
Abstract
Background
MicroRNAs (miRNAs) play key roles in diverse developmental processes, nutrient homeostasis and responses to biotic and abiotic stresses. The biogenesis and regulatory functions of miRNAs have been intensively studied in model angiosperms, such as Arabidopsis thaliana, Oryza sativa and Populus trichocarpa. However, global identification of Pinus densata miRNAs has not been reported in previous research.
Results
Here, we report the identification of 34 conserved miRNAs belonging to 25 miRNA families from a P. densata mRNA transcriptome database using local BLAST and MIREAP programs. The primary and/or precursor sequences of 29 miRNAs were further confirmed by RT-PCR amplification and subsequent sequencing. The average value of the minimal folding free energy indexes of the 34 miRNA precursors was 0.92. Nineteen (58%) mature miRNAs began with a 5' terminal uridine residue. Analysis of miRNA precursors showed that 19 mature miRNAs were novel members of 14 conserved miRNA families, of which 17 miRNAs were further validated by subcloning and sequencing. Using real-time quantitative RT-PCR, we found that the expression levels of 7 miRNAs were more than 2-fold higher in needles than in stems. In addition, 72 P. densata mRNAs were predicted to be targets of 25 miRNA families. Four target genes, including a nodal modulator 1-like protein gene, two GRAS family transcription factor protein genes and one histone deacetylase gene, were experimentally verified to be the targets of 3 P. densata miRNAs, pde-miR162a, pde-miR171a and pde-miR482a, respectively.
Conclusions
This study led to the discovery of 34 conserved miRNAs comprising 25 miRNA families from Pinus densata. These results lay a solid foundation for further studying the regulative roles of miRNAs in the development, growth and responses to environmental stresses in P. densata.
Similar content being viewed by others


Background
MicroRNAs (miRNAs), generally 21-24 nt in length, spatiotemporally regulate gene expression at transcriptional and/or posttranscriptional level in most eukaryotes [1]. They play important roles in plant development, nutrient homeostasis, responses to biotic and abiotic stresses and antibacterial reactions [2–5]. Most miRNAs are transcribed by RNA polymerase II (Pol II) from intergenic regions [6]. Like messenger RNAs (mRNAs), primary miRNA (pri-miRNA) transcripts possess 5' caps and 3' poly(A) tails [7]. Pri-miRNAs are processed into imperfect hairpin precursor miRNAs (pre-miRNAs) and then double-stranded miRNA:miRNA* duplexes by Dicer-like 1 (DCL1) protein [8, 9]. The duplexes are exported into the cytoplasm by HASTY. One selected strand of the duplexes (mature miRNA) is recruited by the ARGONAUTE1 (AGO1) protein to form the so-called RNA-induced silencing complex (RISC) [10]. Mature miRNAs guide the complexes to target mRNAs by base complementarity for direct cleavage or translational repression [11].
Identification of conserved and novel miRNAs usually relies on two approaches: bioinformatic prediction and experimental sequencing. In silico prediction of miRNAs includes searching genomic or EST databases for orthologous sequences of known miRNAs and analyzing their pre-miRNA hairpin structures [12]. The limitation of this approach is that only highly conserved miRNAs can be identified. The advent of high-throughput sequencing technologies, such as massively parallel signature sequencing (MPSS), 454 and sequencing-by-synthesis (SBS), has greatly accelerated the discovery of medium-to-low abundant and species-specific miRNAs from diverse plants, e.g., Triticum aestivum, tomato and Oryza sativa [13–25]. For plant species with complete genome information, conserved and novel miRNAs can be conveniently identified based on their alignments to the genome and known miRNAs in the miRBase and analysis of their pre-miRNA stem-loop structures.
Hundreds of miRNAs have been identified and characterized in model angiosperms, such as Arabidopsis, rice and poplar [26–29]. However, few reports involving miRNAs in conifers and other gymnosperms exist. A total of 37 miRNAs from the stem xylem of Pinus taeda have been identified and the expression of 10 miRNA families was significantly repressed in the galled stem [30]. Stage-specific modulation of specific miRNAs and miRNA biogenesis components in zygotic embryos and female gametophytes of P. taeda were demonstrated to play vital roles during embryogenesis and seed development [31]. Morin et al. reported 53 candidate novel miRNA families from Pinus contorta [32]. By sequencing of small RNA libraries constructed from a Taxus chinensis cell line, Qiu et al. found that the expression levels of 14 miRNAs were down-regulated whereas that of 3 miRNAs were up-regulated after treatment with methyl jasmonate [33]. In Pinus abies, 24 novel and 4 conserved miRNAs were identified, and 7 conserved and 9 novel miRNAs were found participating in epigenetic regulation [34].
Pinus densata is an ecologically important conifer. It represents a highly successful case of homoploid hybrid speciation with far-reaching evolutionary consequences [35]. But to date, little is known about its transcriptional sequence information. Global identification of Pinus densata miRNAs has not been reported in previous research. To gain the knowledge of its mRNA transcriptome, we recently performed a high-throughput sequencing of mRNAs isolated from P. densata needles. The present study was designed to identify P. densata miRNAs from the mRNA transcriptome database. Subcloning and sequencing were conducted to further confirm the pri- and/or pre-miRNA sequences. Meanwhile, using real-time RT-PCR, the expression profiles of 10 miRNAs in P. densata seedling tissues were examined. We have also predicted miRNA targets and 4 target mRNAs were experimentally validated by 5' RACE. Our study is the first comprehensive investigation of miRNAs in P. densata, which not only forms a solid base for further study of regulative functions of miRNAs in the development and growth, but also expands our knowledge of conifer miRNAs.
Results
Transcriptome-wide survey of miRNAs in P. densata
Using Illumina sequencing technology, we obtained 3,968,794 raw sequences and 84,950 consensus-genes, including 287 clusters and 84,663 singletons, from a P. densata mRNA library. To identify conserved miRNAs, we used 3,968,794 sequences as query against mature and precursor sequences in the public microRNA database (miRBase version 16), which contains 2952 miRNAs across 43 plant species [36]. A total of 34 conserved miRNAs were identified comprising 25 miRNA families. The sequences of mature, pre- and pri-miRNAs are shown in Table 1 Additional file 1 and Additional file 2 respectively. The length of P. densata miRNA precursors ranged from 76 to 526 nt, with a majority of them (80%) ranging from 67 to 150 nt. It is consistent with that observed in Arabidopsis and rice [37]. The minimal folding free energy indices (MFEIs) of P. densata miRNA precursors varied from 0.54 to 1.28, with an average value of 0.92. It is similar to that of other plant miRNAs, such as Arabidopsis, rice, Glycine max, Medicago truncatula, Saccharum officinarum, Sorghum bicolor and Zea mays [38]. The hairpin structures of P. densata miRNA precursors predicted by MFOLD are shown in Additional file 3 and Additional file 4. The length of P. densata mature miRNAs varied from 19 to 22 nt, with 21 (38%) and 22 (41%) nt ones as the two major size classes (Figure 1). Notably, 19 (58%) miRNAs start with a 5' terminal uridine residue, a characteristic feature of miRNAs recognized by the AGO1 protein. These results imply that the identified P. densata miRNAs may be canonical.

MiRNA length distribution.
Sequences of miRNAs within a family are identical or nearly identical and mismatched nucleotides between two miRNA family members are up to 4 [39]. Through the analysis of the 34 P. densata miRNA precursors, we identified 19 novel mature miRNAs belonging to 14 conserved miRNA families. It includes pde-miR159a, pde-miR169a, pde-miR396a, pde-miR482c, pde-miR482d, pde-miR949a, pde-miR950a, pde-miR952a, pde-miR952b, pde-miR952c, pde-miR1313, pde-miR1314a, pde-miR1448, pde-miR2118a, pde-miR2118b, pde-miR3701, pde-miR3704a, pde-miR3704b and pde-miR3712 (Table 1), of which 17 miRNAs were further validated by subcloning and sequencing except pde-miR396a and pde-miR482c. Interestingly, each miRNA family contains diverse members in P. densata (Figure 2). For example, the pde-MIR482 family has 4 members, whereas only one exists in 19 miRNA families (pde-MIR159, pde-MIR162, pde-MIR169, pde-MIR171, pde-MIR390, pde-MIR396, pde-MIR783, pde-MIR946, pde-MIR947, pde-MIR950, pde-MIR951, pde-MIR1310, pde-MIR1311, pde-MIR1312, pde-MIR1313, pde-MIR1314, pde-MIR1448, pde-MIR3701 and pde-MIR3712).

Numbers of miRNA family members in Pinus densata.
We identified several singleton sequences from the P. densata mRNA transcriptome database highly homologous with the precursors of P. taeda or P. abies miR948, miR1309, miR1315, miR1316 and miR3700. In addition, seven contig sequences identical to or highly homologous to osa-miR156k, osa-miR399a, osa-miR414, pta-miR948, cre-miR1171, pta-miR1309 and pta-miR1316 were also found in the database (Additional file 5). However, they were not able to form canonical secondary hairpin structures. Therefore, we excluded them from the miRNA candidate list.
In this study, we have tried to identify novel miRNAs in P. densata. Because of the lack of sRNA database of P. densata, 166 and 191 small RNAs from Pinus taeda and Picea abies, two evolutionally related species from Pinaceae, were downloaded and used in the analysis as described above [30, 34]. No novel miRNA was found, which could be attributed to insufficient small RNAs and the limited number of mRNAs in the P. densata transcriptome database.
Validation of pri- and pre-miRNA sequences
We carried out subcloning experiments to validate the pri- and pre-miRNA sequences. The pri-miRNA sequences of 25 miRNAs, including pde-miR159a, pde-miR166a, pde-miR166b, pde-miR169a, pde-miR171a, pde-miR482a, pde-miR482b, pde-miR482d, pde-miR783, pde-miR946a, pde-miR947, pde-miR949a, pde-miR949b, pde-miR951, pde-miR952a, pde-miR1310, pde-miR1311, pde-miR1312a, pde-miR1313, pde-miR1448, pde-miR2118a, pde-miR2118b, pde-miR3701, pde-miR3704a and pde-mi3712, and the pre-miRNA sequence of pde-miR950 were experimentally confirmed. Three novel pri-miRNA sequences, pde-miR952b, pde-miR952c and pde-miR3704b were identified when sequencing the clones for pde-miR952a and pde-miR3704a. Seven of the 29 validated sequences were identical to the sequences obtained from Illumina sequencing, whereas 18 of which had less than 10 mismatched nucleotides and 4 of which had more than 10 mismatched nucleotides, which might be partially attributed to sequence assembly mistakes during the Illumina sequencing. The pre-miRNA sequences of 5 miRNAs, pde-miR162a, pde-miR390a, pde-miR396a, pde-miR482c and pde-miR1314a could not be amplified from total RNAs of two-month-old seedling stems, although we repeated our experiments. It could be due to their tissue-specific expressions or limited information for primer design.
Intriguingly, the precursor sequences of pde-miR482b and pde-miR2118b were found clustered in a single contig (singletons868998), while the antisense strand of which harbored the precursor sequence of pde-miR3704a (Figure 3). Since the primary sequences of pde-miR482b and pde-miR3704a were confirmed, it might be reasonable to deduce that the three miRNA precursors were genuine precursors. Singletons3959581 was also predicted containing precursor sequences of 3 miRNAs, pde-miR482d, pde-miR1448 and pde-miR2118a. Its sequence was validated by both Illumina sequencing and subcloning. Similar phenomenon was also reported in P. taeda [30].

Pde-miR482b, pde-miR2118b and pde-miR3704a clustered in a contig. The sense and antisense strand of singletons868998 harbor mature sequences of pde-miR482b, pde-miR2118b and pde-miR3704a.
Expression patterns of miRNAs
In order to obtain solid evidence to support the existence and expression of conserved miRNAs in P. densata, we examined the expression profiles of 10 mature miRNAs (pde-miR159a, pde-miR166a, pde-miR171a, pde-miR390a, pde-miR396a, pde-miR946, pde-miR950, pde-miR1311, pde-miR1313 and pde-miR1314b) in needles and stems of two-month-old seedlings, using real-time RT-PCR (Figure 4). Our results demonstrated that 9 of them had higher expression levels in needles than in stems except pde-miR171. The expression levels of 7 miRNAs, including pde-miR159a, pde-miR166a, pde-miR390a, pde-miR946, pde-miR1311, pde-miR1313 and pde-miR1314b, were more than 2-fold higher in needles than in stems, Intriguingly, miR166a, an important miRNA known for the functions in establishment of adaxial/abaxial (dorsoventral) leaf polarity, was expressed more than 9 times higher in needles than in stems [40]. The result suggests that pde-miR166 may play key roles in a variety of physiological processes in P. densata needles.

Transcript profiles of P. densata conserved miRNAs. Samples are needles and stems of 2-month-old seedlings. The expression levels of all miRNAs by real-time PCR were relative to that of 5 S rRNA and normalized. Error bars represent the standard deviations of three PCR replicates of a single reverse transcription reaction. The normalized miRNA levels in stems were arbitrarily set to 1.
Prediction of miRNA targets
To better understand the functions of P. densata miRNAs, we predicted their targets using the Web-based program psRNATarget http://bioinfo3.noble.org/psRNATarget/index.php?function=function3 [41]. A total of 3,968,794 sequences from the P. densata mRNA transcriptome database and 38 mature miRNAs were used as a custom target database and a custom miRNA database respectively. Seventy-two mRNAs were predicted to be targets of 25 miRNA families (Table 2 and Additional file 6), of which 10 (14%) targets were homologous to previously validated or predicted miRNA targets in A. thaliana, O. sativa, P. trichocarpa, P. taeda, P. abies and/or T. chinensis (Table 2). It includes DCL1 targeted by pde-miR162, GRAS family transcription factor cleaved by pde-miR171, Class III HD-Zip protein HDZ33 regulated by pde-miR166 and CC-NBS-LRR resistance-like protein sliced by pde-miR2118. Searching target enrichment in the gene ontology (GO) http://www.geneontology.org/ showed that these conserved targets were involved in a variety of physiological processes in plants. The number of conserved targets of each miRNA family varied from 1 to 3. Pde-MIR2118 family had three conserved targets. Pde-MIR171 family had two conserved targets, while pde-MIR162 and pde-MIR166 families each had only one conserved target.
The other 62 mRNAs targeted by 22 P. densata miRNA families showed no similarity to other plant miRNA targets. These targets were predicted to play essential roles in multiple physiological processes. The CC-NBS-LRR resistance-like protein and disease resistance protein targeted by pde-MIR2118 family miRNAs appeared to be involved in defense response. Other putative targets include anion exchanger family protein, ATP binding protein and chaperone ClpB. Forty-two predicted target mRNAs encode hypothetical or unknown function proteins. No targets were found for 5 miRNA families, including pde-MIR390, pde-MIR1310, pde-MIR1311, pde-MIR3701 and pde-MIR3704. It could be due to insufficient mRNAs in the mRNA transcriptome database.
Validation of miRNA-guided cleavage of mRNA
Mature miRNAs can direct RISC complexes to slice target mRNAs or inhibit their translations through nucleotide complementarity [1]. The cleavage site primarily locates to the 10th miRNA nucleotide from the 5'-end. To verify that miRNAs can regulate their target mRNA expression in P. densata by cleavage, we carried out a modified RLM-5' RACE experiment, using total RNAs extracted from seedlings (see Methods) [42].
In the present study, we successfully detected the cleavage sites in four predicted target genes of P. densata (Figure 5). Singletons10015, singletons83401, singletons11093 and singletons7264 were confirmed to be targets of pde-miR171a, pde-miR162a and pde-miR482a, respectively. We observed a shorter or longer cleaved sequence for three putative targets, singletons83401, singletons11093 and singletons7264, after 5' RACE analysis. It could be attributed to secondary siRNA in the 21-nucleotide register with the cleavage site for miRNAs, as reported by Ronemus and De Paola [43, 44]. Singletons10015 is similar to plant proteins coded by GRAS family transcription factor protein, while singletons83401, singletons11093 and singletons7264 coded for proteins homologous to actin binding protein, nodal modulator 1-like protein and histone deacetylase, respectively.

Detection of miRNA-mediated mRNA cleavage. Partial mRNA sequences from target genes were aligned with corresponding miRNAs. Each top strand (blue) represents a miRNA complementary site in the target mRNA and each bottom strand (red) represents the miRNA. G:U wobble pairing (circles) and Watson-Crick pairing (vertical dashes) are indicated. Red arrows indicate the 5' termini of the degraded mRNA fragments isolated from Pinus densata, which is identified from cloned 5' RACE products, with the frequency of clones shown.
Discussion
Conserved miRNAs in P. densata
Previous studies have identified thousands of miRNAs in angiosperms and some of them have been well-characterized [45, 46]. Few miRNAs from gymnosperms have been reported to date [30–34]. P. densata is an ecologically important conifer in East Asia. It is an ideal model plant to study homoploid hybrid speciation. However, only 399 EST sequences are available in the public databases to date. No miRNAs from P. densata has been reported in previous research. In order to obtain global mRNAs from P. densata, we recently sequenced an mRNA library constructed from total RNAs of needles using the Illumina high-throughput sequencing technology. Since pri-miRNA sequences also possess poly(A) tails and thus can be isolated and sequenced as mRNAs, it allows us to identify candidate pri-miRNA sequences from the P. densata mRNA transcriptome database by bioinformatics tools [1]. In this study, a total of 34 conserved miRNAs belonging to 25 miRNA families were identified from P. densata. The primary and/or precursor sequences of 29 miRNAs were further confirmed by subcloning and sequencing. In addition, 19 novel mature miRNAs belonging to 14 conserved miRNA families were found through the analysis of their precursor sequences. The 34 pre-miRNA sequences could form hairpin structures as predicted by MFOLD. They had an average MFEI value of 0.92 (Table 1). Nineteen mature miRNAs (58%) had a 5' terminal uridine residual, which is a characteristic feature of typical miRNAs. These results indicate that the 34 candidate miRNAs may be canonical. The miRNAs identified in the present study can definitely provide useful information for further study on their regulative functions and biogenesis of them.
Among the 21 miRNA families conserved between dicots and monocots, 8 of which were identified in P. densata. It includes pde-MIR159, pde-MIR162, pde-MIR166, pde-MIR169, pde-MIR171, pde-MIR390, pde-MIR396 and pde-MIR399. It indicates that the ancient miRNA regulatory system is well-developed in the common ancestors of gymnosperms and angiosperms [47]. Compared with P. taeda and P. abies miRNA families listed in miRBase version 16, five new families (pde-MIR162, pde-MIR169, pde-MIR399, pde-MIR1448 and pde-MIR2118) were only found in P. densata [30, 34]. Three P. taeda miRNA families, including pta-MIR319, pta-MIR398 and pta-MIR408, were not found in P. densata. It could be due to the simplicity of tissue used for Illumina sequencing.
We also found a few contigs highly homologous to 12 conserved miRNAs. They were excluded for further analysis because they were not able to form canonical hairpin structures. With whole genome sequences and larger EST databases, more miRNAs will be identified from P. densata.
Expression profiles of miRNAs
In the present study, we found the expression levels of 7 conserved miRNAs were more than 2-fold higher in needles than in stems, using real time RT-PCR. It suggests that these conserved miRNAs may play specific roles in a variety of physiological processes in needles. Previous publications have shown that many miRNAs have important functions in the establishment of leaf polarity and virus-induced leaf curling. Zma-miR166a is an important miRNA known for establishing the adaxial/abaxial (dorsoventral) leaf polarity by repressing the expression of class III homeodomain leucine zipper (HD-ZIPIII) transcription factors in maize [48]. The accumulation of miR159 was observed to increase with the days post inoculation (dpi) of tomato leaf curl New Delhi virus (ToLCNDV) agroinfection in tomato cv Pusa Ruby [49]. In our study, pde-miR159 and pde-miR166a were found highly expressed in P. densata needles. Meanwhile, Singletons59617, coding for a protein highly homologous to Class III HD-Zip protein HDZ33, was predicted to be the target of pde-miR166a. These results suggest that the two miRNAs may also involve in needle polarity establishment and antiviral reaction in P. densata.
MiRNA targets and their putative functions
To define and elucidate the putative functions for a miRNA in plant, a necessary step is to predict and validate its target mRNAs. Currently, the most efficient tool available for this purpose is the bioinformatics approach, which is based on perfect or near perfectly complementarity between miRNAs and their targets. In this study, we predicted miRNA targets in P. densata using an online miRNA target search program, psRNATarget [41]. Our results showed that P. densata miRNA targets encoded not only indispensable transcription factors, but also non-transcriptional factor proteins involving in diverse physiological processes. For example, HD-ZIP and GRAS family transcription factors, which are important to root and nodule development in Medicago truncatula and nutrient homeostasis in maize, were predicted to be targets of pde-MIR166 and pde-MIR171, respectively [50, 51]. Non-transcriptional factor proteins, such as DCL1 and CC-NBS-LRR resistance-like protein, were predicted to be targets of pde-MIR162 and pde-MIR2118, respectively [52]. DCL1 is one of the essential components involving in the miRNA biogenesis. The prediction of DCL1 to be the target of pde-miR162 implies that the miRNA biogenesis process is self-regulated in P. densata.
To validate the miRNA targets in P densata, we performed a modified RLM-5' RACE experiment. In the present study, 4 miRNA targets, including singletons10015, singletons83401, singletons11093 and singletons7264 were confirmed to be targets of pde-miR171a, pde-miR162a and pde-miR482a, respectively. These results suggest that miRNAs can regulate the expression of their targets by cleavage in P. densata.
Conclusions
In summary, we performed a transcriptome-wide identification and characterization of miRNAs from P. densata. A total of 34 conserved miRNAs comprising 25 miRNA families were identified. The primary and/or precursor sequences of 29 miRNAs were confirmed by subcloning and sequencing. Analysis of miRNA precursors revealed 19 pre-miRNA sequences harboring novel mature miRNAs belonging to 14 conserved miRNA families. Seventeen of the novel miRNAs were validated by sequencing. Using real-time quantitative RT-PCR, we found that the expression levels of 7 P. densata miRNAs were more than 2-fold higher in needles than in stems. Utilizing a Web-based program psRNATarget, 72 mRNAs were predicted to be targets of 25 miRNA families. Four mRNAs were experimentally validated to be targets of 3 P. densata miRNAs by RLM-5' RACE. These results suggest that regulative miRNAs exist in ecologically important conifer, P. densata, and may play key roles in P. densata development, growth and response to environmental stress.
Methods
Plant material
P. densata needles were harvested from a mature Pinus densata tree in Linzhi city, Tibet, China and stored at -80°C until use. P. densata seedlings were grown under standard greenhouse conditions.
Total RNA isolation and Illumina sequencing
Using RNAiso-mate for plant tissue and RNAiso plus (Takara, Dalian, Liaoning, China), total RNAs were purified from needles and treated with RNase-free DNase I for 30 min at 37°C (Promega) to remove residual DNA. Total RNA of needles was used for Illumina sequencing of mRNA transcriptome, which was performed at Beijing Genomics Institute (BGI), Shenzhen, China. After removing reads containing only 3' sequencing adapters and reads of low quality (reads containing Ns > 5), transcriptome de novo assembly was carried out with short read assembling program - SOAPdenovo [53].
Identification of P. densata miRNAs
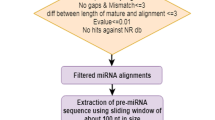
In order to find conserved miRNAs, we aligned 3,968,794 unique sequences from the mRNA transcriptome database of P. densata against the mature and precursor sequences of known plant miRNAs deposited in miRBase version 16 http://www.mirbase.org/, using local BLASTN and MIREAP programs http://sourceforge.net/projects/mireap/ [36, 54]. To discover novel miRNAs, 166 and 191 small RNAs from Pinus taeda and Picea abies were downloaded and used in the analysis as described above [30, 34]. Sequences with an E-value of lower than -2 or a score > 32 were processed for further analysis, allowing for a maximum of 2 nt mismatches. Overlapping contig sequences were used to form longer sequences according to their alignments to known precursor sequences in the miRBase. MFOLD was employed to predict hairpin structures with default parameters http://mfold.bioinfo.rpi.edu/cgi-bin/rna-form1.cgi [55]. Sequences were considered as miRNA precursor sequences if they met the following criteria: the RNA sequence could form an appropriate stem-loop structure, with a mature miRNA sitting in one arm of the hairpin structure; mature miRNAs had less than 6 mismatches with the opposite miRNA* sequences in the other arm; the predicted secondary structure had a minimal folding free energy of less than or equal to -15 kcal/mol, a minimal folding free energy index of more than 0.5, and a 30-70% A + U content [26].
Subcloning and sequencing of pri- and pre-miRNA sequences
Total RNA was isolated from 2-month-old seedling stems as described above. cDNAs were synthesized from 2 μg of purified total RNA in 25-μl reactions, containing 200 U M-MLV reverse transcriptase (Promega, Madison, WI, USA) and 1 μg random nonamer, according to the manufacturer's protocol. The housekeeping gene Actin was used as a positive control. Thirty-eight pairs of primers for P. densata primary or precursor sequences were designed (Additional file 7). PCR amplifications were carried out, using the following thermal cycling conditions: 94°C for 3 min, 35 cycles at 94°C for 30 s, 55°C or 60°C for 15 s and 72°C for 50 s. Amplification fragments were separated on a 2% agarose gel with ethidium bromide (EtBr) staining. Gel-purified PCR fragments were subcloned into pGEM-T Easy Vector (Promega) and sequenced.
Quantitative real-time PCR
Total RNAs were purified from needles and stems of 2-month-old seedlings as described above. Reverse transcription was carried out using 1 μg of total RNA and the NCode miRNA First-Strand cDNA Synthesize Kit (MIRC-50; Invitrogen) following the manufacturer's recommendations. The resulting cDNA was diluted 10 times with sterile water. Quantitative real-time PCR was performed in triplicate reactions using the MX3000P detection system (Stratagene, La Jolla, CA, USA). Ten forward primers were designed based on mature miRNA sequences. If the Tm of a mature miRNA was < 60°C, it would be adjusted by adding Gs or Cs to the 5' end and/or As to the 3' end of the miRNA sequence (Additional file 7) [34]. A 20-bp segment at the 3' end of the 5 s rRNA gene was amplified as an endogenous control to normalize template amounts. Since many miRNA paralogs differ by only 1 nucleotide, we adopted stringent annealing conditions and set the annealing temperature to 65°C for quantitative real-time RT-PCR reactions [56]. Quantitative PCR reactions were conducted in 20 μl volumes containing 2 μl diluted cDNA, 300 nM of each primer, and 10 μl of the Thunderbird SYBR Green PCR Master Mix (Toyobo, Tokyo, Japan) with the following cycling conditions: 95°C for 1 min, 40 cycles at 95°C for 15 s, 65°C for 15 s, and 72°C for 5 s. After amplification, a thermal denaturing cycle at 95°C for 15 s, 65°C for 15 s, and 95°C for 15 s was carried out to determine the dissociation curves and verify the specificity of the amplifications. All expression levels were normalized to the arithmetic mean of the selected 5 S ribosomal RNA gene. Amplification results were analyzed using a comparative Ct method, which uses a formula, 2-ΔΔCt, to achieve results for relative quantification. Ct represents the threshold cycle. The expression level in stems was arbitrarily set to 1 [30].
Prediction of miRNA targets
To identify putative targets of P. densata miRNAs, we used 38 mature miRNAs as custom miRNAs and 3,968,794 sequences in the P. densata mRNA database as custom mRNAs to search for complementary hits using the psRNATarget program http://bioinfo3.noble.org/psRNATarget/ with default parameters. The score system was applied according to Zhang [41]. Sequences with a penalizing score ≤ 3 were chosen as putative targets. We further performed BLASTX searches against the NCBI database to identify putative gene homologs. Similarities with an E-value of less than e-10 were considered a hit.
Validation of miRNA-mediated cleavage of mRNA
To identify cleavage sites in the target mRNAs, a modified RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) was performed using a 5' RACE kit (Takara) [42]. Total RNA was isolated from seedlings as described above. An RNA Oligo adapter was directly ligated to the purified total RNAs (2000 ng) without calf intestinal phosphatase and tobacco acid pyrophosphatase treatment. Twenty pairs of nesting and nested gene-specific primers were designed and applied for PCR amplifications. Six DNA bands with expected sizes were gel purified and cloned into pGEM-T Easy Vector for sequencing. Four of the sequenced DNA bands were identified to be miRNA-guided cleavage products.
Abbreviations
- AGO:
-
Argonaute
- DCL:
-
Dicer-like
- GO:
-
Gene Ontology
- MFEI:
-
Minimal folding free energy index
- miRNA:
-
MicroRNA
- pre-miRNA:
-
Precursor miRNA
- pri-miRNA:
-
Primary miRNA
- RISC:
-
RNA-induced silencing complex
- RT:
-
Reverse transcription.
References
Voinnet O: Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell. 2009, 136 (4): 669-687. 10.1016/j.cell.2009.01.046.
Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS: The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 2009, 138 (4): 750-759. 10.1016/j.cell.2009.06.031.
Abdel-Ghany SE, Pilon M: MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J Biol Chem. 2008, 283 (23): 15932-15945. 10.1074/jbc.M801406200.
Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK: Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005, 123 (7): 1279-1291. 10.1016/j.cell.2005.11.035.
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JD: A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006, 312 (5772): 436-439. 10.1126/science.1126088.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116 (2): 281-297. 10.1016/S0092-8674(04)00045-5.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53. 10.1146/annurev.arplant.57.032905.105218.
Kurihara Y, Watanabe Y: Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA. 2004, 101: 12753-12758. 10.1073/pnas.0403115101.
Kim VN: MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005, 6: 376-385.
Carthew RW, Sontheimer EJ: Origins and mechanisms of miRNAs and siRNAs. Cell. 2009, 136: 642-655. 10.1016/j.cell.2009.01.035.
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O: Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008, 320 (5880): 1185-1190. 10.1126/science.1159151.
Adai A, Johnson C, Mlotshwa S, Archer-Evans S, Manocha V, Vance V, Sundaresan V: Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 2005, 15: 78-91. 10.1101/gr.2908205.
Chi X, Yang Q, Chen X, Wang J, Pan L, Chen M, Yang Z, He Y, Liang X, Yu S: Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS One. 2011, 6 (11): e27530-10.1371/journal.pone.0027530.
Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, Sun Q: Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 2007, 8 (6): R96-10.1186/gb-2007-8-6-r96.
Moxon S, Jing R, Szittya G, Schwach F, Rusholme Pilcher RL, Moulton V, Dalmay T: Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18 (10): 1602-1609. 10.1101/gr.080127.108.
Zhu QH, Spriggs A, Matthew L, Fan L, Kennedy G, Gubler F, Helliwell C: A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008, 18 (9): 1456-1465. 10.1101/gr.075572.107.
Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MP, Moulton V, Dalmay T: High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics. 2008, 9: 593-10.1186/1471-2164-9-593.
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK: Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8: 25-10.1186/1471-2229-8-25.
Subramanian S, Fu Y, Sunkar R, Barbazuk WB, Zhu JK, Yu O: Novel and nodulation-regulated microRNAs in soybean roots. BMC Genomics. 2008, 9: 160-10.1186/1471-2164-9-160.
Xin M, Wang Y, Yao Y, Xie C, Peng H, Ni Z, Sun Q: Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10: 123-10.1186/1471-2229-10-123.
Kwak PB, Wang QQ, Chen XS, Qiu CX, Yang ZM: Enrichment of a set of microRNAs during the cotton fiber development. BMC Genomics. 2009, 10: 457-10.1186/1471-2164-10-457.
Liang C, Zhang X, Zou J, Xu D, Su F, Ye N: Identification of miRNA from Porphyra yezoensis by high-throughput sequencing and bioinformatics analysis. PLoS One. 2010, 5 (5): e10698-10.1371/journal.pone.0010698.
Jagadeeswaran G, Zheng Y, Li YF, Shukla LI, Matts J, Hoyt P, Macmil SL, Wiley GB, Roe BA, Zhang W: Cloning and characterization of small RNAs from Medicago truncatula reveals four novel legume-specific microRNA families. New Phytol. 2009, 184 (1): 85-98. 10.1111/j.1469-8137.2009.02915.x.
Lelandais-Briere C, Naya L, Sallet E, Calenge F, Frugier F, Hartmann C, Gouzy J, Crespi M: Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell. 2009, 21 (9): 2780-2796. 10.1105/tpc.109.068130.
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J: Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62 (6): 960-976.
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP: A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20: 3407-3425. 10.1101/gad.1476406.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC: Highroughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007, 2: e219-10.1371/journal.pone.0000219.
Barakat A, Wall PK, Diloreto S, Depamphilis CW, Carlson JE: Conservation and divergence of microRNAs in Populus. BMC Genomics. 2007, 8: 481-10.1186/1471-2164-8-481.
Klevebring D, Street NR, Fahlgren N, Kasschau KD, Carrington JC, Lundeberg J, Jansson S: A Genome-wide profiling of populus small RNAs. BMC Genomics. 2009, 10: 620-10.1186/1471-2164-10-620.
Lu S, Sun YH, Amerson H, Chiang VL: MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007, 51 (6): 1077-1098. 10.1111/j.1365-313X.2007.03208.x.
Oh TJ, Wartell RM, Cairney J, Pullman GS: Evidence for stage-specific modulation of specific microRNAs (miRNAs) and miRNA processing components in zygotic embryo and female gametophyte of loblolly pine (Pinus taeda). New Phytol. 2008, 179 (1): 67-80. 10.1111/j.1469-8137.2008.02448.x.
Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, Sahinalp SC, Unrau PJ: Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18 (4): 571-584. 10.1101/gr.6897308.
Qiu D, Pan X, Wilson IW, Li F, Liu M, Teng W, Zhang B: High throughput sequencing technology reveals that the taxoid elicitor methyl jasmonate regulates microRNA expression in Chinese yew (Taxus chinensis). Gene. 2009, 436 (1-2): 37-44. 10.1016/j.gene.2009.01.006.
Yakovlev IA, Fossdal CG, Johnsen Ø: MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187 (4): 1154-1169. 10.1111/j.1469-8137.2010.03341.x.
Wang B, Mao J-F, Gao JIE, Zhao WEI, Wang X-R: Colonization of the Tibetan Plateau by the homoploid hybrid pine Pinus densata. Mol Ecol. 2011, 20 (18): 3796-3811. 10.1111/j.1365-294X.2011.05157.x.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36: D154-D158. 10.1093/nar/gkn221.
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA: Conservation and divergence of plant microRNA genes. Plant J. 2006, 46: 243-259. 10.1111/j.1365-313X.2006.02697.x.
Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA: Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci. 2006, 63 (2): 246-254. 10.1007/s00018-005-5467-7.
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X: Criteria for annotation of plant MicroRNAs. Plant Cell. 2008, 20 (12): 3186-3190. 10.1105/tpc.108.064311.
Nogueira FT, Madi S, Chitwood DH, Juarez MT, Timmermans MC: Two small regulatory RNAs establish opposing fates of a developmental axis. Genes Dev. 2007, 21 (7): 750-755. 10.1101/gad.1528607.
Zhang Y: miRU: an automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33: W701-W704. 10.1093/nar/gki383.
Song C, Wang C, Zhang C, Korir NK, Yu H, Ma Z, Fang J: Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics. 2010, 11: 431-10.1186/1471-2164-11-431.
Ronemus M, Vaughn MW, Martienssen RA: MicroRNA-targeted and small interfering RNA-mediated mRNA degradation is regulated by argonaute, dicer, and RNA-dependent RNA polymerase in Arabidopsis. Plant Cell. 2006, 18 (7): 1559-1574. 10.1105/tpc.106.042127.
De Paola D, Cattonaro F, Pignone D, Sonnante G: The miRNAome of globe artichoke: conserved and novel micro RNAs and target analysis. BMC Genomics. 2012, 13: 41-10.1186/1471-2164-13-41.
Chen X: A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004, 303: 202-225. 10.1126/science.1090300.
Lauter N, Kampani A, Carlson S, Goebel M, Moose SP: microRNA172 downgulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci USA. 2005, 102: 9412-9417. 10.1073/pnas.0503927102.
Axtell MJ: Evolution of microRNAs and their targets: are all microRNAs biologically relevant?. Biochim Biophys Acta. 2008, 1779 (11): 725-734.
Nogueira FT, Chitwood DH, Madi S, Ohtsu K, Schnable PS, Scanlon MJ, Timmermans MC: Regulation of small RNA accumulation in the maize shoot apex. PLoS Genet. 2009, 5 (1): e1000320-10.1371/journal.pgen.1000320.
Naqvi AR, Haq QM, Mukherjee SK: MicroRNA profiling of tomato leaf curl New Delhi virus (tolcndv) infected tomato leaves indicates that deregulation of mir159/319 and mir172 might be linked with leaf curl disease. Virol J. 2010, 7: 281-10.1186/1743-422X-7-281.
Boualem A, Laporte P, Jovanovic M, Laffont C, Plet J, Combier JP, Niebel A, Crespi M, Frugier F: MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 2008, 54 (5): 876-887. 10.1111/j.1365-313X.2008.03448.x.
Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li WX: Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One. 2012, 7 (1): e29669-10.1371/journal.pone.0029669.
Xie Z, Kasschau KD, Carrington JC: Negative Feedback Regulation of Dicer-Like1 in Arabidopsis by microRNA-Guided mRNA Degradation. Curr Biol. 2003, 13 (9): 784-789. 10.1016/S0960-9822(03)00281-1.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009, 25 (15): 1966-1967. 10.1093/bioinformatics/btp336.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden T: BLAST+: architecture and applications. BMC Bioinforma. 2009, 10 (1): 421-10.1186/1471-2105-10-421.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13): 3406-3415. 10.1093/nar/gkg595.
Shi R, Chiang VL: Facile means for quantifying microRNA expression by real-time PCR. Biotechniques. 2005, 39 (4): 519-525. 10.2144/000112010.
Acknowledgements
This research was financially supported by Major State Basic Research Development Program by Ministry of Science and Technology of China (2009CB119105), the National High Technology Research and Development Program of China (863 Program, No. 2008AA10Z130), projects from Ministry of Agriculture of China (2009ZX08009-011B and 2009ZX08009-095B) and a grant from National Natural Science Foundation of China (30830010).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
LW designed and carried out the study and drafted the manuscript. HZ and LZ participated in the bioinformatics analysis. SL, ZQ and YZ helped to draft the manuscript. QZ and JL conceived of the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2011_3968_MOESM3_ESM.PDF
Additional file 3: Predicted hairpin structures of P. densata miRNA precursors. The hairpin structures of P. densata miRNA precursors were predicted by MFOLD. Mature miRNAs were marked in yellow. (PDF 457 KB)
12864_2011_3968_MOESM4_ESM.DOC
Additional file 4: Predicted stem-loop structures of precursors containing P. densata microRNA sequences (red and blue). (DOC 140 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wan, LC., Zhang, H., Lu, S. et al. Transcriptome-wide identification and characterization of miRNAs from Pinus densata. BMC Genomics 13, 132 (2012). https://doi.org/10.1186/1471-2164-13-132
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-132