
Abstract
Background
Bacteriophages (phages) are one of the most promising alternatives to traditional antibiotic therapies, especially against multidrug-resistant bacteria. Klebsiella pneumoniae is considered to be an opportunistic pathogen that can cause life-threatening infections. Thus, this study aims at the characterization of a novel isolated phage vB_Kpn_ZC2 (ZCKP2, for short).
Methods
The phage ZCKP2 was isolated from sewage water by using the clinical isolate KP/08 as a host strain. The isolated bacteriophage was purified and amplified, followed by testing of its molecular weight using Pulse-Field Gel Electrophoresis (PFGE), transmission electron microscopy, antibacterial activity against a panel of other Klebsiella pneumoniae hosts, stability studies, and whole genome sequencing.
Results
Phage ZCKP2 belongs morphologically to siphoviruses as indicated from the Transmission Electron Microscopy microgram. The Pulsed Field Gel Electrophoresis and the phage sequencing estimated the phage genome size of 48.2 kbp. Moreover, the absence of lysogeny-related genes, antibiotic resistance genes, and virulence genes in the annotated genome suggests that phage ZCKP2 is safe for therapeutic use. Genome-based taxonomic analysis indicates that phage ZCKP2 represents a new family that has not been formally rated yet. In addition, phage ZCKP2 preserved high stability at different temperatures and pH values (-20 − 70 °C and pH 4 – 9). For the antibacterial activity, phage ZCKP2 maintained consistent clear zones on KP/08 bacteria along with other hosts, in addition to effective bacterial killing over time at different MOIs (0.1, 1, and 10). Also, the genome annotation predicted antibacterial lytic enzymes. Furthermore, the topology of class II holins was predicted in some putative proteins with dual transmembrane domains that contribute significantly to antibacterial activity. Phage ZCKP2 characterization demonstrates safety and efficiency against multidrug-resistant K. pneumoniae, hence ZCKP2 is a good candidate for further in vivo and phage therapy clinical applications.
Similar content being viewed by others

Introduction
Antibiotic resistance is increasing in all parts of the world, posing a serious threat to public health management practices [1]. Antibiotic-resistant strains were only seen in hospitals, but they are now prevalent throughout the world [2]. It is estimated that by 2050 if no new drug is discovered, there will be no effective antibiotics available [3]. Numerous infections have high rates of morbidity, death, and financial expense. One of the most significant public health concerns of the twenty-first century is antimicrobial resistance (AMR), which threatens the effective prevention and treatment of an increasing number of bacterial infections [4]. Resistance to antimicrobials occurs as pathogenic bacteria degrade antibacterial medicines, change their proteins, and alter their membrane permeability to antibiotics [5]. Bacteria that cause common to serious infections have developed resistance to the majority of available antibiotics on the market to varying degrees over several decades [6]. Our capacity to treat common diseases will decrease due to the emergence and spread of drug-resistant bacteria with new resistance mechanisms [7].
The most serious and commonly occurring Gram negative infections that occur in health care settings are caused by Enterobacteriaceae (mostly Klebsiella pneumoniae), Salmonella, and Escherichia coli [8, 9]. Gram negative Klebsiella pneumoniae is one of the most common Gram negative pathogens associated with a wide spectrum of community and hospital-acquired infections, such as urinary tract infection (UTI), pneumonia, intra-abdominal infection, bloodstream infection (BSI), meningitis, and pyogenic liver abscess (PLA) [10]. Globally, the prevalence of extended-spectrum cephalosporin-resistant K. pneumoniae producing extended-spectrum -lactamases (ESBL) has risen dramatically in recent decades [11]. ESBLs hydrolyze the beta lactam ring thereby inactivating the antimicrobial compounds such as penicillin and cephalosporin. The third generation of cephalosporins can hinder the treatment because ESBLs can suppress the oxyimino cephalosporins. [12]. As per Centers for Disease Control and Prevention (CDC) report in 2019, 80% of the 9000 infections that have been reported by carbapenem-resistant Enterobacteriaceae (CRE) in 2013 were caused by AMR K. pneumoniae [13]. CRE are among the top level of the WHO list of antibiotic-resistant “priority pathogens” that pose the greatest threat to human health [14].
A new approach to eradicating the spread of antibiotic-resistant bacteria is required. With the growing concern over antibiotic resistance, interest in phage therapy as a possible solution to the problem that is expanding rapidly. Phage therapy is being used to control pathogenic bacterial infections especially multiple antibiotic-resistant bacterial infections and as potential anti-inflammatory and immunomodulatory agent [15]. Their therapeutic potential in medicine to control MDR pathogens is due to their specificity and potency in inducing lethal effects in the host bacterium by cell lysis [16]. In the context of therapeutics, only virulent phages can be used. Strictly virulent phages can attack particular bacterial strains and contribute to lytic infection associated with metabolic disturbance and cell lysis, which decreases the number of bacterial cells found in the infected human host to a level that presents no danger or harm to the organism [17].
Globally, K. pneumoniae phages have been isolated from various sources, including sewage [18], ponds [19], rivers [20], seas [21], farm wastewater [22], water troughs [23], animal feces [24], and clinical samples [25, 26], with sewage from hospitals being the most common source [27,28,29,30,31,32,33]. The majority of the isolated phages of K. pneumoniae are members of the class Caudoviricetes, which is characterized by being naked (non-enveloped), dsDNA and tailed [34]. Nonetheless, a K. pneumoniae tailless phage with a tectivirus morphotype was also isolated [19]. In addition, many of the isolated K. pneumoniae phages demonstrated anti-capsule and anti-biofilm activity by expressing various types of polysaccharide depolymerases [35, 36, 39].
Prior to the genomic era, the only way to ensure the safety and taxonomy of isolated phages was to use phenotypic assays, which are nearly impossible to reveal everything encoded in the genome [37]. Since DNA sequencing methods have advanced over the last two decades, most laboratories can now sequence the genomes of bacteriophages [38]. Phages for phage therapy can be checked for any unfavorable encoding genes, such as lysogeny-encoding genes and virulence or resistance genes that may be transmitted to bacteria; therefore, genomic analysis and bioinformatics could reduce the efforts required for a safety study [39].
The effectiveness of such a strategy is dependent on two variables: the consistency with which phage resistance develops in vitro and the degree to which the resistance produced in vitro coincides with the resistance produced in vivo [40]. This paper discusses the isolation and characterization of an effective vB_Kpn_ZC2 (ZCKP2) phage against MDR-KP.
Materials and methods
Bacterial growth condition
Thirty sputum-clinical isolates of MDR Klebsiella pneumoniae were employed in this study. The bacterial isolates were collected from the lab stock at Zewail City of Science and Technology. Twenty isolates (KP/01 - KP/20) were previously characterized by Fayez et al. [41], while the remaining ten Klebsiella isolates (K1, K6, K7, K/10, K18, K/20, K24, K25, K/30, and K31) were previously characterized by Zaki et al. [39]. Fresh bacterial cultures were made before each experiment by inoculating one colony from MacConkey agar (Oxoid, England) into 1 mL of TSB in 1.5 mL centrifuge tube and incubated for 16 h at 37°C with shaking (200 rpm).
16S rRNA gene sequencing
PCR amplification and sequencing were performed to confirm the identity of the K. pneumoniae isolate (KP/08) using specific and universal primers for the 16s rRNA gene forward primer (5’-AGAGTTTGATCCTGGCTCAG-3’), and the reverse primer (5’-TACGGYTACCTTGTTACGACTT-3’). Thirty cycles were performed at a denaturation temperature of 94°C for 30 s; annealing at 55°C for 30 s and extension at 72°C for 1 min. The PCR product was run on a 1.5% (w/v) agarose gel to identify its size [42]. Following the manufacturer’s instructions, the amplified 16S rRNA gene fragment was purified using QIAEX II Gel Extraction Kit (QIAGEN, Hilden, Germany) [43]. Finch TV software (https://digitalworldbiology.com/FinchTV/) was used to process the 16S rRNA gene’s acquired nucleotide sequence. The isolated strain was identified using BLASTn (accessed on 9 SEP 2022, at Basic Local Alignment Search Tool, https://blast.ncbi.nlm.nih.gov/Blast.cgi,) against the 16S ribosomal RNA database [44]. The sequence was deposited in the NCBI GenBank database under accession number OP410967.1
Isolation, purification, and amplification of bacteriophage
Six different phages were obtained from sewage water in Giza, Egypt. The water samples were centrifuged at 4000 rpm and the supernatant was filtered from other bacteria using 0.2 µm porous syringe filters [45]. Using enrichment techniques for phage isolation, 10 mL of sewage samples were combined with 1 mL of an overnight culture from KP/08, incubated at 37°C for 4h, then mixed with 1% chloroform and centrifuged at 5000 rpm for 20 minutes while the supernatant was kept. Following this, a spot assay was performed using a mixture of 100 µl of the bacterial host culture and 4 mL of soft agar (0.5% w/v agar) then poured into a TSA plate. From each supernatant 10 µL aliquots were spotted in triplicate on bacterial lawns, and the plates were then incubated at 37°C for 24 h. Phage clear plaques were purified with repeated isolation of a single plaque using sterile micropipette tips. All isolated phages were amplified in liquid culture (TSB), and the lysates were centrifuged at 5,000 g at 4°C for 15 min [46, 47]. Then the supernatant containing phages was centrifuged for 1 h at 15,300 ×g at 4 °C. The pellet was resuspended in SM buffer (100 mM MgSO4.7 H2O; 10 mM NaCl; 50 mM Tris-HCl; pH 7.5) and purified through 0.22 µm syringe filters (Chromtech, Taiwan) 26]. Bacteriophage titers were determined using double agar overlay plaque assays and spotted in triplicate onto bacterial lawns [48]. The isolated phages were enriched and propagated in TSB, 100 mL of host was infected with each phage separately and incubated at 37°C with 120 rpm shaking to increase phage stocks [49].
Phage characterization
Pulsed field gel elecrophorises
For Pulsed Field Gel Electrophoresis (PFGE), DNA was prepared from bacteriophage vB_Kpn_ZC2 (ZCKP2) (1010 PFU/mL) to determine the genome size [50]. Fist, the bacteriophage suspended in agarose plugs were digested with lysis buffer (0.2% w/v SDS [Sigma, Gillingham, UK]; 1% w/ N-Lauryl sarcosine [Sigma, Gillingham, UK]; 100 mM EDTA; 1 mg/mL Proteinase K [Fischer Scientific]), then left overnight at 55 °C. After being washed with a washing buffer, two slices of agarose-containing DNA were placed into the wells that contain 1% w /v agarose gel. By using a Bio-Rad CHEF DRII system, the gel was run in 0.5 X Tris-borate-EDTA, at 200 V at 14 °C for 18 h with a switch time of 30 to 60 s. The genome’s size was calculated by comparing using standard concatenated lambda DNA markers of range 48.5–1,018 kb (Sigma Aldrich, Gillingham, UK).
Examination of phage by Transmission Electron Microscopy (TEM)
The morphology of phage ZCKP2 was examined by glow-discharged (1 min under vacuum) by using TEM at the National Research Center (Cairo, Egypt) [51, 52]. Formvar carbon-coated copper grids (Pelco International) were submerged into phage suspension. (2.5% v/v) glutaraldehyde was used to fix the phage, which was rinsed, and stained using 2% phosphotungstic acid (pH 7.0). After drying, grids were examined using a high-resolutiontransmission electron microscope (JEOL 1230).
Phage host range
The host range of phage ZCKP2 was determined against 30 clinical isolates of MDR K. pneumoniae by using the spot assay in triplicate as previously mentioned [48, 53]. Briefly, 100 µl of freshly prepared culture of each bacterial strain was added to 4 mL of 0.5% top agar, which was subsequently poured onto a base TSA agar. Ten microliters of phage lysate at a titer of 1010 PFU/mL were spotted onto freshly lawns of the bacterial strains and incubated overnight at 37 °C.
Relative efficiency of plating
The relative efficiency of plating (EOP) of the phage ZCKP2 was conducted by counting plaques of clear lysis after plating 10-fold serial dilutions of the phage onto fresh lawns of each susceptible bacterial strain. The plaque enumeration was accomplished by using the spotting assay over a double agar overlay. Then, EOP was calculated by dividing the enumerated PFU on each bacterial strain by the counted PFU on the isolating host. Subsequently, the EOP was classified into high (≥ 0.5), medium (0.5 − 0.1), or low (0.1 – 0.001) [54].
One-step growth curve
The eclipse period, latent period, lysis time and burst size of the ZCKP2 phage were defined by observing dynamic variations in the number of phage particles throughout a replicative cycle (modified from) [55]. Shortly, host strain KP/08 was grown at 37 °C to exponential phase (~ 108 CFU/mL) and incubated with ZCKP2 phage (~ 107 PFU/mL) at an MOI of 0.1. Aliquots of the infected culture were serially diluted at each time point to count the number of phages present using a spotting test [53]. Two aliquots were collected at each time point, the first one was treated with 1% (v/v) chloroform to induce the release of the intracellular phage, while the second one was spotted without chloroform to determine the phage infective center (IC). The ratio of the released phage titer to the original number of infected cells was used to calculate the burst size per infected cell. By deducting the average of the early time point phage titers treated with chloroform (before the end of the eclipse period) from that of the IC early titers, the number of infected cells was calculated [56]. The experiment was conducted in triplicate.
Time-killing curves
The bacterial killing activity was determined for the phage ZCKP2. The experiment lasted for 325 min, using ZCKP2 phage at different MOIs (0.1, 1, 10, and 100) against strain KP/08 at 37 °C to exponential phase (~ 107 CFU/mL) [53]. Throughout the time, the optical density (O.D 600 nm) was measured using 96-well plate (FLUOstar Omega, BMG LABTECH, Ortenberg, Germany). Data were collected at 25 min intervals for around 5.5 h using the MARS Data Analysis Software package (version 3.42). The bacterial culture without phage inoculation was used as a control. The experiments were performed in triplicate.
Phage temperature and pH stability
The stability of bacteriophages over a range of temperatures was evaluated by incubating 20 µl of the phage suspensions in 180 µl SM buffer at -20, 4, 40, 50, 60, 70, 75, and 80 °C for 4 h. The bacteriophage samples were taken after 4 h. of incubation to detect the change in phage titer upon sudden temperature alteration [49]. The titers of released bacteriophages were determined using serial dilutions and spotting using double agar overlay plaque assays. By incubating 10 µl of bacteriophage suspensions in 990 µl of TSB at different pH values (2, 3, 4, 5, 7, 9, 11, 12, and 13), and the viability of the bacteriophage was determined over time. After 4 h of incubation, pH samples were taken to determine the phage titer using serial dilutions and spotting as described before to evaluate the titers of released bacteriophages [53].
Phage genome sequancing and characterization
Genomic DNA was extracted from phage ZCKP2 (1010 PFU/mL) lysates using proteinase K (100 g/mL in 10 mM EDTA pH 8), and then resin purification using the Wizard DNA kit (Promega, UK) in accordance with the manufacturer’s instructions. DNA sequencing was carried out using the Illumina MiSeq platform. Then the Illumina Nextera tagmentation protocol (Illumina, Cambridge, UK) was used for library preparation. The data included 150 bp length paired-end sequences. FASTQC was used to access the sequences’ precision [57]. Using SPAdes [58] and K-mers of 21, 33, 55, 77, and 99, sequences were de novo assembled, producing a 48.2 kbp unique contig.
BLASTn was performed against the nucleotide collection database to find the closely matching phages After that, MEGA-X ages were imported with the best-matched phages [59] to draw a phylogenetic tree using the CLUSTAL-W aligner [60] and the best Maximum Likelihood fit model (GTR: General Time Reversible substitution model, G: Gamma distributed among sites). The NCBI ORF finder search server was used to identify open-reading frames using methionine and alternate initiation codons as the start codon. The putative coding sequences were then identified by comparing the predicted ORFs to the NCBI non-redundant protein sequences (nr) database using BLASTp (CDSs) with considering an e-value cutoff < 10− 7. Moreover, the predicted ORFs and coding sequences were matched to those predicted by PHASTER [61]. An additional round of ORFs prediction and functional annotation was performed on RASTtk [62,63,64] and BV-BRC [65] to increase confidence in the predicted encoding gene.
SnapGene Viewer (GSL Biotech; available at https://www.snapgene.com/; access on 9 SEP 2022) was used to create the circular genomic map. Phage ZCKP2’s annotated whole genome was uploaded to the GenBank database with the accession number NC_071151. Phage ZCKP2 suitability for therapeutic application was assessed using PhageLeads, which checked the genome for temperate markers, antibiotic resistance genes, and virulence genes [66]. DeepTMHMM was used to analyse the putative proteins and detect transmembrane topology [67].
The phylogenetic analysis of the phage ZCKP2 was conducted by different approaches. The phages of high similarity to phage ZCKP2 were identified by BLASTn, and their pairwise intergenomic similarities were computed by the Virus Intergenomic Distance Calculator (VIRDIC), in which the default thresholds for species (> 95%) and genus (> 70%) were applied [68]. Viral Proteomic Tree (ViPTree) was used to generate a proteomic tree of the ZCKP2 genome based on genome-wide sequence similarities computed by tBLASTx [69]. Additionally, the orthologous (signature) genes of the closely related phages were predicted by using CoreGenes 0.5 [70]. Accordingly, conserved phage proteins were used to perform phylogenetic analysis based on the alignment of their amino acid sequences. The conserved proteins were aligned using CLUSTAL-W and analysed by the best Maximum Likelihood fit model in MEGA 11 [71, 72].
Statisical analysis
All experiments were conducted in triplicate, and the results were illustrated in the form of mean ± standard deviation (SD). In this study, GraphPad Prism 9.1.1 software was used to generate graphs and perform all statistical analyses. Both Student’s t-test (two-tailed) and ANOVA tests were used during the work to evaluate the significance of p < 0.05.
Results
16 S rRNA gene sequence
The 16S rRNA sequence for the main bacterial host KP/08 was performed afterward (GenBank Acc. No. OP410967.1). BLASTn of the 16S rRNA sequence had a 99% sequence identity to the K. pneumoniae strain.
Morphology of vB_Kpn_ZC2 phage
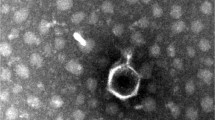
Isolated phage was initially screened against the K. pneumoniae strain through a spot test. A clear zone over a bacterial lawn was observed due to the lytic activity of the phage. On double-layer agar plates, vB_Kpn_ZC2 (ZCKP2) phage produced small but clear plaques of similar morphology (Fig. 1A). TEM micrographs showed phage ZCKP2 with an icosahedral head, a filamentous, cross-banded, and non-contractile tail; these morphological findings are characteristics of siphovirus (Fig. 1B). The phage proportions were measured on virions and the head diameter is ~ 65 nm, and the tail length is ~ 160 nm.

Visualization of the isolated phage ZCKP2. A. Clear plaques of similar morphology for ZCKP2 over double-layer agar. B. Transmission electron microscopic image of phage ZCKP2.
Bacteriophage host range and relative efficiency of plating
Seven bacterial isolates, out of 30 screened isolates, were susceptible to the phage ZCKP2 which produced clear lysis zones on them. The phage ZCKP2 was observed against the susceptible isolates of K. pneumoniae in terms of relative EOP as in Table 1.
One step growth curve
The observed phage replication kinetics in the one-step growth experiment revealed that the ZCKP2 phage had about 22 min eclipse period and a latent period of 25 (± 3) min, followed by 10 min lysis. ZCKP2 phage demonstrated a large burst size of about 650 (± 50) PFU/mL per infected bacterial cell (Fig. 2).

One-step growth curve of phage ZCKP2 at MOI 0.1. The titers phage ZCKP2 was enumerated by spotting assay. Data points represent the mean of PFU/mL at different intervals during the experiment time
Time-killing curves
The time-killing curves displayed a reduction in the optical density O.D at 600 nm for the groups treated with phage, unlike the untreated group. Moreover, the bacteria treated with higher MOIs (10 and 100) presented a faster reduction in the O.D readings (Fig. 3). After the 325 min, the untreated bacteria were at the O.D of 10.8 ± 0.96; at MOI 0.1, the O.D was 0.275 ± 0.05, at MOI 1, the O.D was 0.125 ± 0.01; at MOI 10, the O.D was 0.085 ± 0.01; and at MOI 100, the O.D was 0.095 ± 0.01. At higher MOIs (10 and 100), the bacteria started to display increase in the O.D reading, which might reflect the probable phage resistance.

Time-killing Curve of Klebsiella strain KP/08 using ZCKP2 phage at different MOIs (0.1, 1, 10 and 100) over 325 min in a shaking condition.
Phage temperature and pH stability
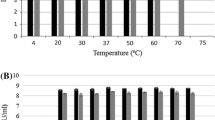
The phage ZCKP2 demonstrated high stability at storage temperature (-20 and 4°C) which was comparable to its stability at incubation temperature (40 °C) (Fig. 4A). Regarding thermal stability (above 40 °C), the phage was stable with a slight reduction in its titer at range of 50–60 °C. The phage dramatically reduced at temperature above 70 °C that even became undetectable at 80 °C.
The most optimum pH was 7.0, in addition, the phage ZCKP2 demonstrated acceptable stability with limited titer reduction at range of pH 4.0 to 9 (Fig. 4B). However, the phage did not tolerate the higher acidity (≤ pH 3.0) and alkalinity (≥ pH 11.0) conditions since no phage titers were determined.

Violin plots of the thermal and pH stability of phage ZCKP2. The phage stability is represented as enumerated titer (log10 of PFU/mL). A. Thermal stability: phage titer at -20 °C was compared to other temperatures’ phage titers. B. pH stability: phage titer at pH 7.0 was compared to phage titers at the other pH values. The mean of phage titer triplicates was calculated, and statistically analyzed by t-test at a significance level of P < 0.05.
Genome annotation and bioinformatics analysis
Pulse Field Gel Electrophoresis (PFGE) estimated the size of the double-stranded DNA genome of the phage ZCKP2 at about 48.2 kbp. The phage ZCKP2’s whole genome was sequenced and added to the GenBank database (GenBank Acc. No. NC_071151). The sequenced reads of phage ZCKP2 were assembled as one contig of size 48,258 bp, a G + C content of 47.5%, and sixty-nine ORFs. The functional genes are highlighted on the genomic map (Fig. 5). Twenty-eight putative proteins have been ascribed roles, including DNA replication/transcription/repair proteins, DNA packaging proteins, structural proteins, and cell lysis proteins. The list of the putative protein-coding genes was manually curated and listed in Supplementary Table S1. Further genomic analysis uncovered one tRNA-Arg gene with the anticodon sequence TCT.

A genomic map of phage ZCKP2, and the predicted coding sequences with a signed functions are labelled on the genomic map.
None of the predicted ORFs encodes any lysogenic phage-related proteins, such as transposases or integrases. Also, PhageLeads screened the phage genome, and no genes were predicted to have the potential for the temperate life cycle, antibiotic resistance, or bacterial virulence. The latter results indicate the safety and applicability of phage ZCKP2 for therapeutic purposes. The putative proteins were analyzed using DeepTMHMM for transmembrane domains (TMDs), which were predicted in five putative proteins (ORFs: 11, 15, 16, 28, and 35; Figure S1). The topology of two TMDs was detected in a putative holin (ORF 15), as described in Fig. 6.

Predicted transmembrane topology using the DeepTMHMM tool of putative holin (ORF 15). Red blocks represent the predicted transmembrane domains, while the pink line and blue line represent the domains inside and outside the membrane, respectively. The Y-axis represents the prediction probability, while the X-axis represents the amino acids sequence position.
Phylogenetic analysis
VIRDIC computed the intergenomic similarity of phage ZCKP2 and the top-matched phages, and only Klebsiella phage ZCKP8 was within the genus threshold (93% intergenomic similarity). Therefore, phage ZCKP2 and Klebsiella phage ZCKP8 were clustered into the same genus but different species (Fig. 7). Likewise, the proteomic tree inferred that phage ZCKP2 is closely related to Klebsiella phage ZCKP8. Additionally, the proteomic tree grouped the phage ZCKP2 with unclassified siphoviruses in a separate clade from siphoviruses of the family Drexlerviridae (Fig. 8). The genomes of the closely related phages (ViPTree score SG > 0.64) were aligned and compared to ZCKP2 (Fig. 9). The whole genome comparison highlighted the differences between the genomes, and particularly, between phage ZCKP2 and the closest phage (ZCKP8). Therefore, phage ZCKP2 may be novel based on results from whole-genome comparison and VIRIDIC’s intergenomic similarity.
The pan-genome analysis of phage ZCKP2 was conducted against closely related phages, representatives from the family Drexlerviridae and outgroup phages (myoviruses and podoviruses) (Supplementary Tables S2-S6). The pan genome analysis revealed that phage ZCKP2 and closely related phages of SG above 0.5 shared 24 orthologous genes. As summarized in Supplementary Table S7, the number of orthologs increased with higher SG, reaching a maximum of 69 in the case of the closest phage, ZCKP8 (SG = 0.92). Conversely, the number of orthologs dropped to seven when phage ZCKP2 was compared to phages from the family Drexlerviridae.
Based on the pan-genome analysis, the proteins of the most prominent signature genes were selected to conduct further phylogenetic analysis (Fig. 10A-D). The signature genes included major capsid protein, terminase large subunit, single-strand DNA-binding protein, and DNA polymerase III. In the four trees, phage ZCKP2 was clustered with other unclassified siphoviruses, but particularly Klebsiella phage ZCKP8, phage 6991, phage VLCpiS13a, phage VLCpiS13b, and phage VLCpiS13c were common to all clusters. Representatives of the family Drexlerviridae were separately clustered from ZCKP2 in all inferred protein-based phylogenetic trees. Consequently, phage ZCKP2 most likely represents a new family with other unclassified siphoviruses and shares the same genus as ZCKP8.

Virus Intergenomic Distance Calculator (VIRIDIC) heatmap for the intergenomic similarity between ZCKP2 and top-matched phages on BLASTn.

Proteomic tree generated by ViPTree of phage ZCKP2. (A) Circular proteomic tree of phage ZCKP2, top BLASTn hits, and related phages of RefSeq genomes. (B) Rectangular tree represents a subset of the closely related phages from circular tree.

Whole-genome alignment and comparison between the phage ZCKP2 and closely related phages.

Phylogenetic tree of the aligned amino acid sequence of signature proteins, (A) major capsid protein, (B) terminase large subunit, (C) single-strand DNA-binding protein, and (D) DNA polymerase III. The analysis was conducted by the Maximum likelihood method with a bootstrap of 100 replicates, in MEGA11.
Discussion
Klebsiella pneumoniae is an important opportunistic pathogen that regularly causes nosocomial infections and contributes to substantial morbidity and mortality. Currently, K. pneumoniae is showing a high resistance to a wide-ranging spectrum of drugs including beta-lactam antibiotics, fluoroquinolones, and aminoglycosides [41, 73, 94]. Commonly, antimicrobial resistance is associated with the proliferation of transmissible plasmids and the acquisition of resistance genes that generally occur via horizontal gene transfer, which may also include virulence factors [74, 94].
Bacteriophage therapy is one such approach that can be used as an alternative to antibiotics. Conventionally, phage therapy relies on the use of naturally occurring bacterial parasites which are incapable of reproducing on their own (i.e., they are non-living) and are entirely dependent on a bacterial host for their existence by infecting and lysing them [75]. Their therapeutic potential in medicine to control MDR pathogens is due to their specificity and potency in inducing lethal effects in the host bacterium by cell lysis [76]. Interestingly, phage vB_Kpn_ZC2 (ZCKP2) infected 7 out of 30 bacterial strains, demonstrating a relatively narrow host range. However, ZCKP2 has shown that the phage may lyse several Klebsiella strains. The most vulnerable bacterial host is intended to be used in the classic isolation method for enrichment. However, current research indicates that using a large number of hosts during the isolation process increases the likelihood of separating phages with greater host ranges.
The evaluation of the phage growth curve is considered the essential part to be demonstrated when it is applied as a therapeutic agent. The short latent period and large burst size (> 200 PFU/cell) of phage ZCKP2 are compared to a few published Klebsiella siphoviruses [22, 77] and also, a few phages had medium burst size [39, 78, 79].
Time-killing curves were done to study the antibacterial activity of ZCKP2 phage against its host KP/08 by infecting the bacteria at exponential phase for 325 min with phage at different MOIs in a shaking condition to achieve a faster rate of bacterial growth and the homogeneous distribution of the nutrition [80]. The curves showed that the ZCKP2 phage inhibited the bacterial growth in a MOI-dependent manner, where the higher MOIs showed the highest reduction value and the bacteria started to display an increase in the O.D reading, which might reflect the probable phage resistance. These results are consistent with the previous phages, in which the ZCKP2 phage displayed similar activity as the ZCKP1 [52], ZCKP8 [41], vB_KpnS_Kp13 [81] and P545 [82], but different activity from vB_KpnS_MK54 that employed a different methodology for determining the optimal MOI. Unlike, ZCKP1, vB_KpnS_Kp13, P545 and ZCKP2 phages, vB_KpnS_MK54 phage has an optimal MOI of 0.01. Respectively, further investigations are needed to study and standardize the methodology of determining the optimal MOI [83].
The resistance to environmental stress assay was performed to investigate the phage application of primary conditions and prospects [84]. Phage stability following exposure to varying temperatures and pH was determined. The phage was stable at -20 °C and 4 °C. Although the titer of the phages was slightly reduced after 4 h of exposure at 40 °C, this phage was stable at temperatures ranging from 20 °C to 50 °C. Incubation at more than 60 °C for 4 h was lethal to the phage, thus completely inactivating them. Such characteristics aligned with the results reported in a previous study with the phage VpKK5. The phage BPA43 was tested for its pH stability and the phage was also found to be stable from pH 4.0 to 9.0 and was most active at pH 5.0 whilst it lost its activity completely at pH 3.0, 11, 12, and 13 [85].
The whole genome analysis facilitates the characterization of novel phages and readily fills the gaps resulting from in vitro analysis. The genomic characterization identified a cluster of two adjacent genes (ORFs 14 and 15) encoding putative holin and lysozyme. Holins form holes in the inner membrane of the bacterial cytoplasmic membrane; these holes allow lysozymes to leak into the periplasmic space; consequently, the lysozymes can reach and effectively break down the bacterial rigid barrier ‘peptidoglycan’ [86]. The predicted cluster of the genes encoding holin and lysozyme reveals the possible mechanism by which ZCKP2 lyses the infected bacterial cells to free the newly formed progeny at the end of the lytic cycle. In silico analysis of the phage did not detect any genes related to lysogeny, antibiotic resistance or bacterial virulence. Accordingly, ZCKP2 genomic analysis strongly suggests the safety and applicability of using a phage as a therapeutic agent [37].
Identifying regions of transmembrane in the putative proteins would indicate possible functions of these proteins. DeepTMHMM tool predicted two hydrophobic transmembrane domains in the putative holin (ORF 15, 81 amino acids) that comply with the topology of class II holins. Class II holins are characterized by double transmembrane and 60 to 185 amino acid residues [87]. Likewise, the putative tail length measuring protein (ORF 35) had two transmembrane domains. The latter prediction complies with this protein function as it facilitates the transfer of viral DNA to the infected bacterial cytoplasm through forming a channel within the bacterial cell membranes [88]. Also, double transmembrane topology was predicted in the hypothetical protein (ORF 28), suggesting that this protein could be a novel antimicrobial [89]. Future research on ZCKP2 would isolate and investigate this hypothetical protein activity.
Different approaches were applied for the phylogenetic analysis of ZCKP2, and all these approaches had quite similar results. VIRIDIC classified ZCKP2 is the same genus as ZCKP8, but the intergenomic similarity was below the species clustering threshold. VIRIDIC classification is highly reliable since it follows the algorithm used by ICTV [68]. Similarly, proteomic tree phylogeny inferred that ZCKP2 is closely related to ZCKP8 and other siphoviruses, but distantly related to members of family Drexlerviridae. Proteomic trees reveal information about the evolutionary history of phages [90, 91], which is useful for studying more distant relationships [92, 95]. According to the International Committee on Taxonomy of Viruses (ICTV) 2018–2019 update, the host type and genomic characteristics (GC content, genome size, number of coding sequences) of the family Drexlerviridae [93] are similar to those of phage ZCKP2; however, proteomic and pan-genome evolutionary analyses clustered the phage in a different unrated family.
Conclusion
Phage therapy gains more scientific attention as a potential alternative to antibiotics. Here, the paper evaluated the potential activity and safety of phage vB_Kpn_ZC2 (ZCKP2) upon employing it to control the growth of K. pneumoniae. The findings suggest that phage ZCKP2 is a potential candidate for further study in vivo to confirm its safety and efficiency in the field of phage therapy. In addition, further studies can be done to augment the phage K. pneumoniae infections with other phages (a phage cocktail) or antibiotics to increase its host range and target more bacteria in different applications.
Data Availability
The dataset presented in this study can be found in NCBI GenBank. The 16s rRNA sequence was deposited under the accession number OP410967.1. In addition, the annotated genome sequence of the phage ZCKP2 was deposited under the accession numbers GenBank Acc. No. NC_071151.
Change history
05 June 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12985-023-02083-2
References
Collignon P. Antibiotic resistance: are we all doomed?: antibiotic resistance: are we all doomed? Intern Med J. 2015;45:1109–15.
Ventola CL. The Antibiotic Resistance Crisis. Pharm Ther. 2015;40:277–83.
Vivas R, Barbosa AAT, Dolabela SS, Jain S. Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb Drug Resist. Mary Ann Liebert, Inc., publishers; 2019;25:890–908.
Prestinaci F, Pezzotti P, Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog Glob Health. 2015;109:309–18.
Dahal RH, Chaudhary DK. Microbial Infections and Antimicrobial Resistance in Nepal: current Trends and Recommendations. Open Microbiol J. 2018;12:230–42.
Abdelsattar AS, Makky S, Nofal R, Hebishy M, Agwa MM, Aly RG, et al. Enhancement of wound healing via topical application of natural products: in vitro and in vivo evaluations. Arab J Chem. 2022;15:103869.
Zaman SB, Hussain MA, Nye R, Mehta V, Mamun KT, Hossain N. A Review on Antibiotic Resistance: Alarm Bells are Ringing. Cureus. 2020;9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5573035/
Rossolini GM, Arena F, Pecile P, Pollini S. Update on the antibiotic resistance crisis. Curr Opin Pharmacol. 2014;18:56–60.
CDC. Antibiotic-resistant Germs: New Threats [Internet]. Cent. Dis. Control Prev. 2021. Available from: https://www.cdc.gov/drugresistance/biggest-threats.html
Vading M, Nauclér P, Kalin M, Giske CG. Invasive infection caused by Klebsiella pneumoniae is a disease affecting patients with high comorbidity and associated with high long-term mortality. PLoS ONE. 2018;13:e0195258.
Iredell J, Brown J, Tagg K. Antibiotic resistance in Enterobacteriaceae: mechanisms and clinical implications. BMJ. 2016;352:h6420.
Paterson DL, Bonomo RA. Extended-spectrum beta-lactamases: a clinical update. Clin Microbiol Rev. 2005;18:657–86.
Solomon SL, Oliver KB. Antibiotic resistance threats in the United States: stepping back from the Brink. Am Fam Physician. 2014;89:938–41.
Willyard C. The drug-resistant bacteria that pose the greatest health threats. Nature. 2017;543:15–5.
Górski A, Międzybrodzki R, Jończyk-Matysiak E, Żaczek M, Borysowski J. Phage-specific diverse effects of bacterial viruses on the immune system. Future Microbiol Future Medicine. 2019;14:1171–4.
Dufour N, Debarbieux L. [Phage therapy: a realistic weapon against multidrug resistant bacteria]. Med Sci. 2017;33:410–6.
Kutter E, De Vos D, Gvasalia G, Alavidze Z, Gogokhia L, Kuhl S, et al. Phage therapy in clinical practice: treatment of human infections. Curr Pharm Biotechnol. 2009;11:69–86.
Sofy AR, El-Dougdoug NK, Refaey EE, Dawoud RA, Hmed AA. Characterization and full genome sequence of Novel KPP-5 lytic phage against Klebsiella pneumoniae responsible for recalcitrant infection. Biomedicines. 2021;9:342.
Singh A, Singh AN, Rathor N, Chaudhry R, Singh SK, Nath G. Evaluation of bacteriophage cocktail on Septicemia caused by colistin-resistant Klebsiella pneumoniae in mice Model. Front Pharmacol. 2022;13:778676.
Anand T, Virmani N, Kumar S, Mohanty AK, Pavulraj S, Bera BC, et al. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J Glob Antimicrob Resist. 2020;21:34–41.
Cui Z, Shen W, Wang Z, Zhang H, Me R, Wang Y, et al. Complete genome sequence of Klebsiella pneumoniae phage JD001. J Virol. 2012;86:13843.
Pertics BZ, Cox A, Nyúl A, Szamek N, Kovács T, Schneider G. Isolation and characterization of a Novel Lytic bacteriophage against the K2 Capsule-Expressing Hypervirulent Klebsiella pneumoniae strain 52145, and identification of its functional depolymerase. Microorganisms. 2021;9:650.
Luo Z, Geng S, Lu B, Han G, Wang Y, Luo Y, et al. Isolation, genomic analysis, and preliminary application of a bovine Klebsiella pneumoniae bacteriophage vB_Kpn_B01. Front Vet Sci. 2021;8:622049.
Harb L, Boeckman J, Newkirk H, Liu M, Gill JJ, Ramsey J. Complete genome sequence of the Novel Klebsiella pneumoniae phage Marfa. Microbiol Resour Announc. 2019;8:e00748–19.
Komisarova EV, Kislichkina AA, Krasilnikova VM, Bogun AG, Fursova NK, Volozhantsev NV. Complete nucleotide sequence of Klebsiella pneumoniae bacteriophage vB_KpnM_KpV477. Genome Announc. 2017;5:e00694–17.
Solovieva EV, Myakinina VP, Kislichkina AA, Krasilnikova VM, Verevkin VV, Mochalov VV, et al. Comparative genome analysis of novel podoviruses lytic for hypermucoviscous Klebsiella pneumoniae of K1, K2, and K57 capsular types. Virus Res. 2018;243:10–8.
Li F, Tian F, Nazir A, Sui S, Li M, Cheng D, et al. Isolation and genomic characterization of a novel Autographiviridae bacteriophage IME184 with lytic activity against Klebsiella pneumoniae. Virus Res. 2022;319:198873.
Pu M, Han P, Zhang G, Liu Y, Li Y, Li F, et al. Characterization and comparative Genomics analysis of a New Bacteriophage BUCT610 against Klebsiella pneumoniae and Efficacy Assessment in Galleria mellonella Larvae. Int J Mol Sci Multidisciplinary Digital Publishing Institute. 2022;23:8040.
Bai J, Zhang F, Liang S, Chen Q, Wang W, Wang Y, et al. Isolation and characterization of vB_kpnM_17 – 11, a novel phage efficient against Carbapenem-Resistant Klebsiella pneumoniae. Front Cell Infect Microbiol. 2022;12:897531.
Chen X, Tang Q, Li X, Zheng X, Li P, Li M, et al. Isolation, characterization, and genome analysis of bacteriophage P929 that could specifically lyase the KL19 capsular type of Klebsiella pneumoniae. Virus Res. 2022;314:198750.
Han K, Zhu Y, Li F, Li M, An X, Song L et al. Genomic Analysis of Bacteriophage BUCT86 Infecting Klebsiella Pneumoniae.Microbiol Resour Announc. 11:e01238-21.
Habibinava F, Soleimani M, Sabouri S, Zargar M, Zolfaghari MR. Isolating and sequencing vB_Kpn_3, a lytic bacteriophage against multidrug-resistant Klebsiella pneumoniae. Future Microbiol. 2022;17:235–49.
Fang Q, Feng Y, McNally A, Zong Z. Characterization of phage resistance and phages capable of intestinal decolonization of carbapenem-resistant Klebsiella pneumoniae in mice. Commun Biol. 2022;5:48.
Herridge WP, Shibu P, O’Shea J, Brook TC, Hoyles L. Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J Med Microbiol. 2020;69:176–94.
Chhibber S, Nag D, Bansal S. Inhibiting biofilm formation by Klebsiella pneumoniae B5055 using an iron antagonizing molecule and a bacteriophage. BMC Microbiol. 2013;13:174.
Jamal M, Hussain T, Das CR, Andleeb S. Characterization of Siphoviridae phage Z and studying its efficacy against multidrug-resistant Klebsiella pneumoniae planktonic cells and biofilm. J Med Microbiol Microbiology Society. 2015;64:454–62.
Philipson CW, Voegtly LJ, Lueder MR, Long KA, Rice GK, Frey KG, et al. Characterizing phage genomes for therapeutic applications. Viruses. 2018;10:188.
Klumpp J, Fouts DE, Sozhamannan S. Next generation sequencing technologies and the changing landscape of phage genomics. Bacteriophage. 2012;2:190–9.
Zaki BM, Fahmy NA, Aziz RK, Samir R, El-Shibiny A. Characterization and comprehensive genome analysis of novel bacteriophage, vB_Kpn_ZCKp20p, with lytic and anti-biofilm potential against clinical multidrug-resistant Klebsiella pneumoniae. Front Cell Infect Microbiol. 2023;13. Available from: https://www.frontiersin.org/articles/https://doi.org/10.3389/fcimb.2023.1077995
Hesse S, Rajaure M, Wall E, Johnson J, Bliskovsky V, Gottesman S, et al. Phage resistance in Multidrug-Resistant Klebsiella pneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. Volume 11. mBio. American Society for Microbiology; 2020. pp. e02530–19.
Fayez MS, Hakim TA, Agwa MM, Abdelmoteleb M, Aly RG, Montaser NN, et al. Topically Applied Bacteriophage to control Multi-Drug resistant Klebsiella pneumoniae infected Wound in a rat model. Volume 10. Antibiotics. Multidisciplinary Digital Publishing Institute; 2021. p. 1048.
Srivastava S, Singh V, Kumar V, Verma PC, Srivastava R, Basu V, et al. Identification of regulatory elements in 16S rRNA gene of Acinetobacter species isolated from water sample. Bioinformation. 2008;3:173–6.
Abraham O-SJ, Miguel T-S, Inocencio H-C, Blondy C-C. A quick and effective in-house method of DNA purification from agarose gel, suitable for sequencing. 3 Biotech. 2017;7:180.
Treves DS. Review of three DNA analysis applications for Use in the Microbiology or Genetics Classroom. J Microbiol Biol Educ American Society for Microbiology. 2010;11:186–7.
Townsend EM, Kelly L, Gannon L, Muscatt G, Dunstan R, Michniewski S, et al. Isolation and characterization of Klebsiella Phages for Phage Therapy. PHAGE Ther Appl Res. 2021;2:26–42.
Islam MdS, Zhou Y, Liang L, Nime I, Liu K, Yan T, et al. Application of a phage cocktail for control of Salmonella in Foods and reducing Biofilms. Viruses. 2019;11:841.
Abdelsattar AS, Abdelrahman F, Dawoud A, Connerton IF, El-Shibiny A. Encapsulation of E. coli phage ZCEC5 in chitosan–alginate beads as a delivery system in phage therapy. AMB Express. 2019;9:87.
Mazzocco A, Waddell TE, Lingohr E, Johnson RP. Enumeration of bacteriophages by the direct plating plaque assay. Methods Mol Biol Clifton NJ. 2009;501:77–80.
Abdelrahman F, Rezk N, Fayez MS, Abdelmoteleb M, Atteya R, Elhadidy M, et al. Isolation, characterization, and genomic analysis of three Novel E. coli Bacteriophages that effectively infect E. coli O18. Microorganisms. Volume 10. Multidisciplinary Digital Publishing Institute; 2022. p. 589.
Senczek D, Stephan R, Untermann F. Pulsed-field gel electrophoresis (PFGE) typing of Listeria strains isolated from a meat processing plant over a 2-year period. Int J Food Microbiol. 2000;62:155–9.
Makky S, Rezk N, Abdelsattar AS, Hussein AH, Eid A, Essam K, et al. Characterization of the biosynthesized Syzygium aromaticum-mediated silver nanoparticles and its antibacterial and antibiofilm activity in combination with bacteriophage. Results Chem. 2023;5:100686.
Taha OA, Connerton PL, Connerton IF, El-Shibiny A. Bacteriophage ZCKP1: A Potential Treatment for Klebsiella pneumoniae Isolated From Diabetic Foot Patients. Front Microbiol. Frontiers; 2018;9. Available from: https://www.frontiersin.org/articles/https://doi.org/10.3389/fmicb.2018.02127/full#B54
Rezk N, Abdelsattar AS, Elzoghby D, Agwa MM, Abdelmoteleb M, Aly RG, et al. Bacteriophage as a potential therapy to control antibiotic-resistant Pseudomonas aeruginosa infection through topical application onto a full-thickness wound in a rat model. J Genet Eng Biotechnol. 2022;20:133.
Mirzaei MK, Nilsson AS. Isolation of phages for phage therapy: a comparison of Spot tests and efficiency of plating analyses for determination of host range and efficacy. Volume 10. PLOS ONE. Public Library of Science; 2015. p. e0118557.
Kutter E, Sulakvelidze A. BACTERIOPHAGES: BIOLOGY AND APPLICATIONS.:5.
Entrepreneurial Orientation and Business Performance: An Assessment of past Research and Suggestions for the Future - Andreas Rauch, Johan Wiklund, G.T. Lumpkin, Michael Frese., 2009. Available from: https://journals.sagepub.com/doi/full/https://doi.org/10.1111/j.1540-6520.2009.00308.x
LaMar D.FastQC. 2015
SPAdes: A. New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing | Journal of Computational Biology [Internet]. [cited 2022 Oct 15]. Available from: https://www.liebertpub.com/doi/abs/https://doi.org/10.1089/cmb.2012.0021
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35:1547–9.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: Rapid annotations using Subsystems Technology. BMC Genomics. 2008;9:75.
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep Nature Publishing Group. 2015;5:8365.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014;42:D206–214.
Olson RD, Assaf R, Brettin T, Conrad N, Cucinell C, Davis JJ, et al. Introducing the bacterial and viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023;51:D678–89.
Yukgehnaish K, Rajandas H, Parimannan S, Manickam R, Marimuthu K, Petersen B, et al. PhageLeads: Rapid Assessment of Phage therapeutic suitability using an Ensemble Machine Learning Approach. Viruses. Volume 14. Multidisciplinary Digital Publishing Institute; 2022. p. 342.
Hallgren J, Tsirigos KD, Pedersen MD, Armenteros JJA, Marcatili P, Nielsen H et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv; 2022. p. 2022.04.08.487609. Available from: https://www.biorxiv.org/content/https://doi.org/10.1101/2022.04.08.487609v1
Moraru C, Varsani A, Kropinski AM. VIRIDIC—A Novel Tool to calculate the intergenomic similarities of Prokaryote-Infecting Viruses. Viruses. Volume 12. Multidisciplinary Digital Publishing Institute; 2020. p. 1268.
Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, Goto S. ViPTree: the viral proteomic tree server. Bioinformatics. 2017;33:2379–80.
Davis P, Seto D, Mahadevan P. CoreGenes5.0: an updated user-friendly webserver for the determination of Core genes from sets of viral and bacterial genomes. Viruses. 2022;14:2534.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Improved General Amino Acid Replacement Matrix. | Molecular Biology and Evolution | Oxford Academic. Available from: https://academic.oup.com/mbe/article/25/7/1307/1041491
Ferreira RL, da Silva BCM, Rezende GS, Nakamura-Silva R, Pitondo-Silva A, Campanini EB, et al. High prevalence of Multidrug-Resistant Klebsiella pneumoniae harboring several virulence and β-Lactamase encoding genes in a brazilian Intensive Care Unit. Front Microbiol. 2018;9:3198.
Derakhshan S, Najar Peerayeh S, Bakhshi B. Association between Presence of virulence genes and Antibiotic Resistance in Clinical Klebsiella Pneumoniae isolates. Lab Med. 2016;47:306–11.
Phage therapy. : An alternative to antibiotics in the age of multi-drug resistance - PMC. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5547374/
Dufour N, Debarbieux L. [Phage therapy: a realistic weapon against multidrug resistant bacteria]. Med Sci MS. 2017;33:410–6.
Wintachai P, Naknaen A, Thammaphet J, Pomwised R, Phaonakrop N, Roytrakul S, et al. Characterization of extended-spectrum-β-lactamase producing Klebsiella pneumoniae phage KP1801 and evaluation of therapeutic efficacy in vitro and in vivo. Sci Rep. 2020;10:11803.
Feng J, Gao L, Li L, Zhang Z, Wu C, Li F, et al. Characterization and genome analysis of novel Klebsiella phage BUCT556A with lytic activity against carbapenemase-producing Klebsiella pneumoniae. Virus Res. 2021;303:198506.
Tabassum R, Shafique M, Khawaja KA, Alvi IA, Rehman Y, Sheik CS, et al. Complete genome analysis of a Siphoviridae phage TSK1 showing biofilm removal potential against Klebsiella pneumoniae. Sci Rep Nature Publishing Group. 2018;8:17904.
Juergensmeyer MA, Nelson ES, Juergensmeyer EA. Shaking alone, without concurrent aeration, affects the growth characteristics of Escherichia coli. Lett Appl Microbiol. 2007;45:179–83.
Horváth M, Kovács T, Koderivalappil S, Ábrahám H, Rákhely G, Schneider G. Identification of a newly isolated lytic bacteriophage against K24 capsular type, carbapenem resistant Klebsiella pneumoniae isolates. Sci Rep Nature Publishing Group. 2020;10:5891.
Li M, Guo M, Chen L, Zhu C, Xiao Y, Li P, et al. Isolation and characterization of Novel Lytic Bacteriophages infecting Epidemic Carbapenem-Resistant Klebsiella pneumoniae strains. Front Microbiol. 2020;11:1554.
Lu B, Yao X, Han G, Luo Z, Zhang J, Yong K, et al. Isolation of Klebsiella pneumoniae phage vB_KpnS_MK54 and pathological Assessment of Endolysin in the treatment of Pneumonia mice Model. Front Microbiol. 2022;13:854908.
Krasowska A, Biegalska A, Augustyniak D, Łoś M, Richert M, Łukaszewicz M. Isolation and characterization of phages infecting Bacillus subtilis. BioMed Res Int Hindawi. 2015;2015:e179597.
Anand T, Virmani N, Kumar S, Mohanty AK, Pavulraj S, Bera BCh, et al. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J Glob Antimicrob Resist. 2020;21:34–41.
Cahill J, Young R. Phage lysis: multiple genes for multiple barriers. Adv Virus Res. 2019;103:33–70.
Vukov N, Scherer S, Hibbert E, Loessner MJ. Functional analysis of heterologous holin proteins in a λ∆S genetic background. FEMS Microbiol Lett. 2000;184:179–86.
Xu J, Xiang Y. Membrane penetration by bacterial viruses. J Virol. 2017;91:e00162–17.
Spruit CM, Wicklund A, Wan X, Skurnik M, Pajunen MI. Discovery of three toxic proteins of Klebsiella phage fHe-Kpn01. Viruses. 2020;12:544.
Rohwer F, Edwards R. The phage proteomic tree: a genome-based taxonomy for phage. J Bacteriol. 2002;184:4529–35.
Hendrix RW. Bacteriophage genomics. Curr Opin Microbiol. 2003;6:506–11.
Grose JH, Casjens SR. Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology. 2014;468–470:421–43.
Adriaenssens EM, Sullivan MB, Knezevic P, van Zyl LJ, Sarkar BL, Dutilh BE, et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV bacterial and archaeal viruses Subcommittee. Arch Virol. 2020;165:1253–60.
Osama DM, Zaki BM, Khalaf WS, Mohamed MY, Tawfick MM, Amin HM. Occurrence and Molecular Study of Hypermucoviscous/Hypervirulence Trait in Gut Commensal K. pneumoniae from Healthy Subjects. Microorganisms. 2023;11(3):704.
Zaki BM, Mohamed AA, Dawoud A, Essam K, Hammouda ZK, Abdelsattar AS, El-Shibiny A. Isolation, screening and characterization of phage. 2023.
Acknowledgements
The authors would like to thank Reem El-Menyawy and Nouran Rezk for her help in finalizing the manuscript and thanks to Fatma Abdelrahman for extracting phage DNA and sequencing.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) grant #41909 in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.S.F. and A.E.-S.; methodology, M.S.F., T.A.H., K.E., A.S., S.M.; soft-ware, B.M.Z., M.A., A.S.A.; validation, M.S.F. and A.E.-S.; writing—original draft preparation, M.S.F., T.A.H., S.M., B.M.Z., K.E., A.S., A.S.A., and A.E.-S.; writing—review and editing, M.S.F., B.M.Z., and A.E.-S.; supervision A.E.-S.; funding acquisition, A.E.-S. All authors have read and agreed to the published version of the manuscript.”
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fayez, M.S., Hakim, T.A., Zaki, B.M. et al. Morphological, biological, and genomic characterization of Klebsiella pneumoniae phage vB_Kpn_ZC2. Virol J 20, 86 (2023). https://doi.org/10.1186/s12985-023-02034-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-023-02034-x