
Abstract
Background
High-throughput sequencing technology is capable to identify novel short RNAs in plant species. We used Solexa sequencing to find new microRNAs in one of the model legume species, barrel medic (Medicago truncatula).
Results
3,948,871 reads were obtained from two separate short RNA libraries generated from total RNA extracted from M. truncatula leaves, representing 1,563,959 distinct sequences. 2,168,937 reads were mapped to the available M. truncatula genome corresponding to 619,175 distinct sequences. 174,504 reads representing 25 conserved miRNA families showed perfect matches to known miRNAs. We also identified 26 novel miRNA candidates that were potentially generated from 32 loci. Nine of these loci produced eight distinct sequences, for which the miRNA* sequences were also sequenced. These sequences were not described in other plant species and accumulation of these eight novel miRNAs was confirmed by Northern blot analysis. Potential target genes were predicted for most conserved and novel miRNAs.
Conclusion
Deep sequencing of short RNAs from M. truncatula leaves identified eight new miRNAs indicating that specific miRNAs exist in legume species.
Similar content being viewed by others


Background
Gene expression is regulated at several layers in plants to ensure optimal temporal and spatial accumulation of proteins. One of the latest discovered regulatory layers involves short RNA (sRNA) molecules 21–24 nucleotides in length that act post-transcriptionally [1]. There are surprisingly many different sRNAs in plant cells indicating an extensive role for these molecules [2]. Plant sRNAs are produced from double stranded RNA (dsRNA) by one of the four Dicer-like proteins (DCL1-4). The different DCL proteins process dsRNAs generated by diverse pathways [1]. MicroRNAs (miRNAs) are produced from partially complementary dsRNA precursor molecules (pre-miRNA) [3]. Pre-miRNAs are originally single stranded RNAs with hairpin structures and recognized by DCL1 [4]. The other large class of plant sRNAs is small interfering RNAs (siRNAs). siRNAs are processed from dsRNAs usually generated by one of the RNA Dependent RNA Polymerases (RDRs). Trans-acting siRNA (ta-siRNAs) precursors are generated by RDR6 [5, 6] and heterochromatin siRNA precursors are made by RDR2 [7]. Another group of siRNAs, the natural antisense siRNAs (nat-siRNAs), are processed from dsRNA produced by overlapping antisense mRNAs [8].
MiRNAs are the best characterized sRNAs in plants [9]. The primary transcript (pri-miRNA) is transcribed by RNA polymerase II and contains an imperfect hairpin structure. DCL1 trims this hairpin structure producing the pre-miRNA and then a second cleavage by DCL1 produces the miRNA/miRNA* duplex [10]. This molecule has a two nucleotide 3' overhang at each side of the duplex and contains a few mismatches [9]. One of the strands of the miRNA/miRNA* duplex is integrated into RISC (RNA induced silencing complex). This strand is called mature miRNA and the partially complementary miRNA* strand gets degraded, although in most cases the miRNA* strand also accumulates at a lower level [9]. RISC finds specific mRNAs because the incorporated mature miRNA can anneal to partially complementary target sites [3]. Target sites show near perfect matches to plant miRNA sequences and initially it was thought that all target mRNAs are cleaved by RISC [3]. Recently it was shown that the translation of plant mRNAs is also suppressed without a cleavage [11].
Most plant miRNA families have been identified by traditional Sanger sequencing method in model species with known genome sequences (Arabidopsis, rice and poplar) and most miRNAs are conserved across plant families [12]. However, some miRNAs are species/family specific and Allen et al. [13] suggested that these "young" miRNAs have evolved recently, in contrary to the conserved miRNAs ("old" miRNAs). Since non-conserved miRNAs are often accumulated at a lower level than conserved miRNAs, traditional small-scale sequencing primarily reveals conserved miRNAs. Establishment of high-throughput technologies has allowed the identification of several non-conserved or lowly expressed miRNAs through deep sequencing, e.g. in Arabidopsis, wheat and tomato [14–17]. Here we describe the deep sequencing of short RNAs extracted from M. truncatula leaves and the experimental validation of eight novel miRNAs.
Results
Deep sequencing of M. truncatula short RNAs
Two separate cDNA libraries of short RNAs were generated from Medicago truncatula leaves and both libraries were sequenced by Solexa (Illumina). The first PCR product was quantified by nanodrop and 5 pM was loaded to Solexa, which yielded 5942 clusters and 872,048 sequence reads. The second sample was also quantified on polyacrylamide gel because the capacity of Solexa is higher than this. Since the nanodrop gave an approximately 50 times higher concentration than quantification on a gel we concluded that the nanodrop overestimates the concentration of the PCR product. To obtain more sequences, we loaded four times of the amount that we would have loaded based only on the nanodrop reading and this yielded 23301 clusters and 3,076,823 sequence reads. It is worth mentioning that loading based on only quantification on gel would have led to overloading the Solexa and would have produced many but unreliable sequences.
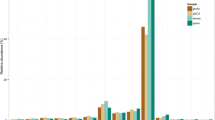
The two sets of reads were combined and analysed together (Table 1) using the miRCat pipeline we have developed earlier [18]. The size distribution of sequence reads showed that the 24 nt class was the most abundant group of sRNAs followed by the 21 nt sequences and then the 23, 22 and 20 nt reads (Figure 1). The almost four million reads represented 1,563,959 distinct sequences suggesting that the library is still not saturated. Out of the four million reads 2,168,937 matched to the genome without any mismatches, representing 619,175 sequences.

Size distribution of sequenced short RNAs.
Conserved miRNAs
First we looked for known miRNAs by comparing our library to known miRNAs from other plant species. 174,504 reads corresponding to 25 conserved miRNA families showed perfect matches to known miRNAs. We analysed the number of reads for conserved miRNAs and miR156, 159 and 166 were represented most frequently in the library (Table 2). Allowing one or two mismatches between sequences in our library and sequences in miRBase increased the number of conserved miRNA families in M. truncatula to 31 (Table 2). Next we predicted target genes and putative targets were identified for 27 out of 31 conserved families (Additional File 1). No targets were found in M. truncatula for miR163, miR169, miR394 and miR894. We found homologs of known miRNA target genes for several conserved M. truncatula miRNAs, such as SBP for miR156, NAC for miR160, AGO1 for miR168, bZIP for miR165, GRAS for miR171, AP2 for miR172 and low affinity sulphur transporter for miR395. On the other hand we predicted many genes with unknown function and hypothetical genes for miRNA targeting (Additional File 1) and careful analysis of these potential targets will contribute to our understanding of the role of miRNAs in legume plants.
Novel miRNAs
The flanking regions of the 2,168,937 sequence reads matching the genome were subjected to secondary structure analysis. We used the strict criteria suggested by Jones-Rhoades et al. [9] to identify potential miRNA loci. 32 short RNA producing loci corresponding to 26 sequence reads could be folded into step-loop structures (Additional File 2). Nine of these loci produced eight distinct sequences, for which we also sequenced the miRNA* sequences (Table 3). These eight sequences were considered as novel legume specific miRNAs based on Rajagopalan et al. [15], who demonstrated that loci producing sequenced mature miRNAs and miRNA* sequences are miRNA loci. The other 23 loci producing 18 sRNAs were considered as putative new miRNA genes since at the moment there is not sufficient evidence to classify them as miRNA genes. Accumulation of all eight new miRNAs and most miRNA*s were validated by Northern blot analysis (Figure 2).

We also predicted target genes for putative miRNAs (Additional file 3) and the new miRNAs with sequenced miRNA* (Additional file 4). Five out of the eight new miRNAs have potential targets in the available M. truncatula genome. It is interesting that two of these five new miRNAs potentially target disease resistance genes suggesting a role for miRNAs in the regulation of biotic stress response.
Discussion
Deep sequencing of M. truncatula short RNAs
Most conserved plant miRNAs were identified by traditional Sanger sequencing. Due to the relatively small number of sequences generated this method primarily discovered conserved miRNAs. Recent progress in sequencing technologies allows deep sequencing of large libraries of short RNAs [2, 7, 17, 19–23]. The longest reads are obtained by the 454 technology, which currently gives reads of 250–300 base pairs (bp). However, this technology yields much less reads than other techniques (about 400 000 per sample) [17]. The Solexa platform (Illumina) generates shorter reads (up to 35 bp) but yields 1–3 million reads per sample [22]. The other very high-throughput technique, massively parallel sequencing (MPSS), gives more reads than Solexa but the reads are even shorter, only 17 bp [23]. miRNAs are only 21–23 nt sequences therefore even MPSS can identify them, although this technology generates shorter than 21 bp reads. Because of this it is not the ideal choice for miRNA discovery but it is useful for profiling conserved miRNAs. We decided to use the Solexa platform to test whether new miRNAs can be found in the model legume M. truncatula.
We found the quantification of PCR product problematic because the nanodrop overestimated the concentration and using quantitative markers on polyacrylamide gel seemed to underestimate the concentration. The difference between the two approaches was 50 fold. Loading 5 pM sample according to the nanodrop result yielded relatively low number of sequences (872,048 reads), well below the capacity of Solexa. However, loading 50 times more would have given poor quality sequences due to overloading. We decided to load four times of the amount suggested by the nanodrop and this approach yielded sufficient number (more than 3 million) and good quality reads. Many short sequences did not map to the genome but it is not known what the explanation is for that. One possibility is that the rate of sequencing error is high and many short reads contains mistakes. The other possibility is that there are polymorphisms between the species we used and the one that was used for the genome sequencing. It is also possible that plants used in these experiments were infected symptomless pathogens and genomes of those organisms contaminated our libraries.
First we searched for conserved miRNAs in the combined, almost four million reads from the two separate sRNA libraries. MiRNAs showing perfect matches to 25 conserved families were found in our combined library, which is more than was identified using bioinformatics approaches [24–26]. Allowing up to two mismatches further increased this number to 31 families. This work experimentally verified all conserved miRNAs predicted by [[24, 25] and [26]]. However, none of the predicted new miRNAs [26] were found and the eight new miRNAs we found were not predicted by any of the three studies [[24, 25] and [26]] illustrating the benefit of the sequencing approach. Target prediction for conserved miRNAs found several genes showing homology to genes validated in other species. However, several predicted targets are new without known function indicating that more work is required to elucidate the role of miRNAs in legumes.
New miRNAs
Validation of non-conserved miRNAs can be achieved through several ways. One possibility is providing evidence of biogenesis characteristic of miRNAs. This involves the identification of stem-loop structure of the potential pre-miRNAs and sequencing of the miRNA* molecules [9]. In the absence of evidence for biogenesis, functional data can complement the predictions based on structural analysis. MiRNA mediated cleavage is located in a specific site within the target site and this can be identified by 5'RACE analysis. We have found eight new non-conserved miRNAs supported by biogenesis data since the miRNA* strands were sequenced and detected by Northern blot analysis. We also found 18 other putative miRNAs based on structural data. However, we did not sequence the miRNA* strands for these and we do not have functional proof therefore at the moment these are only putative miRNAs.
The role of miRNAs in development and abiotic stress response is well documented. Several transcription factors involved in development are targeted by miRNAs [27]. Other miRNAs play a role in nutrient assimilation and responses to drought, cold and other abiotic stresses [1, 28]. Reports of miRNAs involved in biotic stress response are less common. Navarro et al [29] showed that bacterial flagellin expression induced miR393 and F-box auxin receptor genes were regulated by miR393. Two of the new miRNAs are predicted to target disease resistance genes suggesting a more extensive role for miRNAs in biotic stress response. Targets of the other new miRNAs also include beta-glucan-binding protein, peptidyl-prolyl cis-trans isomerise and a hypothetical protein. The biological importance of the potential regulation of these genes by miRNAs needs further investigation.
Conclusion
High-throughput sequencing analysis of sRNAs from M. truncatula leaves identified eight new miRNAs, which were not found in other species. Sequencing sRNAs from specific tissues of legume plants by deep sequencing is expected to reveal more new miRNAs.
Methods
Cloning of M. truncatula sRNAs and Northern blot analysis
RNA was extracted using the miRVana kit (Ambion) from the aerial part (including stems and leaves) of nine week old Medicago truncatula Gaertn. cv. Jemalong seedlings. Plants were grown in pots with soil in a growth chamber having a photoperiod of 16/8 h day/night, a thermoperiod of 25/18°C day/night and humidity of 40%. Small RNA fraction between 19–24 nt was isolated from 15% denaturing polyacrylamide gel and 15 μg was ligated to adaptors without de-phosphorylating and re-phosphorylating [30]. The short RNAs were converted to DNA by RT-PCR and the DNA was sequenced on a Solexa machine (Illumina). 15 μg of RNA extracted from M. truncatula leaves was analysed by Northern blot as described by Pall et al. [31].
Sequence analysis
The Medicago truncatula genome sequence (version 2.0) was downloaded from the Medicago Sequencing Resource website http://www.medicago.org/genome/downloads/Mt2/ and sequence reads were mapped to the genome using PatMaN [32]. miRNA candidates were generated by miRCat (http://srna-tools.cmp.uea.ac.uk/mircat/; [33]) using default parameters. DFCI Medicago Gene Index v 9.0 was used for the target predictions: ftp://occams.dfci.harvard.edu/pub/bio/tgi/data/Medicago_truncatula/MTGI.release_9.zip. Target searches were performed on the srna-tools website: http://srna-tools.cmp.uea.ac.uk/targets/[33].
The sequences can be found in GEO under GPL7704 platform (High-throughput sequencing of small RNAs in Medicago truncatula), series GSE13761, samples GSM346592 and GSM346593.
References
Phillips J, Dalmay T, Bartels D: The role of small RNAs in abiotic stress. FEBS Letters. 2007, 581 (19): 3592-7. 10.1016/j.febslet.2007.04.007.
Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ: Elucidation of the small RNA component of the transcriptome. Science. 2005, 309: 1567-1569. 10.1126/science.1114112.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297. 10.1016/S0092-8674(04)00045-5.
Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP: MicroRNAs in plants. Genes Dev. 2002, 16: 1616-1626. 10.1101/gad.1004402.
Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS: SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNA in Arabidopsis. Genes Dev. 2004, 18: 2368-2379. 10.1101/gad.1231804.
Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, Hilbert JL, Bartel DP, Crete P: Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell. 2004, 16: 69-79. 10.1016/j.molcel.2004.09.028.
Lu C, Kulkarni K, Souret FF, MuthuValliappan R, Tej SS, Poethig RS, Henderson IR, Jacobsen SE, Wang W, Green PJ, Meyers BC: MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Research. 2006, 16: 1276-1288. 10.1101/gr.5530106.
Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK: Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005, 123: 1279-1291. 10.1016/j.cell.2005.11.035.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53. 10.1146/annurev.arplant.57.032905.105218.
Kurihara Y, Watanabe Y: Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA. 2004, 101: 12753-12758. 10.1073/pnas.0403115101.
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O: Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008, 320 (5880): 1185-90. 10.1126/science.1159151.
Axtell MJ, Bartel DP: Antiquity of microRNAs and their targets in land plants. Plant Cell. 2005, 17: 1658-1673. 10.1105/tpc.105.032185.
Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC: Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004, 36: 1282-1290. 10.1038/ng1478.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC: High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE. 2007, 2 (2): e219-10.1371/journal.pone.0000219.
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP: A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20: 3407-25. 10.1101/gad.1476406.
Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, Sun Q: Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biology. 2007, 8 (6): R96-10.1186/gb-2007-8-6-r96.
Moxon S, Jing R, Szittya G, Schwach F, Rusholme Pilcher RL, Moulton V, Dalmay T: Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Research. 2008,
Moxon S, Schwach F, MacLean D, Dalmay T, Studholme DJ, Moulton V: A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008,
Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, Jacobsen SE: Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nature Genetics. 2006, 38: 721-725. 10.1038/ng1804.
Berezikov E, Thuemmler F, van Laake L, Kondova I, Bontrop R, Cuppen E, Plasterk R: Diversity of microRNAs in human and chimpanzee brain. Nature Genetics. 2006, 38: 1375-1377. 10.1038/ng1914.
Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP: Large-Scale Sequencing Reveals 21U-RNAs and Additional MicroRNAs and Endogenous siRNAs in C. elegans. Cell. 2006, 127: 1193-1207. 10.1016/j.cell.2006.10.040.
Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP, Tizard ML: A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Research. 2008, 18 (6): 957-64. 10.1101/gr.074740.107.
Lu C, Kulkarni K, Souret FF, MuthuValliappan R, Tej SS, Poethig RS, Henderson IR, Jacobsen SE, Wang W, Green PJ: MicroRNAs and other small RNAs enriched in the Arabidopsis RNAdependent RNA polymerase-2 mutant. Genome Research. 2006, 16 (10): 1276-1288. 10.1101/gr.5530106.
Sunkar R, Jagadeeswaran G: In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biology. 2008, 8: 37-50. 10.1186/1471-2229-8-37.
Zhou ZS, Huang SQ, Yang ZM: Bioinformatic identification and expression analysis of new microRNAs from Medicago truncatula. Biochemical and Biophysical Research Communications. 2008, 374: 538-542. 10.1016/j.bbrc.2008.07.083.
Wen J, Frickey T, Weiller GF: Computational prediction of candidate miRNAs and their targets from Medicago truncatula non-protein-coding transcripts. In Silico Biol. 2008, 8 (3-4): 291-306.
Kidner CA, Martienssen RA: The developmental role of microRNA in plants. Curr Opin Plant Biol. 2005, 8 (1): 38-44. 10.1016/j.pbi.2004.11.008.
Sunkar R, Zhu JK: Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004, 16 (8): 2001-19. 10.1105/tpc.104.022830.
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JD: A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006, 312 (5772): 436-9. 10.1126/science.1126088.
Rathjen T, Pais H, Sweetman D, Moulton V, Munsterberg A, Dalmay T: High throughput sequencing of microRNAs in chicken somites. BMC Genomics. 2008,
Pall GS, Codony-Servat C, Byrne J, Ritchie L, Hamilton A: Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Research. 2007, 35 (8): e60-10.1093/nar/gkm112.
Prufer K, Stenzel U, Dannemann M, Green REE, Lachmann M, Kelso J: PatMaN: rapid alignment of short sequences to large databases. Bioinformatics. 2008, 24 (13): 1530-1. 10.1093/bioinformatics/btn223.
Moxon S, Schwach F, Maclean D, Dalmay T, Studholme DJ, Moulton V: A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008, 24 (19): 2252-3. 10.1093/bioinformatics/btn428.
Acknowledgements
The work was supported by the BBSRC (TD) and FCT (MPSF) through the ERA-NET Plant Genomics initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SG cloned the short RNAs and carried out most of the Northern blot analysis; SM analysed the sequences and was supervised by VM; DMS grew the plants and prepared the RNA; RJ carried out some of the Northern blot analysis; MPSF and TD designed and coordinated the study; TD analysed the results and wrote the manuscript.
Gyorgy Szittya, Simon Moxon contributed equally to this work.
Electronic supplementary material
12864_2008_1786_MOESM1_ESM.xls
Additional file 1: Predicted targets of conserved M. truncatula miRNAs. The data provided describes the predicted target genes of conserved miRNAs found in M. truncatula. (XLS 128 KB)
12864_2008_1786_MOESM2_ESM.pdf
Additional file 2: Predicted secondary structures of new and putative M. truncatula . The data shows the predicted secondary structures of validated and putative new M. truncatula miRNAs. (PDF 92 KB)
12864_2008_1786_MOESM3_ESM.xls
Additional file 3: Predicted targets of putative miRNAs. This table provides a list of predicted target genes of potential but not validated M. truncatula miRNAs. (XLS 124 KB)
12864_2008_1786_MOESM4_ESM.doc
Additional file 4: Predicted targets of new validated M. truncatula miRNAs. This table provides a list of predicted target genes of new M. truncatula miRNAs with sequenced miRNA*. (DOC 52 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Szittya, G., Moxon, S., Santos, D.M. et al. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 9, 593 (2008). https://doi.org/10.1186/1471-2164-9-593
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-593