Additionally, we discovered that constitutive activation of HacA results in the down regulation of the AmyR transcription factor and the AmyR regulon, which includes the most abundantly produced extracellular glycoproteins, thereby reducing import of new proteins into the ER. The down-regulation of the AmyR regulon revealed by the genome wide expression analysis was phenotypically confirmed as the HacACA mutant displayed a strongly reduced growth phenotype on starch plates.
Results
Construction and analysis of a strain expressing a constitutively activated form of hacA
To obtain an A. niger strain with a constitutively activated HacA (HacACA) transcription factor, the wild-type hacA gene was replaced by the spliced form of hacA that lacks the 20 nucleotide intron. For the construction of a reference strain and a strain only expressing the hacA induced form, plasmids pHacWT and pHacCA were used [Additional file 1 (A and B)]. Transformants with the correct integration pattern for each plasmid were selected after Southern blot analysis (data not shown) and the absence of the intron was confirmed in the HacACA strain [Additional file 1 (C and D)]. Growth assays were performed with both strains at different temperatures (Figure 1A and B). At each temperature tested, radial growth rate (colony size) of the HacACA strain was reduced compared to the HacAWT strain, and this growth impairment was more pronounced at 37 and 42 °C (Figure 1A). Differences in phenotype between both strains were also apparent as HacACA showed a delay in growth and conidiation in comparison to HacAWT (Figure 1B). As no phenotypic differences were found between our reference strain HacAWT and N402 (data not shown), we conclude that the phenotypic effects observed in HacACA are due to the presence of only the UPR-induced form of hacA. The effects of having a constitutive activation of the UPR are different from the absence of a functional UPR. The deletion of the HacA transcription factor in A. niger has a profound effect on growth and morphology of the fungus, resulting in smaller and more compact colonies that hardly form conidia [30, 47].

Growth and phenotypic profiles of HacAWTand HacACAstrains. (A) Differences on colony size (diameter) of HacAWT and HacACA strains growing at different temperatures. 104 spores were spotted on solid CM plates and growth was monitored for 6 days. (B) Strains phenotype on CM after 3 and 6 days of growth at 30 °C. HacACA phenotype is characterized by a slower growth/colony size as well as a delay in sporulation compared to the HacAWT. Bars indicate standard deviations from three individual measurements.
Physiological consequences of the constitutive hacA activation in batch cultivations
Growth of batch cultures of the A. niger HacAWT and HacACA strains was characterized as filamentous and highly reproducible. The growth kinetics of a representative culture of each strain is shown in Figure 2 and results from all cultures are given in the supplemental material [Additional file 2. Cultures of the HacAWT strain exhibited exponential growth with a specific growth rate (μ) of 0.22 ±0.01 h-1 (n = 4) from exit of lag phase to depletion of glucose (Figure 2A). Initial growth of HacACA was similar to that of the HacAWT; it was exponential with a μ of 0.21 ±0.01 h-1 (n = 3). However, after 21–22 h of batch cultivation, when half of the glucose was consumed, the growth kinetics shifted from exponential to apparently linear (Figure 2B). It was not clear from the relatively few determinations of biomass concentration whether growth was truly linear in the second phase but this was strongly supported by analysis of the growth-dependent alkali addition (inset Figure 2A, B). We established a concordance between growth and alkali added to maintain constant pH in the cultures (not shown), and used this as an indirect measure of growth as described previously by Iversen et al. [48]. Linearity was then confirmed by log-transformation of alkali addition rates using the computer recorded titrant addition data and the LOS program [49]. During exponential growth, growth yield on substrate (Yxs) was comparable in both strains: 0.53 ± 0.02 for HacAWT and 0.52 ± 0.04 for HacACA.

Growth profiles of one of the triplicate A. niger HacAWT(A) and HacA CA(B) batch cultures. Dry weight biomass concentration (gDWkg-1) as a function of time (h) illustrates the growth of the cultures. The maximum specific growth rate for each culture was determined from the slope (α) of the ln transformation of biomass (Cbiomass) in the exponential growth phase as a function of time (h), as well from log transformation of alkali addition as a function of time (h). Dash-line represents the end of the exponential growth phase (depletion of glucose). Arrows indicate time-points where mycelium was harvested for transcriptomic analysis.
Impact of the constitutive activation of hacA on the transcriptome of A. niger
Three independent bioreactor cultures with the HacAWT strain were performed. From each cultivation experiment, biomass was harvested from the mid-exponential growth phase (biomass concentration 1.5 gr/kg (Figure 2A)) and used for RNA extraction and subsequent microarray analysis (time point 1; HacAWT-1; [glucose] = 5.8 g/L). Likewise, for the HacACA strain three bioreactor cultivations were performed and biomass was harvested from each culture and RNA was isolated from the mid-exponential time point (time point 1; HacACA-1; [glucose] = 5.7 g/L) (Figure 2B). Global transcription profiles were determined in triplicate for mid-exponential growth phase of HacAWT strain cultures and at the corresponding biomass concentration for the HacACA strain cultures, represented by the arrows in Figure 2A and 2B. For the HacACA cultures, RNA was extracted from two additional time points subsequent to the shift to linear growth and the RNA was also analyzed (time point 2 and 3; HacACA-2; [glucose] = 3.5 g/L and HacACA-3; [glucose] = 1.2 g/L) (Figure 2B). Thus, the data set in this study consists of four groups of triplicate biological replicates of HacAWT and HacACA at three time points (HacACA-1, HacACA-2 and HacACA-3). The reproducibility of triplicate array analyses was high with a mean coefficient of variation (CV) ranging from 0.12 to 0.14 for transcripts rated as present or marginal.
The number of differentially-expressed genes (FDR <0.005) in a pair-wise comparison are given in Table 1. In response to constitutive activation of hacA at time point 1 (HacACA-1), 1235 genes were differentially expressed. The number of differentially expressed genes increased when comparing the later time points (HacACA-2 and HacACA-3) to the wild-type strain to give a total number of 1698 and 1978 differentially expressed genes. Table 1 also shows that the transcriptomic differences between the different time points of the constitutive HacA strain were relatively minor (48 and 179 differentially expressed genes comparing HacACA-2 vs. HacACA-1 and HacACA-3 vs. HacACA-1 respectively). Comparison of HacACA-2 with HacACA-3 revealed very similar transcriptomes and with the stringent FDR of <0.005, no differentially expressed genes were detected. As a start to analyse the expression data, Venn diagrams were made to identify genes that were differentially expressed in HacACA at all three time points when compared to the wild-type strain. As shown in Figure 3A, 616 genes were up-regulated in the constitutive HacA strain at all three time points and 433 genes were down-regulated (Figure 3B). A complete list of all expression data and the FDR-values for the pair-wise comparison of the different strains and time points is given in [Additional file 3].

Analysis of differentially expressed genes in HacACAat all three time points in comparison to HacAWT. Venn diagrams of the number of overlapping and non-overlapping induced (A) or repressed (B) genes on A. niger HacACA mutant strain at different time points in comparison to HacAWT strain
From the 616 up-regulated genes [Additional file 4 we were able to retrieve 598 upstream regions. These upstream regions were analysed for the presence of UPRE sequences (5’-CAN(G/A)NTGT/GCCT-3’, [32]). From the up-regulated genes in the HacACA strain, we found 47 genes that contained at least one UPRE sequence within the 400 bp region up-stream their start codon [Additional file 5. Compared to the frequency of UPRE in the 400 bp up-stream region of the remaining non up-regulated genes (457 out of 13156) a statistical significant enrichment (p ≤ 5.4 × 10-7) was assessed with the Fisher's exact test (one-sided). Although this analysis indicates a statistical enrichment for genes containing a HacA binding site in the promoter region of HacA induced genes, it shows that only about 10% of the HacACA induced genes contain a putative HacA binding site. It suggests that either the currently used HacA binding consensus site is too stringent and that additional sequences allow HacA to bind, or that additional transcription factors are involved in the induction in response to the constitutive activation of HacA. The data set of HacA induced genes with a putative UPRE site include genes related to protein folding (as previously described [32]), lipid metabolism, transport within the cell, glycosylation, ER quality control as well as a large set of genes that code for hypothetical and unknown function proteins [Additional file 5.
Identification of biological processes enriched in the transcriptomic profiles of the HacACA strain
To obtain an overview of the processes affected at the transcriptional level between the HacAWT and the HacACA-1 mutant, overrepresented GO-terms among differentially expressed genes were identified. For this analysis, we used the Fisher's exact test Gene Ontology annotation tool (FetGOat) [50]. Network maps of related GO-terms (Biological Processes), over- or under-represented in the HacACA strain, are given in Additional file 6 and Additional file 7. In Additional file 8 and Additional file 9, the results of the GO-enrichment analysis are given. To analyse the results, two complementary approaches were taken. Firstly, we rationally defined GO-terms of higher order that include several GO-terms. Secondly, we looked specifically at GO-terms that are terminal in the network, as these annotations are the most detailed (see Additional file 6). These approaches enabled us to identify four major categories of genes to describe the most relevant up-regulated biological processes in the HacACA strain (Figure 4). The four main categories of genes included those related to I) ER translocation and protein folding [Additional file 10, II) intracellular vesicle trafficking [Additional file 11, III) protein glycosylation [Additional file 12 and IV) lipid metabolism [Additional file 13. These four main categories are further described in the following section.

Functional classification of differentially expressed genes in HacACA. Representation of the main significant induced and repressed biological processes in the HacACA mutant strain in comparison to HacAWT strain.
In the HacACA strain we found enriched GO-terms linked to ER processes, such as those related to entry in the ER: signal particle recognition, cleavage of signal sequence, and translocation (e. g. Sec61 and related subunits). In addition to the processes that mediate the recognition, targeting and entering of proteins into the ER, enriched GO-terms also included a large number of genes involved in the subsequent events of protein folding and quality control. The genes related to protein folding included the well known HacA targets such as bipA pdiA tigA and prpA[32]. After being synthesized and folded properly in the ER, proteins are packed in vesicles and transported to the Golgi and from there on, further transported to reach their final intra- or extra-cellular destination. Our analysis identified a number of genes that encode proteins that take part in the vesicle/trafficking machinery such as those involved in ER-to-Golgi (COPII associated components), Golgi-to-ER (COPI transport vesicles, Sec components) and Golgi to endosome transport. Additionally, genes involved in exocytosis were also induced (Figure 4). GO-terms related to processes involving protein glycosylation, were up-regulated in the HacACA strain. The processes include genes involved in sugar nucleotide synthesis, oligosaccharyl synthesis (ALG-genes) and transfer (OST-complex) of the preassembled oligosaccharide to certain asparagine residues (N-glycosylation). In addition, genes related to the addition of O-glycans (genes homologous to the S. cerevisiae Pmt-family and Kre2-family of mannosyltransferases) were up-regulated. Finally, several genes related to the synthesis and transfer of glycosylphosphatidylinositol (GPI) anchors to proteins were found to be up-regulated. Additional file 12 lists the differentially expressed genes with a proposed function in relation to protein glycosylation or GPI-anchor attachment. In addition, the constitutive activation of HacA has a pronounced effect on the transcription of genes involved in phospholipid metabolism and includes proteins that are homologous to proteins involved in ergosterol biosynthesis as well as proteins involved in the metabolism of fatty acids and inositol [Additional file 13. Categories containing fewer GO-terms included terms related to intracellular pH regulation and terms related to glutathione catabolic processes [Additional file 14.
Concerning the biological processes over-represented in the down-regulated set of genes we found one major category linked to the central metabolic pathways (Figure 4 and Additional file 15). This category includes genes within glycolysis/gluconeogenesis; alcohol catabolic/metabolic process; carboxylic acid cycle and carbon metabolic/catabolic metabolism. Categories containing fewer GO-terms included terms related to transporters and response to oxidative stress. The down-regulation of genes in central metabolic pathways may reflect the growth limitation observed in the HacACA mutant (Figures. 1 and 2).
Common and different features of the constitutive activation of HacA and the UPR induction by chemicals or heterologous protein expression
To gain a broader overview of the impact of a constitutive activation of HacA on A. niger we compared our data set (HacACA-1/HacAWT) with the data of Guillemette and co-workers ([37]; [Additional file 3 and Additional file 16) in which the genome-wide transcriptional protein secretion-related stress responses was analyzed. In this study [37], transcriptional targets of the UPR pathway were identified by treatment of A. niger with the ER-disturbing chemical agents tunicamycin and dithiothreitol (DTT) and using a strain producing the recombinant tissue plasminogen activator (t-PA) as a model for heterologous protein production. As shown in Figure 5, in the induced set of genes, 13 genes are commonly unregulated in both studies (all conditions) and 81 genes are differentially expressed in HacACA-1/HacAWT in at least two of the three conditions performed by Guillemette et al. [37]. These 94 commonly induced genes include all the genes identified in the Guillemette et al.’ study related to protein folding, translocation/signal peptidase complex and glycosylation and most of the genes that belong to the categories of vesicle trafficking and lipid metabolism [Additional file 16. However, more genes belonging to each of these categories have been identified in the HacACA-1/HacAWT comparison (Figure 4 and Additional file 10, Additional file 11, Additional file 12, Additional file 13). Unique genes found in at least two of the conditions tested (56) and not in our data set relate mainly to the categories of cellular transport, stress related, amino acid metabolism, carbohydrate metabolism and unclassified genes.

Analysis of differentially expressed genes in HacACA(this study) and Guillemette’ study [[37]] . Venn diagrams of the number of overlapping and non-overlapping induced or repressed genes of A. niger HacACA-1/HacAWT(A) and genes from Guillemette et al. (2007) induced (or repressed) at least in two conditions (B) or induced in all conditions (C).
For the repressed set of genes we found 45 common genes to our study and Guillemette et al. [37] which are evenly distributed throughout the categories established by the authors (Additional file 6 in [37]). The fact that the number of commonly down-regulated genes is small between the two studies suggests important differences and heterogeneous responses to the induction of the UPR indirectly (chemicals and heterologous protein) and the manipulation of the transcription factor that regulates this pathway in the overall cell metabolism.
The constitutive activation of HacA triggers the induction of ERAD genes
Secretory proteins that fail to fold properly usually accumulate in the ER and are sooner or later targeted to destruction by the proteasome, a process termed ER-associated degradation (ERAD) [51]. Genes encoding proteins that are putatively involved in ERAD have been identified in the A. niger genome [52, 53] and the expression of these genes was examined in the microarray data set. As highlighted in Table 2, the expression of several putative ERAD components was induced in the HacACA mutant. For instance, the der1 homologue (derA, An01g00560), involved in transport of unfolded proteins out of the ER [54], is 4.0-fold induced; hrd3 (hrdC, An03g04600), involved in recognition and presentation of the substrate for degradation [55], is 3.3-fold induced. The mifA (An01g14100) gene, a homologue of mammalian herp1/mif1 protein and suggested as the link between the UPR and ERAD pathways [56], is 3.1-fold induced. Furthermore, mns1 (mnsA, An18g06220), a mannosidase that by removal of 1,2 α-mannose units targets the substrate to degradation [57], is 4.2-fold induced. In comparison to Travers et al. [14], our study allowed us to unravel the regulation of other ERAD related genes in relation to UPR, such as mns1 mif1, a DSK2 homologue An08g09000, putatively encoding a ubiquitin-like protein) (1.8-fold induction) and another putative α-mannosidase (An12g00340, 3.2-fold induced).
Constitutive activation of HacA leads to the down-regulation of the AmyR regulon
Although an increase in expression of secretion related processes (folding, glycosylation, vesicle transport) is observed in the HacACA strain, the expression of several genes encoding secreted proteins is down-regulated [Additional file 15. In addition, expression of the AmyR transcription factor was repressed under these conditions (−3.3 fold, FDR < 10-5). Starch is a polymeric carbon source consisting of glucose units joined together by alpha1,4- and alpha1,6-glycosidic bonds and naturally synthesized by plants. A. niger is able to degrade starch by secreting various amylases that convert starch into maltose and glucose [58]. The transcription of these amylolytic enzymes is mediated by AmyR [59]. The AmyR regulon has been defined and consists of several alpha-glucosidases as well as two sugar transporters [58, 60]. Our transcriptome profiles show that the enzymes and sugar transporters in the AmyR regulon are commonly down-regulated (Table 3).
The down-regulation of genes involved in starch degradation and uptake suggested that the HacACA mutant’s growth may be severely affected on starch as sole carbon source. In order to test this, we performed growth tests of HacACA together with HacAWT and a ΔamyR strain in which the AmyR-encoding gene has been deleted [58] on solid media containing starch or its derivatives in a range of different complexity (Figure 6).

Effects of the constitutive activation of the UPR on the utilization of starch and starch related carbon sources. The wild-type strain (HacAWT), the strain containing a constitutive active form of hacA (HacACA) and the AmyR disruptant (ΔamyR) strain were grown on MM containing 1% of the different carbon sources indicated at 30 °C for 3 days.
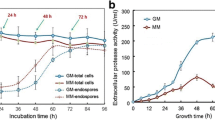
As predicted from the transcriptomic data and similar to the ΔamyR strain, HacACA was unable to grow on the plate containing starch as sole carbon source. With the aim of testing if this reduced growth was specific for growth on starch or if it would apply to other complex carbohydrates, we performed a similar test on other polymers, inulin, xylan and pectin and respective monomeric substrates, fructose, xylose and galacturonic acid (Figure 7). In addition, growth of the HacACA strain was analysed on milk-plates (Figure 7). These results show that the HacACA strain is growth impaired when challenged to assimilate nutrients from complex substrates. Although this was not so evident when grown on inulin, growth of the HacACA strain was clearly further reduced on xylan, pectin and milk-plates suggesting that the down-regulation of extracellular enzyme expression is not limited to the amylolytic genes, but also for xylanolytic, pectinolytic and proteolytic genes.

Effects of the constitutive activation of the UPR on the utilization of different polimeric and monomeric carbon sources. The wild-type strain (HacAWT), the strain containing a constitutive active form of hacA (HacACA) the amyR disruptant (ΔamyR) and inuR disruptant (ΔinuR) strains were grown on MM containing 1% of the different carbon sources or 1% dried skim milk at 30 °C for 4 days.
Discussion
Genome-wide gene expression variations upon constitutive activation of HacA
Using a defined A. niger strain bearing a constitutively active form of HacA (HacACA), the key regulator of the UPR pathway in eukaryotic cells, together with Affymetrix GeneChips technology, we have defined a large set of HacA-responsive genes. Unlike other studies, in which the hacA mRNA splicing is stimulated by the presence of unfolded proteins in the ER by chemicals or by expression of heterologous proteins [29, 37], we used a different approach by creating a strain lacking the 20 nt intron in the hacA gene. To minimize additional effects of expressing the constitutive form of hacA, the hacACA gene was targeted to its endogenous locus. This contrasts to previous studies in which the constitutive hacA was expressed from a highly-expressed promoter [43] or expressed from the pyrG locus [30]. The microarray data revealed, even under stringent criteria (False Discovery Rate at q < 0.005), a large number of differentially-expressed genes (1235 to 1978) upon HacA activation (Table 1). The transcriptomic data obtained in our study reflects the consequences of a constitutive activation of the HacA transcription factor that results in the induction of many genes associated with the secretory pathway (Figure 4) and related to ER translocation, glycosylation, folding, quality control, ERAD, GPI anchor biosynthesis, vesicle-mediated transport between organelles (ER-Golgi), lipid metabolism, endocytosis and vacuolar sorting. Because of the highly defined conditions (both the defined mutants and the bioreactor controlled cultivations), this study revealed new categories of differentially-expressed genes as well as a much larger number of genes related to each category. Our data are however consistent with previous UPR-related studies in fungal and mammalian cells where many secretory functions are up-regulated by Hac proteins, either directly or indirectly [14, 33–35, 37].
Our results from the transcriptomic study also revealed that constitutive activation had a negative effect on central metabolism as well as on the production of extracellular enzymes. Although a clear growth reduction was observed for the HacACA strain on milk-plates (Figure 7), none of the main extracellular proteases (PepA, PepB, PepD or PepF)) [8] was shown to be transcriptionally down-regulated under the bioreactor growth conditions (glucose and ammonium) (Additional file 17). Possibly, the effect of downregulation of these enzymes in the HacACA strain is only occurring during inducing conditions, which might explain the reduced growth on milk-plates. The expression level of prtT, which encodes the transcriptional activator of extracellular proteases [8] was significantly down-regulated in the HacACA strain (Additional file 17), but this has apparently no effect of the four target genes indicated above.
As the global mechanisms for energy generation and cell development are arrested or directed towards up-regulation of the protein secretion machinery, this might account for the unbalanced growth observed in HacACA in comparison to the HacAWT (Figure 2). These results suggest an implication for heterologous protein production if the protein causes ER stress. Studies on increasing heterologous protein production by enhancing UPR targets are contradictory and vary according to the protein expressed. Although protein-specific effects are likely, most studies were not controlled for the levels of chaperones or foldases co-expressed and it has been shown that there is an optimum level of both BipA [61] and PdiA [62].
GO-enrichment analysis on the induced set of genes showed that all the well-known UPR target genes related to folding are represented in the HacACA data set, and include genes encoding the chaperone BipA, and homologues of LhS1p (An01g13220), P58PK (An11g11250) and Scj1p (An05g00880), as well as the protein disulfide isomerases PdiA, PrpA and TigA . Glycosylation also appeared as one of the enriched categories. Several aspects of protein glycosylation including the categories of oligosaccharide-lipid assembly, oligosaccharyl transferase complex, UDP-glucose transport, O-linked glycosylation and GPI anchor biosynthesis (Figure 4), were up-regulated indicating that the cell responds to ER stress by increasing the capacity to glycosylate proteins. The induction of genes associated with lipid metabolism [Additional file 13 suggests a proliferation of the ER to bear the increase of proteins that reside in this organelle, as also indicated in UPR studies of S. cerevisiae[14].
The elimination of unfolded proteins from the ER involves the ERAD pathway [51]. Travers et al. [14] demonstrated that up-regulation of ERAD-related genes in S. cerevisiae is part of the UPR. These ERAD genes include DER1 and HRD3, UBC7, the ubiquitin-related DOA4, the proteasome-related PEX4 and translocon-related SEC61 [14]. From the ERAD components defined in A. niger[52], 11 out of 20 genes are induced in the HacACA strain (Table 2). Furthermore, analysis of the 400 bp of the up-stream regions of derA (An15g00640), sec61 (An03g04340) and An04g06990 (high similarity with a human 1,2-mannosidase) revealed that these genes contain at least one UPRE sequence [Additional file 5. These results support the connection between the two pathways, as previously suggested [14, 53, 63, 64], although the mechanistic connection between the two pathways is unresolved. We compared our data sets with those in Guillemette et al. [37] and found broad agreement with a wide range of up-regulated genes under ER stress conditions. However, Guillemette et al. [37] showed trigger-specific responses that do not complicate our analyses with HacACA. Additionally, we find several putative translation initiation factors [Additional file 4, An18g06260 (highly homologous to the mammalian eIF3), repressed in HacACA-1 and putative elongation factors An11g10630, An14g01030, An16g06850, An16g05260, An01g06230, An06g01710, An02g12320, An02g12420 and An04g01940 repressed in the other time points (HacACA-2 and/or HacACA-3).
New leads on the RESS mechanism
The accumulation of misfolded protein in the ER leads to a selective down-regulation of genes encoding secreted proteins in fungi and plants [44–46, 64]. This phenomenon is termed REpression under Secretion Stress (RESS). In these studies, associated with the UPR activation by chemical induction is the down-regulation of transcription encoding extracellular enzymes that include cellulases and xylanases in T. reesei[44] and glucoamylase in A. niger[46] amongst other genes encoding secreted proteins [37]. The mechanism by which the down-regulation is mediated is unknown, but glaA promoter studies in A. niger indicated that a promoter region between 1 and 2 kb upstream of translational start is important and a direct mediation of RESS through the UPR was questioned [46]. RESS has been recognized as an effort from the cells to prevent the entry and overload of newly synthesized proteins into the already “full” ER [44, 46, 64]. In our study, the activation the UPR by introducing the constitutive active form of the HacA transcription factor lead to the down-regulation not only of glucoamylase (glaA), but also other genes coding for starch-degrading enzymes that include acid α-amylase (aamA), α-glucosidases A and B (agdA and agdB) and α-amylase C (amyC), and additional sugar transporters (Table 3). In addition, the expression of the transcriptional activator of starch degrading enzymes is down-regulated (3.3-fold) in the HacACA strain. It has been shown previously that the AmyR transcription factor is induced (2.6-fold) upon the shift from xylose to maltose medium, suggesting that this down-regulation is biologically relevant. The down-regulation of the AmyR regulon and sugar transporters (Table 3) had a clear phenotypic effect resulting in the inability of the HacACA strain to grow on starch (Figure 6). Growth assays on other polymeric substrates (Figure 7) suggested that the down-regulation might not to be specific for starch but is relevant to other sugar polymers including xylan (Figure 7). Several scenarios can be envisioned by which the constitutive activation of HacA could result in down-regulation of secreted enzymes. We speculate that HacA activation leads to inactivation of the transcriptional factor such as AmyR (starch regulator), and possibly XlnR (xylan regulator). The inactivation results in down-regulation of the entire regulon of the transcription factor. However, a direct effect of HacA-mediated effects on individual promoters cannot be excluded. It will be of interest for future studies to determine the molecular mechanism that results in the down-regulation of AmyR and AmyR target genes in response to HacA activation.
Relation between yeast, filamentous fungi and mammalian UPR counterparts
The mammalian ER contains three types of transmembrane proteins – IRE1P, PERK and ATF6 – which sense the accumulation of unfolded proteins and are responsible to activate three different branches of the UPR pathway (reviewed in [65]). Most of the players in the IRE1P pathway are conserved in fungi [66] in which, by activation of the transcription factor Hac1p/HacA, there is an induction of expression of UPR target genes related to the folding machinery [20, 29], but proteins homologous to PERK and ATF6 seem to be absent from fungal systems.
To prevent the influx of proteins into the ER in mammalian cells, a mechanism of translation attenuation is activated that is mediated by PERK. This transcription factor mediates the phosphorylation of eIF2 (eukarytotic translation initiation factor) which in turn leads to the arrest of protein translation. The eIF2 is also required for the translation of selective mRNAs such as the Activating Transcription Factor-4 (ATF4) [67]. ATF4 is involved in the regulation of UPR genes involved in ERAD, metabolism and apoptosis [68]. Gcn4p/CpcA are the ATF4 homologues of S. cerevisiae and filamentous fungi, respectively. Both S. cerevisiae and A. niger lack an obvious PERK homologue. Gcn2p phosphorylates eIF2 leading to a global reduction on protein synthesis and stimulation of Gcn4 translation, that has been shown to control amino acid biosynthesis [69]. Although this resembles the PERK function, Gcn2p–eIF2 phosphorylation is only attributed to amino acid starvation and not to ER stress [70, 71]. In S. cerevisiae, the involvement of Gcn2p and Gcn4p in the UPR has been shown [72]. In our transcriptomic profiles, a gcn2 homologue (An17g00860) is not differentially expressed, whereas cpcA (An01g07900) shows ≈ 2 fold higher expression in comparison with the wild-type strain. According to our results the activation of cpcA is likely to occur in a Gcn2p-independent way and it is tempting to speculate that in filamentous fungi a similar PERK-eIF2-ATF4 pathway may exist. ATF4 is involved in glutathione biosynthesis [73] and glutathione-S-transferases have been shown to be up-regulated under ER stress conditions [74]. According to our data, the homologue to human glutathione-S-transferase 3 (An12g03580) is 2-fold induced in HacACA-1 and 2.6 fold induced at the later time points. What we also observe is that as in the case of ATF4-regulated genes, not all the genes involved in glutathione metabolism are affected under secretion stress situation [73], as for example asparagine synthase (An01g07910) or glutathione reductase (An03g03660) that are not differentially expressed. Similar results have been observed in T. reesei[35].
Another interesting observation is the 4-fold induction of the human homologue RNA-activated protein kinase inhibitor P58 (An11g11250). In mammals, P58 is induced via ATF6, a transcription factor also involved in the regulation of UPR chaperones and apoptosis (no homologue in fungi), and it is an important component on the regulation of PERK-eIF2-ATF4 pathway, attenuating the UPR [75]. The up-regulation of P58 has been shown in studies characterizing the UPR under different conditions [37, 38]; however, the role and (putative) involvement of a fungi P58 homologue in this pathway remains to be elucidated. ATF6, that induces XBP1 (HacA homologue), also possesses the ability to enhance lipid biosynthesis and expansion of the ER [76]. The identification of these potential regulatory genes involved in mediating the HacA response in this study has given multiple new leads for further research to better understand the mechanism of how A. niger reacts to secretion stress.
Conclusions
The combination of a genetic defined constitutively activated HacA transcription factor mutant and controlled bioreactor cultivation conditions have provided a solid basis for a genome-wide expression analysis to study the response of A. niger towards ER stress. Comparison of the transcriptome obtained form the constitutive HacA mutant to previous studies in which ER stress was induced by chemical treatments or the expression of a heterologous protein revealed a consistent up-regulation of genes associated with the secretory pathway. Because of the highly defined conditions and reduced heterogeneity in our cultures, this study revealed new categories of differentially expressed genes as well as a larger number of genes related to individual categories. We also show that constitutively activation of the HacA transcription factor has a negative effect on the expression and consequently the production of extracellular enzymes. We conclude that activation of HacA induces a dual response to cope with ER stress: increasing the folding capacity of the cell by the up-regulation of genes related to secretion processes in the ER on the one hand and reducing the import of new proteins into the ER by reducing the expression of genes encoding secreted proteins on the other hand.
Methods
Strains and culture conditions
Aspergillus niger strains used throughout study (Table 4) were cultivated in minimal medium (MM) [77] containing 1% (w/v) of glucose (or other as indicated) as a carbon source, 7 mM KCl, 11 mM KH2PO4, 70 mM NaNO3, 2 mM MgSO4, 76 nM ZnSO4, 178 nM H3BO3, 25 nM MnCl2, 18 nM FeSO4, 7.1 nM CoCl2, 6.4 nM CuSO4, 6.2 nM Na2MoO4, 174 nM EDTA; or in complete medium (CM) containing, in addition to MM, 0.1% (w/v) casamino acids and 0.5% (w/v) yeast extract. When required, 10 mM uridine was added. The glucose minimal medium used for bioreactor cultivations has been previously described [78]. For the protease assay, strains were cultivated in MM containing 1% (w/v) dried skim milk and 0.05% Triton X100. Plates were incubated for 4 days at 30° and protease activity was verified by the appearance of a clear halo around the colony.
Construction of the constitutive active hacA strain and the hacA reference strain
To replace to endogenous hacA gene on the hacA locus with a constitutive activated allele of the hacA gene, a replacement cassette was constructed. As a control, a similar replacement cassette was made with the wild-type hacA gene. To construct the hacA reference strain, three PCR fragments consisting of the hacA gene including promoter and terminator regions, the Aspergillus oryzae pyrG selection marker and a hacA terminator region were cloned into pBluescript-SK. Subsequently, this plasmid was used as template to introduce the mutations that led to a constitutive active hacA allele by site directed mutagenesis (according to Quick Change II site directed mutagenesis protocol, Stratagene). To construct the wild-type hacA replacement construct the A. niger hacA gene (accession number: AY303684), including about 0.6 kb promoter and 0.6 kb of terminator regions, was amplified by PCR using N402 genomic DNA as template and primers NC8 and NC11 (Table 5) to which Not I and Xho I restriction sites were added, respectively. The amplified gene was cloned into pTZ57R/T (Fermentas) and sequenced. The hacA terminator region (≈1 kb) was amplified by PCR using N402 genomic DNA as template and primers NC1 and NC2, to which Sal I and Kpn I restriction enzymes were added, respectively. The fragment was cloned into pGEM-T easy (Promega) and sequenced. For PCR amplification, Phusion™ High-Fidelity PCR Kit (Finnzymes) was used according to manufacturer’s instructions. The AopyrG gene (≈2 kb) was PCR amplified using pAO4-13 [80] as template DNA and primers NC7 and pAOpyrG-GA5rev, to which Xho I and Sal I restriction sites were added, respectively. The fragment was cloned into pGEM-T easy (Promega) and sequenced. The fragments corresponding to the hacA terminal region and pyrG were digested from the plasmids using the respective restriction enzymes mentioned above and cloned in a 3-way ligation step into pBlue-SK, previously digested with Xho I-Kpn I to give pBS-pyrG-3’hac. To obtain the final construct, the hacA gene was digested from pTZ57R/T using Not I-Xho I and cloned into pBS-pyrG-3’hac, previously digested with the same enzymes. The final construct, named pHAC, was linearized with Not I and transformed into the A. niger MA70.15 strain. Transformants with a targeted integration of the construct at the hacA locus were screened by Southern blot analysis. To obtain a strain only expressing the constitutively active hacA gene, a construct was made lacking the 20 nucleotide intron (see introduction for details) using the site-directed mutagenesis technique. Mutagenic oligonucleotide primers NC31 and NC32 (Table 5) were designed, surrounding each side of the intron region. PCR was performed using PfuUltra HF DNA polymerase (Stratagene), the pHAC (≈10 ng) as template and conditions as follows: initial denaturation of 1 min at 95 °C, 18 cycles of 30 sec denaturation at 95 °C, annealing at 55 °C for 30 sec and elongation for 8 min and 30 sec at 68 °C. Afterwards, PCR products were digested with Dpn I for one hour at 37 °C, for destruction of parental methylated and hemi-methylated plasmid DNA. The mixture was directly used for E. coli transformation. Plasmid pConstHac was analyzed by restriction enzymes and sequencing, confirming the absence of the 20 nt intron. This construct was linearized with Not I and then transformed into A. niger MA70.15. Southern analysis of putative transformants carrying the wild-type hacA and the constitutively active hacA was performed by digesting the genomic DNA with Nhe I and probing with a 0.6 kb probe corresponding to the hacA 3’-flanking region. Transformants NC1.1 containing expressing the wild-type hacA and NC2.1 expressing the activated hacA form at the endogeneous hacA locus were chosen for further studies and these strains are here referred as the HacAWT (wild-type) and HacACA (Constitutive Active) strains, respectively. The absence of the intron in the NC2.1 strain was further confirmed by PCR analysis using genomic DNA as template, together with primers phac1 and phac2 (Table 5) using Taq polymerase (Fermentas).
Bioreactor cultivation conditions
Conidia for inoculation of bioreactor cultures were harvested from solidified CM with a sterile detergent solution containing 0.05% (w/v) Tween80 and 0.9% (w/v) NaCl. Batch cultivation of HacAWT and HacACA was initiated by inoculating 5 L MM with conidial suspension to give 109 conidia L-1. Glucose was sterilized separately and added to sterile MM to give a final concentration of 0.75% (w/v). During cultivation at 30 °C, pH 3 was maintained by computer-controlled addition of 2 M NaOH or 1 M HCl. Sterile air was supplied at 1 L min-1 through a ring-sparger. Dissolved oxygen tension was above 40% of air saturation at any time, ensuring sufficient oxygen for growth. After spore germination 0.01% (v/v) polypropyleneglycol P2000 was added as antifoam agent. Submerged cultivation was performed with 6.6 L BioFlo3000 bioreactors (New Brunswick Scientific, NJ, USA). A more detailed description of the medium and batch cultivation protocol is given in Jørgensen et al. [78].
Biomass concentration and substrate determination
Dry weight biomass concentration was determined by weighing lyophilized mycelium separated from a known mass of culture broth. Culture broth was filtered through GF/C glass microfibre filters (Whatman). The filtrate was collected and frozen for use in solute analyses. The mycelium was washed with demineralised water, rapidly frozen in liquid nitrogen and stored at −80 °C until lyophilization. Glucose was determined according to the method of Bergmeyer et al. [83] with a slight modification: 250 mM triethanolamine (TEA) was used as buffer (pH7.5).
RNA isolation and quality control
Mycelium intended for gene-expression analyses was separated from culture medium and frozen in liquid nitrogen within 15–20 s from sampling RNA was extracted from mycelium and quickly frozen in liquid nitrogen using Trizol reagent (Invitrogen). Frozen ground mycelium (≈200 mg) was directly suspended in 800 μl Trizol reagent and vortexed vigorously for 1 min. After centrifugation for 5 min at 10000 × g, 450 μl of the supernatant was transferred to a new tube. Chloroform (150 μl) was added and after 3 min incubation at room temperature, samples were centrifuged and the upper aqueous phase was transferred to a new tube to which 400 μl of isopropanol was added, followed by 10 min incubation at room temperature and centrifugation for 10 min at 10000 × g. The pellet was washed with 75% (v/v) ethanol and finally dissolved in 100 μl H2O. RNA samples for micro-array analysis were additionally purified on NucleoSpin RNA II columns (Machery-Nagel) according to the manufacturer’s instructions. RNA quantity and quality was determined on a Nanodrop spectrophotometer.
Microarray analysis
Probe synthesis and fragmentation were performed at ServiceXS (Leiden, The Netherlands) according to the GeneChip Expression Analysis Technical Manual [82]. DSM (Delft, The Netherlands) proprietary A. niger GeneChips were hybridised, washed, stained and scanned as described in the GeneChip Expression Analysis Technical Manual [84]. The 3’ to 5’ signal ratio of probe sets of internal control genes, like gpdA (glyceraldehyde-3-phosphate dehydrogenase), pkiA (pyruvate kinase), hxk (hexokinase) and actin, were below 3 on all 12 arrays.
Transcriptomic data analysis
Bioconductor, a collection of open source and open development packages for the statistical programming language R, was used for data analyses [85, 86]. The transcriptomic data set comprises 12 arrays representing independent triplicates for each of the following four conditions: HacAWT, HacACA-1, HacACA-2 and HacACA-3. Using the robust multi-array analysis (RMA) package [87], RMA expression values were computed from the perfect match probes only. Background correction, normalization and probe summarization steps were performed according to the default settings of the RMA package. Defining the following contrast matrix (HacACA-1 - HacAWT, HacACA-2 - HacAWT, HacACA-3 - HacAWT), three sets of differentially expressed genes were determined by moderated t-statistics using the Limma package [88]. The Benjamini and Hochberg False Discovery Rate [89] (FDR) was controlled at q < 0.005. A minimal fold change criterion was not applied for the identification of differentially expressed genes, as fold changes are not necessarily related to biological relevance [90, 91]. RMA expression values (log2 scale) for each array, mean expression values (normal scale) for each condition, fold-changes and FDR q-values for each of the three comparisons as well as classifiers for the moderated t-statistics are summarized in [Additional file 3. Results are presented as the relative fold change in a linear scale. To make the interpretation more intuitive, we have expressed the relative reduction in transcript level (down-regulated genes) with a “-” (minus). Microarray data described in this study is available at the GEO database under accession number GSE39070.
Enrichment analysis of Gene Ontology (GO) terms
Controlling the FDR at q < 0.05, over-represented GO terms in sets of differentially expressed genes were determined with the Fisher's exact test Gene Ontology Annotation tool (FetGOat) [50].
References
Jeenes DJ, Mackenzie DA, Roberts IN, Archer DB: Heterologous protein production by filamentous fungi. Biotechnol Genet Eng Rev. 1991, 9: 327-367.
Gouka RJ, Punt PJ, van den Hondel CA: Efficient production of secreted proteins by Aspergillus: progress, limitations and prospects. Appl Microbiol Biotechnol. 1997, 47: 1-11. 10.1007/s002530050880.
Braaksma M, Smilde AK, van der Werf MJ, Punt PJ: The effect of environmental conditions on extracellular protease activity in controlled fermentations of Aspergillus niger. Microbiology. 2009, 155: 3430-3439. 10.1099/mic.0.031062-0.
Nemoto T, Maruyama J, Kitamoto K: Improvement of heterologous protein production in Aspergillus oryzae by RNA interference with alpha-amylase genes. Biosci Biotechnol Biochem. 2009, 73: 2370-2373. 10.1271/bbb.90177.
Lubertozzi D, Keasling JD: Developing Aspergillus as a host for heterologous expression. Biotechnol Adv. 2009, 27: 53-75. 10.1016/j.biotechadv.2008.09.001.
Conesa A, Punt PJ, van Luijk N, van den Hondel CA: The secretion pathway in filamentous fungi: a Biotechnological view. Fungal Gene and Biol. 2001, 33: 155-171. 10.1006/fgbi.2001.1276.
Jacobs DI, Olsthoorn MM, Maillet I, Akeroyd M, Breestraat S, Donkers S, van der Hoeven RA: van den Hondel, et al.: Effective lead selection for improved protein production in Aspergillus niger based on integrated genomics. Fungal Genet Biol. 2009, 46: S141-152. 10.1016/j.fgb.2008.08.012.
Punt PJ, Schuren FH, Lehmbeck J, Christensen T, Hjort C, van den Hondel CA: Characterization of the Aspergillus niger prtT, a unique regulator of extracellular protease encoding genes. Fungal Genet Biol. 2008, 45: 1591-1599. 10.1016/j.fgb.2008.09.007.
Yoon J, Kimura S, Maruyama J, Kitamoto K: Construction of quintuple protease gene disruptant for heterologous protein production in Aspergillus oryzae. Appl Microbiol Biotechnol. 2009, 82: 691-701. 10.1007/s00253-008-1815-5.
Wösten HA, Moukha SM, Sietsma JH, Wessels JG: Localization of growth and secretion of proteins in Aspergillus niger. J Gen Microbiol. 1991, 137 (8): 2017-23. 10.1099/00221287-137-8-2017.
Taheri-Talesh N, Horio T, Araujo-Bazán L, Dou X, Espeso EA, Peñalva MA, Osmani SA, Oakley BR: The tip growth apparatus of Aspergillus nidulans. Mol Biol Cell. 2008, 19: 1439-1449. 10.1091/mbc.E07-05-0464.
Sagt CM, Müller WH, van der Heide L, Boonstra J, Verkleij AJ, Verrips CT: Impaired cutinase secretion in Saccharomyces cerevisiae induces irregular endoplasmic reticulum (ER) membrane proliferation, oxidative stress, and ER-associated degradation. Appl Environ Microbiol. 2002, 68: 2155-2160. 10.1128/AEM.68.5.2155-2160.2002.
Rakestraw A, Wittrup KD: Contrasting secretory processing of simultaneously expressed heterologous proteins in Saccharomyces cerevisiae. Biotechnol Bioeng. 2006, 93: 896-905. 10.1002/bit.20780.
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P: Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000, 101: 249-258. 10.1016/S0092-8674(00)80835-1.
Gonzalez TN, Walter P: Ire1p: a kinase and site-specific endoribonuclease. Methods Mol Biol. 2001, 160: 25-36.
Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F: Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008, 132: 89-100. 10.1016/j.cell.2007.10.057.
Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K: Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell. 2003, 14: 2559-2569. 10.1091/mbc.E02-11-0708.
Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P: On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005, 102: 18773-18784. 10.1073/pnas.0509487102.
Gardner BM, Walter P: Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011, 333 (6051): 1891-1894. 10.1126/science.1209126.
Sidrauski C, Walter P: The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997, 90: 1031-1039. 10.1016/S0092-8674(00)80369-4.
Kawahara T, Yanagi H, Yura T, Mori K: Unconventional splicing of HAC1/ERN4 mRNA required for the unfolded protein response. Sequence-specific and non-sequential cleavage of the splice sites. J Biol Chem. 1998, 273: 1802-1807.
Valkonen M, Penttilä M, Saloheimo M: The ire1 and ptc2 genes involved in the unfolded protein response pathway in the filamentous fungus Trichoderma reesei. Mol Genet Genomics. 2004, 272: 443-451. 10.1007/s00438-004-1070-0.
Shen X: Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K. Kaufman RJ: Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001, 107: 893-903.
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D: IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. Erratum in: Nature 420:202. 2002, 415: 92-96.
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ: IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16: 452-466. 10.1101/gad.964702.
Plongthongkum N, Kullawong N, Panyim S, Tirasophon W: Ire1 regulated XBP1 mRNA splicing is essential for the unfolded protein response (UPR) in Drosophila melanogaster. Biochem Biophys Res Commun. 2007, 354: 789-794. 10.1016/j.bbrc.2007.01.056.
Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ: The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006, 103: 14343-14348. 10.1073/pnas.0606480103.
Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P: The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009, 457: 687-693. 10.1038/nature07661.
Mulder HJ, Saloheimo M, Penttila M, Madrid SM: The transcription factor HacA mediates the unfolded protein response in Aspergillus niger, and up-regulates its own transcription. Mol Genet Genomics. 2004, 271: 130-140. 10.1007/s00438-003-0965-5.
Mulder HJ, Nikolaev I: HacA-dependent transcriptional switch releases hacA mRNA from a translational block upon endoplasmic reticulum stress. Eukaryot Cell. 2009, 8: 665-675. 10.1128/EC.00131-08.
Saloheimo M, Wang H, Valkonen M, Vasara T, Huuskonen A, Riikonen M, Pakula T, Ward M, Penttilä M: Characterization of secretory genes ypt1/yptA and nsf1/nsfA from two filamentous fungi: induction of secretory pathway genes of Trichoderma reesei under secretion stress conditions. Appl Environ Microbiol. 2004, 70: 459-467. 10.1128/AEM.70.1.459-467.2004.
Mulder HJ, Nikolaev I, Madrid SM: HacA, the transcriptional activator of the unfolded protein response (UPR) in Aspergillus niger, binds to partly palindromic UPR elements of the consensus sequence 5'-CAN(G/A)NTGT/GCCT-3'. Fungal Genet Biol. 2006, 43: 560-572. 10.1016/j.fgb.2006.02.005.
Lee AH, Iwakoshi NN, Glimcher LH: XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003, 23: 7448-59. 10.1128/MCB.23.21.7448-7459.2003.
Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM: XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004, 1: 81-93.
Arvas M, Pakula T, Lanthaler K, Saloheimo M, Valkonen M, Suortti T, Robson G, Penttilä M: Common features and interesting differences in transcriptional responses to secretion stress in the fungi Trichoderma reesei and Saccharomyces cerevisiae. BMC Genomics. 2006, 7: 32-10.1186/1471-2164-7-32.
Sims AH, Gent ME, Lanthaler K, Dunn-Coleman NS, Oliver SG, Robson GD: Transcriptome analysis of recombinant protein secretion by Aspergillus nidulans and the unfolded-protein response in vivo. Appl Environ Microbiol. 2005, 71: 2737-2747. 10.1128/AEM.71.5.2737-2747.2005.
Guillemette T, van Peij NN, Goosen T, Lanthaler K, Robson GD, van den Hondel CA, Stam H, Archer DB: Genomic analysis of the secretion stress response in the enzyme-producing cell factory Aspergillus niger. BMC Genomics. 2007, 8: 158-10.1186/1471-2164-8-158.
Jørgensen TR, Goosen T, Hondel CA, Ram AF, Iversen JJ: Transcriptomic comparison of Aspergillus niger growing on two different sugars reveals coordinated regulation of the secretory pathway. BMC Genomics. 2009, 10: 44-10.1186/1471-2164-10-44.
Robinson AS, Hines V, Wittrup KD: Protein disulfide isomerase overexpression increases secretion of foreign proteins in Saccharomyces cerevisiae. Biotechnology. 1994, 12: 381-384. 10.1038/nbt0494-381.
Harmsen MM, Bruyne MI, Raué HA, Maat J: Overexpression of binding protein and disruption of the PMR1 gene synergistically stimulate secretion of bovine prochymosin but not plant thaumatin in yeast. Appl Microbiol Biotechnol. 1996, 46: 365-370. 10.1007/BF00166231.
Shusta EV, Raines RT, Plückthun A, Wittrup KD: Increasing the secretory capacity of Saccharomyces cerevisiae for production of single-chain antibody fragments. Nat Biotechnol. 1998, 16: 773-777. 10.1038/nbt0898-773.
Valkonen M, Penttilä M, Saloheimo M: Effects of inactivation and constitutive expression of the unfolded- protein response pathway on protein production in the yeast Saccharomyces cerevisiae. Appl Environ Microbiol. 2003, 69: 2065-2072. 10.1128/AEM.69.4.2065-2072.2003.
Valkonen M, Ward M, Wang H: Penttilä M. Saloheimo M: Improvement of foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the unfolded-protein response. Appl Environ Microbiol. 2003, 69: 6979-6986.
Pakula TM, Laxell M, Huuskonen A, Uusitalo J, Saloheimo M, Penttilä M: The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. J Biol Chem. 2003, 278: 45011-45020. 10.1074/jbc.M302372200.
Martínez IM, Chrispeels MJ: Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell. 2003, 15: 561-576. 10.1105/tpc.007609.
Al-Sheikh H, Watson AJ, Lacey GA, Punt PJ, MacKenzie DA, Jeenes DJ, Pakula T, Penttilä M, Alcocer MJ, Archer DB: Endoplasmic reticulum stress leads to the selective transcriptional downregulation of the glucoamylase gene in Aspergillus niger. Mol Microbiol. 2004, 53: 1731-1742. 10.1111/j.1365-2958.2004.04236.x.
Carvalho ND, Arentshorst M: Jin Kwon M, Meyer V, Ram AF: Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol. 2010, 87: 1463-1473. 10.1007/s00253-010-2588-1.
Iversen JJL, Thomsen JK, Cox RP: On-line growth measurements in bioreactors by titrating metabolic proton exchange. Appl Microbiol Biotechnol. 1994, 42: 256-262. 10.1007/BF00902726.
Poulsen BR, Ruiter G, Visser J, Iversen JJ: Determination of first order rate constants by natural logarithm of the slope plot exemplified by analysis of Aspergillus niger in batch culture. Biotechnol Lett. 2003, 25: 565-571. 10.1023/A:1022836815439.
Nitsche BM, Crabtree J, Cerqueira GC, Meyer V, Ram AF, Wortman JR: New resources for functional analysis of omics data for the genus Aspergillus. BMC Genomics. 2011, 12: 486-10.1186/1471-2164-12-486.
Nishikawa SI, Brodsky JL, Nakatsukasa K: Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem. 2005, 137: 551-555. 10.1093/jb/mvi068.
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, et al,Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007, 25: 221-231. 10.1038/nbt1282.
Carvalho ND, Arentshorst M: Kooistra R, Stam H, Sagt CM, van den Hondel CA, Ram AF: Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger. Appl Microbiol Biotechnol. 2011, 89: 357-373. 10.1007/s00253-010-2916-5.
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA: A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2001, 429: 841-847.
Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH: Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci. 1999, 112: 4123-4134.
van Laar T, van der Eb AJ, Terleth C: Mif1: a missing link between the unfolded protein response pathway and ER-associated protein degradation?. Curr Protein Pept Sci. 2001, 2: 169-190. 10.2174/1389203013381189.
Tremblay LO, Herscovics A: Cloning and expression of a specific human alpha 1,2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology. 1999, 9: 1073-1078. 10.1093/glycob/9.10.1073.
Yuan XL, van der Kaaij RM, van den Hondel CA, Punt PJ, van der Maarel MJ, Dijkhuizen L, Ram AF: Aspergillus niger genome-wide analysis reveals a large number of novel alpha-glucan acting enzymes with unexpected expression profiles. Mol Genet Genomics. 2008, 279: 545-561. 10.1007/s00438-008-0332-7.
Petersen KL, Lehmbeck J, Christensen T: A new transcriptional activator for amylase genes in Aspergillus. Mol Gen Genet. 1999, 262: 668-676. 10.1007/s004380051129.
VanKuyk PA, Benen JA, Wösten HA, Visser J, de Vries RP: A broader role for AmyR in Aspergillus niger: regulation of the utilisation of D-glucose or D-galactose containing oligo- and polysaccharides. Appl Microbiol Biotechnol. 2012, 93 (1): 285-93. 10.1007/s00253-011-3550-6.
Lombraña M, Moralejo FJ, Pinto R, Martín JF: Modulation of Aspergillus awamori thaumatin secretion by modification of bipA gene expression. Appl Environ Microbiol. 2004, 70: 5145-5152. 10.1128/AEM.70.9.5145-5152.2004.
Moralejo FJ, Watson AJ, Jeenes DJ, Archer DB, Martín JF: A defined level of protein disulfide isomerase expression is required for optimal secretion of thaumatin by Aspegillus awamori. Mol Genet Genomics. 2001, 266: 246-253. 10.1007/s004380100550.
de Virgilio M: Kitzmüller C, Schwaiger E, Klein M, Kreibich G, Ivessa NE: Degradation of a short-lived glycoprotein from the lumen of the endoplasmic reticulum: the role of N-linked glycans and the unfolded protein response. Mol Biol Cell. 1999, 10: 4059-4073.
Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L: Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38: 5075-5087. 10.1093/nar/gkq256.
Malhotra JD, Kaufman RJ: The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007, 18: 716-731. 10.1016/j.semcdb.2007.09.003.
Niwa M, Sidrauski C, Kaufman RJ, Walter P: A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999, 99: 691-702. 10.1016/S0092-8674(00)81667-0.
Vattem KM, Wek RC: Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004, 101: 11269-11274. 10.1073/pnas.0400541101.
Fels DR, Koumenis C: The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006, 5: 723-728.
Hinnebusch AG: Gene-specific translational control of the yeast GCN4 gene by phosphorylation of eukaryotic initiation factor 2. Mol Microbiol. 1993, 10: 215-223. 10.1111/j.1365-2958.1993.tb01947.x.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D: Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000, 5: 897-904. 10.1016/S1097-2765(00)80330-5.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D: Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000, 6: 1099-1108. 10.1016/S1097-2765(00)00108-8.
Patil CK, Li H, Walter P: Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2004, 2: E246-10.1371/journal.pbio.0020246.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D: An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003, 11: 619-633. 10.1016/S1097-2765(03)00105-9.
Gilmore WJ, Kirby GM: Endoplasmic reticulum stress due to altered cellular redox status positively regulates murine hepatic CYP2A5 expression. J Pharmacol Exp Ther. 2004, 308: 600-608.
van Huizen R, Martindale JL, Gorospe M, Holbrook NJ: P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem. 2003, 278: 15558-15564. 10.1074/jbc.M212074200.
Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ, Brewer JW: ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. 2009, 122: 1626-1636. 10.1242/jcs.045625.
Bennett JW, Lasure L: More gene manipulations in fungi. 1991, Academic,
Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CA, Ram AF: Transcriptomic insights into the physiology of Aspergillus niger approaching a specific growth rate of zero. Appl Environ Microbiol. 2010, 76: 5344-5355. 10.1128/AEM.00450-10.
Bos CJ, Debets AJ, Swart K, Huybers A, Kobus G, Slakhorst SM: Genetic analysis and the construction of master strains for assignment of genes to six linkage groups in Aspergillus niger. Curr Genet. 1988, 14: 437-443. 10.1007/BF00521266.
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CA, Ram AF: Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol. 2007, 128: 770-775. 10.1016/j.jbiotec.2006.12.021.
Yuan XL, Roubos JA, van den Hondel CA, Ram AF: Identification of InuR, a new Zn(II)2Cys6 transcriptional activator involved in the regulation of inulinolytic genes in Aspergillus niger. Mol Genet Genomics. 2008, 279: 11-26. 10.1007/s00438-007-0290-5.
de Ruiter-Jacobs YM, Broekhuijsen M, Unkles SE, Campbell EI, Kinghorn JR, Contreras R, Pouwels PH, van den Hondel CA: A gene transfer system based on the homologous pyrG gene and efficient expression of bacterial genes in Aspergillus oryzae. Curr Genet. 1989, 16: 159-163. 10.1007/BF00391472.
Bergmeyer HU, Bernt E, Schmidt F, Stork H, Glucose. In Methods of Enzymatic Analysis: Volume 3. Edited by Bergmeyer HU. 1974, Academic, New York, 1196-1201.
Affymetrix (2002): Affymetrix. 2002, ExpressionAnalysis Technical Manual, , [http://www.affymetrix.com/support/technical/manuals.affx]
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J,Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-10.1186/gb-2004-5-10-r80.
Team RDC (2010): R: A language and environment for statistical computing. 2010, R Foundation for Statistical Computing, Vienna, Austria
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP: Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31: e15-10.1093/nar/gng015.
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. 2004, 3: Article3, Stat Appl Genet Mol Biol
Benjamini Y, Hochberg Y: Controling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B. 1995, 57: 289-300.
Van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ: Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006, 7: 142-10.1186/1471-2164-7-142.
Van den Berg RA, Braaksma M, van der Veen D, van der Werf MJ, Punt PJ, van der Oost J, de Graaff LH: Identification of modules inAspergillus nigerby gene co-expression network analysis. Fungal Genet Biol. 2010, 47: 539-50. 10.1016/j.fgb.2010.03.005.
Acknowledgements
This project was carried out within the research programme of the Kluyver Centre for Genomics of Industrial Fermentation, which is part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NDSPC constructed the Aspergillus strains used in this study. NDSPC, TJR and MA carried out and analysed the bioreactor cultivations. BMN carried out the statistical analysis of the transcriptomic data. NDSPC, TRJ, BMN, DBA and AFJR interpreted the transcriptomic data. NDSPC, TRJ, MA, CAMJJvdH, and AFJR designed the experiments. All authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4244_MOESM1_ESM.ppt
Additional file 1: Construction plasmids and confirmation of a reference strain and a strain only expressing the hacA induced form. Schematic representation of the plasmids pHAC (A) and pConstHac (B) (Note: fragment sizes are not on scale). (C) Sequence alignment of pHAC and pConstHAC showing the absence of the 20 nt intron on pConstHac. (D) PCR amplification of gDNA of HacAWT (NC1.1) and HacACA (NC2.1) transformants. Primers were designed about 100 bp upstream and 100 bp downstream of the hacA intron region, giving rise to a band of 200 bp for HacACA and 220 bp for HacAWT. Sizes of the DNA Marker (M) are indicated. (PPT 334 KB)
12864_2012_4244_MOESM2_ESM.ppt
Additional file 2: Growth profiles of A. niger HacAWT(A, B, C) and HacACA(D, E, F) triplicate batch cultures. Dry weight biomass concentration (gDWkg-1) as a function of time (h) illustrates the growth of the cultures. The maximum specific growth rate for each culture was determined from the slope (α) of the ln transformation of biomass (Cbiomass) (lnX) in the exponential growth phase as a function of time (h), as well from log transformation of alkali addition as a function of time (h). Dash-line represents the end of the exponential growth phase (depletion of glucose). (PPT 268 KB)
12864_2012_4244_MOESM3_ESM.xls
Additional file 3: Complete list of all differentially expressed genes in HacACA. Expression data and the FDR-values for the pair wise comparison of the different strains and time points. (XLS 6 MB)
12864_2012_4244_MOESM4_ESM.doc
Additional file 4: Overview of the 616 HacACAup-regulated genes in the 3 time points. Subset of all differentially expressed genes (Additional file 3). (DOC 530 KB)
12864_2012_4244_MOESM5_ESM.doc
Additional file 5: HacACAup-regulated genes that contain at least one UPRE sequence. Subset of all differentially expressed genes (Additional file 3). (DOC 102 KB)
12864_2012_4244_MOESM6_ESM.pdf
Additional file 6: Network maps of related up-regulated GO-terms. Results of the GO-enrichment analysis of biological processes of all differentially expressed genes in HacACA-1/HacAWT. (PDF 49 KB)
12864_2012_4244_MOESM7_ESM.pdf
Additional file 7: Network maps of related down-regulated GO-terms. Results of the GO-enrichment analysis of biological processes of all differentially expressed genes in HacACA-1/HacAWT. (PDF 43 KB)
12864_2012_4244_MOESM8_ESM.doc
Additional file 8: GO analysis of biological processes enriched in the up-regulated set of genes in HacACA. Subset of all differentially expressed genes (Additional file 3). (DOC 52 KB)
12864_2012_4244_MOESM9_ESM.doc
Additional file 9: GO analysis of biological processes enriched in the down-regulated set of genes in HacACA. Subset of all differentially expressed genes (Additional file 3). (DOC 50 KB)
12864_2012_4244_MOESM10_ESM.doc
Additional file 10: Expression values of selected genes related to enriched GO terms of ER associated processes. Subset of all differentially expressed genes (Additional file 3). (DOC 106 KB)
12864_2012_4244_MOESM11_ESM.doc
Additional file 11: Expression values of selected genes related to enriched GO terms associated with vesicle transport within the cell. Subset of all differentially expressed genes (Additional file 3). (DOC 106 KB)
12864_2012_4244_MOESM12_ESM.doc
Additional file 12: Expression values of selected genes related to enriched GO terms associated with glycosylation processes. Subset of all differentially expressed genes (Additional file 3) (DOC 118 KB)
12864_2012_4244_MOESM13_ESM.doc
Additional file 13: Expression values of selected genes related to enriched GO terms associated with lipid metabolic processes. Subset of all differentially expressed genes (Additional file 3). (DOC 79 KB)
12864_2012_4244_MOESM14_ESM.doc
Additional file 14: Expression values of selected genes related to the GO terms “hydrolase activity”, “glutathione catabolic processes” and “vacuolar acidification”. Subset of all differentially expressed genes (Additional file 3). (DOC 42 KB)
12864_2012_4244_MOESM15_ESM.doc
Additional file 15: Expression values of selected down-regulated genes related to enriched GO terms. Subset of all differentially expressed genes (Additional file 3). (DOC 155 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Carvalho, N.D., Jørgensen, T.R., Arentshorst, M. et al. Genome-wide expression analysis upon constitutive activation of the HacA bZIP transcription factor in Aspergillus niger reveals a coordinated cellular response to counteract ER stress. BMC Genomics 13, 350 (2012). https://doi.org/10.1186/1471-2164-13-350
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-350