
Abstract
Background
Mannhemia haemolytica is a Gram-negative bacterium and the principal etiological agent associated with bovine respiratory disease complex. They transform from a benign commensal to a deadly pathogen, during stress such as viral infection and transportation to feedlots and cause acute pleuropneumonia commonly known as shipping fever. The U.S beef industry alone loses more than one billion dollars annually due to shipping fever. Despite its enormous economic importance there are no specific and accurate genetic markers, which will aid in understanding the pathogenesis and epidemiology of M. haemolytica at molecular level and assist in devising an effective control strategy.
Description
During our comparative genomic sequence analysis of three Mannheimia haemolytica isolates, we identified a number of genes that are unique to each strain. These genes are "high value targets" for future studies that attempt to correlate the variable gene pool with phenotype. We also identified a number of high confidence single nucleotide polymorphisms (hcSNPs) spread throughout the genome and focused on non-synonymous SNPs in known virulence genes. These SNPs will be used to design new hcSNP arrays to study variation across strains, and will potentially aid in understanding gene regulation and the mode of action of various virulence factors.
Conclusions
During our analysis we identified previously unknown possible type III secretion effector proteins, clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated sequences (Cas). The presence of CRISPR regions is indicative of likely co-evolution with an associated phage. If proven functional, the presence of a type III secretion system in M. haemolytica will help us re-evaluate our approach to study host-pathogen interactions. We also identified various adhesins containing immuno-dominant domains, which may interfere with host-innate immunity and which could potentially serve as effective vaccine candidates.
Similar content being viewed by others


Background
Mannhemia haemolytica is a weakly haemolytic, Gram-negative bacterium and the principal casual agent associated with the respiratory-disease complex in ruminants. M. haemolytica is a normal commensal of the upper respiratory tract and tonsillar crypts in healthy ruminants. However, in the case of animals with compromised pulmonary defense mechanisms and stress, it can migrate into the lungs and cause acute fibrinous pleuropneumonia or pasteurellosis, commonly known as "shipping fever" [1–3]. Young animals are more susceptible than adults leading to sudden death with or without clinical signs [4]. Outbreaks of Pasteurellosis caused by M. haemolytica result in substantial economic losses to the global cattle industry and accounts for 30% of the total cattle deaths worldwide [5–7]. This impact is particularly devastating to the North American cattle and sheep industries [5, 6]. The U.S beef industry alone loses more than one billion dollars annually to shipping fever [8]. In addition, M. haemolytica infection results in collateral losses to other domestic and wild ruminants.
Despite its enormous economic importance, there are no specific and accurate genetic markers to precisely understand the pathogenesis and epidemiology of M. haemolytica at molecular level. Commonly used genotyping techniques such as16 S rRNA sequence phylogeny, DNA:DNA hybridization, pulse field gel electrophoresis and restriction fragment length polymorphisms are unreliable, time consuming and cannot be correlated to pathogenesis or species specificity of an isolate [9–11]. The molecular basis of virulence mechanisms of M. haemolytica is fragmentary, due to the complex gene regulatory machinery involved during the expression of virulence and virulence-associated factors in host tissues [12]. In addition, until our recent publication of the genome sequences of two M. haemolytica serotype A2 strains, only one genome sequence from M. haemolytica, serotype A1 was available in the GenBank [13, 14]. As a result, there was clearly a dearth of knowledge about the range and diversity of potential virulence factors in different strains of M. haemolytica.
The current classification of M. haemolytica relies on serotyping based on external capsular polysaccharides and twelve different serotypes have been identified. Furthermore, these serotypes do not conform to the classical Koch's postulates on microbial pathogenesis due to a great deal of genome plasticity and frequent serotype switching [15]. Of these, A1 and A2 are the most prevalent serotypes and are normal residents of the upper respiratory tracts of healthy cattle and sheep worldwide [16, 17]. They are generally, but not exclusively, species-specific in their ability to cause pneumonia [16, 17]. Bovine pneumonic pasteurellosis is mostly caused by M. haemolytica serotype A1, while the serotype A2 causes pneumonia in sheep. With the objectives of identifying the species specificity of serotypes at the molecular level, single nucleotide polymorphisms (SNPs) that may be associated with fibronecrotizing pneumonia, and identifying additional virulence factors, we sequenced two M. haemolytica A2 serotypes from two different ruminant species. One was isolated from the pneumonic lungs of domestic sheep (Ovis aries) and designated as Ovine (O), while the other isolate from cattle (Bos taurus), was designated as Bovine (B). We performed a genome-wide comparative sequence analysis between these strains and the genome sequence of M. haemolytica bovine serotype A1, PHL213 which we designated as A1.
During this investigation, we identified all the previously described virulence factors including lipopolysaccharide (LPS) biosynthesis, iron acquisition, complex carbohydrate biosynthesis, capsular polysaccharides and adhesion biosynthesis genes in both the B and O genomes. We used our data to identify genes that are unique to each strain as well as SNP variation between the strains with a focus on non-synonymous SNPs in known virulence factors. In pathogenic bacteria, SNPs serve as evolutionary markers and those present in virulence factors may aid in defining the host specificity at molecular level [18]. We identified clustered regularly interspaced short palindromic repeats (CRISPR), CRISPR-associated sequences (Cas) and a previously undetected possible type III effector protein secretory pathway, which may be implicated in modulating M. haemolytica pathogenesis in a species-specific manner. We identified high confidence SNPs (hcSNPs) within the leukotoxin (Lkt) operon across all three isolates but only one hcSNP between bovine isolates in the open reading frames (ORFs) encoding O-sialoendoglycopeptidase, a key enzyme which determines the host-specific colonization of the bacteria. Along with these genotypic markers, we identified various adhesins containing hedgehog/intein and hep/hag immuno-dominant domains, which could potentially be used to engineer vaccine strains.
Construction and content
Genome sequencing and assembly
M. haemolytica serotype A2 isolated from pneumonic lungs of domestic sheep (Ovis aries) and cattle (Bos taurus), were grown overnight in brain heart infusion broth at 37°C/200 rpm. The next day, cells were harvested and the total genomic DNA was extracted using QuickExtract™ Bacterial DNA Extraction Kit (Epicenter Biotechnologies) following the manufacturer's instructions. Genomic libraries for sequencing were prepared from 5 μg of total genomic DNA. The sequencing reaction was performed using the 454 pyrosequencing technology and run on Genome Sequencer FLX Instrument (Software 1.0.53) following the manufacturer's instructions (Hoffmann-La Roche Ltd) [19]. The raw data was assembled using the Newbler Assembler Software (Genome Sequencer 20, Version 1.0.53), with default parameters.
Gene prediction and annotation
The assembled contig sequences were processed by our in-house pipeline for gene prediction and annotation using the genome of serotype A1 as a guide. We adapted the protocol previously developed by The Institute for Genomic Research (J. Craig Venter Institute). Briefly, Glimmer3, Exonerate and tRNAscan SE tools were used to predict protein-, rRNA- and tRNA-coding genes respectively [20–22]. All protein-coding genes were then annotated by using the RAST annotation pipeline http://rast.nmpdr.org/[23]. Each protein sequence was also BLAST searched against Clusters of Orthologous Groups of proteins (COGs) database using the NCBI BLAST package [24, 25]. Then a COG identification number was assigned to each gene if the best BLASTP hit exhibits at least 80% sequence coverage in both query and hit sequences and at least 30% protein sequence identity. Finally, protein-coding genes were analyzed to identify putative frameshift mutations using BLAST Extend-Repraze http://ber.sourceforge.net/. Genes containing frameshift mutations were considered as putative pseudogenes. Unique genes of each genome were identified by BLAST searching each gene sequence to the genome of another strain with an E-value cutoff of 1e-6. A gene was considered unique if no significant hit was reported. Novel computational methods were used to detect evidence of secreted effector proteins for the type III secretion system and web based software (http://www.genome.jp/, KEGG pathway) was used to detect secretion system components [26].
Identification of high confidence single nucleotide polymorphisms (hcSNPs)
We developed a protocol that utilizes raw 454 read data (Hoffmann-La Roche Ltd) to identify hcSNPs between two genomes. Given two sets of 454 reads (readA and readB) from different genomes, we first used the newbler assembler to create two independent sets of contigs, named as contigA and contigB. The contig sequence from one genome was used as a reference while the reads from the other genome were aligned based on them using gsMapper software. Specifically, two gsMapper runs were carried out, where readA was mapped to contigB in one run and readB to contigA in another run. The gsMapper software utilizes a reference sequence to aid the assembly of raw read data. In addition to sequence assembly, this software gives a "high confidence difference (HCD)" file that summarizes all regions, where the sequence alignment shows differences between reference and multiple read sequences spanning that region. From "readA mapped to contigB" gsMapper run, readA sequences spanning a HCD region (e.g., HCD1) were checked to identify regions within contigA sequence, where they were assembled by the previous newbler run. If this particular region also appears in the HCD output file from the "readB mapped to contigA" gsMapper run, where readB sequences spanning this HCD have been assembled to the region where the previous HCD1 is found, we considered this HCD to be an hcSNP. This protocol essentially allows us to screen for differences between two genomes that were supported by multiple raw sequencing reads. For the comparison of B and O strains, each hcSNPs are supported by at least 10 reads from each genome, where at least 80% of the reads spanning that region show the difference. For comparison of B or O to previously sequenced A1 serotype, we were limited by the lack of raw data from A1. Hence, we were able to compare only to the consensus sequence of A1 as reported in the GenBank. As a result, hcSNPs from comparisons to A1 are only supported by multiple reads from either B or O genome. We anticipate that the error rate in such an assembly will range from 1 part in 10,000 to about 1 part in 40,000. For a genome of approximately 2.2 Mb in length, we anticipate approximately 55-220 sequence errors in the assembly. Hence sequence differences between B/O and A1 will be a combination of both real sequence differences and errors in the assembly of A1. Finally, the hcSNP data is filtered based on contig base quality score (at least 60) as well as whether or not the hcSNP is found on a homopolymer region of 3 bases or more to reduce false positive error.
Phylogenetic trees construction
We identified conserved genes common in all nine genomes based on their DNA sequence similarity using a combination of BLASTCLUST and BLAST software [25]. This resulted in a list of 28 genes, most of which coded for 30 S and 50 S ribosomal subunit proteins. The concatenated sequences of these 28 conserved genes (average length of 17,638 bp) were then aligned using ClustalW version 2 with default parameters [27]. PHYLIP program version 3.6 was used to construct the tree using the F84 evolutionary model and neighbor-joining method http://evolution.genetics.washington.edu/phylip/getme.html[28]. Finally, the Phylodendron software was used to draw the tree [29].
Utility and discussion
Gene content analysis
We obtained 20× coverage for B and O genomes after high-throughput sequencing using the 454- pyrosequencing technology. The assemblies produced 84 contigs for the B (average contig length of 30.2 kb) and 144 contigs for the O (average contig length of 17.9 kb) genomes respectively. The average contig length selected for cut off was >300 bp. Based on our previous experience, at this coverage, the gaps in the sequence are likely to be small and typically associated with repeat regions. The total number of base pairs in the contigs were 2,478,004 for B and 2,584,200 for O, which is comparable to the only other previously sequenced M. haemolytica A1 draft genome with a 8× coverage (2,569,125 bp) [14]. The complete annotation of B and O strains with additional details can be found in the GenBank (accession numbers ACZY00000000 and ACZX00000000, project numbers 40173 and 40171 respectively) [13]. The overall GC content of the M. haemolytica genomes is approximately 41%. Using an automated gene finding and annotation pipeline (see construction and content), we identified 2,550 open reading frames (ORFs) in the B and 2,682 ORFs in the O genome. The number of genes identified in these genomes are comparable to the previously reported M. haemolytica A1 genome (2,695 ORFs, GenBank accession number AASA00000000). Table 1 summarizes the basic features of the B and O genomes. We found 82-100% (average = 99.2%) overall nucleotide identity among the 1,664 protein encoding genes that are at least 300 bp in length in all 3 strains (Figure 1), which suggests that most of these common genes are highly conserved among these three isolates.

Distribution of homologous genes that is common in A1, B and O genomes across DNA sequence identity range. DNA sequences of 1,664 homologous protein-coding genes with length of at least 300 bp are considered in this analysis. This figure shows that most of the coding genes are highly conserved between across 3 strains Mannheimia haemolytica with an average DNA sequence identity of 99.2%.
By performing an all genes against all genomes analysis, we found that the A1, B and O genomes have 233, 38 and 62 unique genes respectively (a full list of A1, B and O strain specific genes can be found in the Additional file 1, Table S1, Additional file 2, Table S2 and Additional file 3, Table S3 respectively). Among the A1 specific genes 57% are hypothetical proteins and 20% are bacteriophage encoded proteins. The A1 genome has a few functionally important enzymes such as the UDP N acetylglucosamine 2 epimerase (MHA_0505), capsule biosynthesis protein (MHA_0507) and sialyltransferase (MHA_1780), indicating their possible role in conferring serotype specificity. Similarly, 57% of the B genome specific genes are hypothetical proteins and 29% are phage proteins. Among the B specific genes, enzymes such as peptidoglycan transglycolyase (COK_0539) involved in peptidoglycan biosynthesis and beta hexosamidase (COK_2260), a glycoside hydrolase involved in trimming carbohydrate decorations and pathogenesis are unique and noteworthy. On the other hand, the O genome accounts for 70% hypothetical proteins, of which less than 2% originate from phages. The only specific protein in the O genome, is the silent information regulator (Sir2, COI_1007), a homolog of transcription regulator, which is critical in maintaining bacterial replication and gene expression.
In summary, we found that the vast majority of these strain specific genes do not exhibit significant sequence similarity to other proteins of known function and cannot be categorized into any Clusters of Orthologous Groups (COG, Figure 2). However, the B and O strain specific genes appear to resemble integrated phage genomes. The pair wise comparison of gene content from one serotype in relation to the other reveals that about 9-12% of the genes are variable between A1 and A2 (B and O). Comparatively, a smaller number of variable genes (2-6%) are found between B and O, indicating that these two genomes share a more similar gene content in relation to each other than to A1 (Table 2). This degree of strain specificity and variable gene pool is an important group for further study, since bacteria frequently acquire virulence factors via horizontal gene transfer [30].

Strain specific gene in each Clusters of Orthologous Groups, COG category.
SNPs among virulence genes
Pathogenicity of M. haemolytica is due to a repertoire of exotoxins, endotoxins, and host immune-modulating proteins produced by it. Therefore, to characterize the potentially interesting SNPs in the M. haemolytica genome, we performed a SNPs analysis between these three genomes. In brief, we mapped individual reads from each strain to the consensus sequence of the other strains and identified SNPs that meet certain quality criteria (see construction and content). These SNPs are dubbed as high confidence single nucleotide polymorphisms (hcSNPs). The availability of individual reads from the strains of interest allowed us to compare the B and O genomes in both the directions (e.g., reads from B mapped to the consensus sequence of O and vice versa), which enabled us to filter the results and identify hcSNPs that meet the quality criteria for both mapping directions. These hcSNP's have high sequence coverage in both strains and consistently vary between strains in the individual reads.
To estimate the rate of false positives in our SNP detection methods and to obtain a quantitative estimate of the overall error of 454 sequencing and assembly, we have done the following: 1) We have previously sequenced two strains of Pseudomonas aeruginosa (unpublished data) to a very high depth of coverage (approximately 40×). 2) To estimate the precision in independently sequenced and assembled genomes, we have subsampled (without replacement), the reads from one these strains to create independent assemblies at 20× coverage (see Additional file 4, Table S4). 3) Assembled these sampled reads using the newbler assembler and mapped the trimmed contigs from these two independent assemblies onto each other to determine the precision, e.g., the differences that one obtains through sequencing and assembly errors when sequencing the same DNA. 4) Our results show that the difference between the two independent sequencing and assemblies was 1 bp in 17,681 bp (a total of 368 differences in a total of 6,506,615 aligned basepairs) and most (280 out of 368 or 76%) of these differences is in areas of homopolymer repeats. Since the B and O M. haemolytica genomes were sequenced to a similar depth (about 20×), we estimate a similar level of precision in the assemblies of the O and B genomes, e.g., approximately 1 difference in 17,681 bp due to sequencing and assembly errors. Furthermore, we estimate the precision in non-homopolymer repeat regions to be approximately 1 bp in 74 kb. The SNP's that we have called between the O and B genomes occur at a rate of approximately 1SNP per 646 bp. Since we have filtered out SNP's in homopolymer regions, an estimate of the false positive rate in our SNP calls is of order 1% (646 bp/74 kb = 0.008).
For the SNP comparisons between the O/B genomes and the A1 genome it is a bit more difficult to estimate the false positive rate, since we don't have the raw read data that went into the assembly of A1. However, we do have quality value estimates for each position in the assembly ftp://ftp.hgsc.bcm.tmc.edu/pub/data/MhaemolyticaPHL213/MhaemPHL213-10Aug2006-scaffolds.qual and we filtered SNPs to eliminate regions in the A1 assembly that are of Q-value <60. Even with this stringent filtering, we identified 13,705 and 9,968 hcSNPs between B and A1 and O and A1 respectively (or 1SNP/180 bp for B/A1 and 1SNP/259 bp for O/A1). Given that typical precision rates for assemblies from eight fold Sanger sequencing data are of order 1 bp in 20,000-100,000 bp, we assume that the A1 assembly is accurate to at least 1 bp in 20,000 [31]. Hence, we estimate that the rate of false positive for SNP detection in comparing the O/B genomes to the A1 genome is also of order 1%.
Table 3 summarizes the total number of synonomous and non-synonomous hcSNPs across all three isolates with additional files 1, 2 and 3 showing individual hcSNP genes with substitutions across each genome. In the following sections, we discuss the sequence variations which we identified in known virulence and virulence associated factors.
Leukotoxin hcSNPs and sequence variation
The leukotoxin (Lkt) secreted by M. haemolytica is a well studied, chief virulence factor [32–34]. Lkt is also produced by other members of gram-negative bacteria including Aggregatibacter actinomycetemcomitans, Actinobacillus pleuropneumoniae and Escherichia coli[35, 36]. Lkt belongs to the RTX (repeats-in-toxin) family of pore-forming exotoxins. Although these toxins have broad target cell specificity, M. haemolytica Lkt is specific for ruminant macrophages, neutrophils and all leukocyte subsets [37]. The β2 integrins LFA-1, Mac-1 and CR4 expressed by ruminant polymorphonuclear leukocytes (PMNs), serve as receptors for M. haemolytica Lkt [38–42]. These receptors exhibit a high degree of plasticity by binding to Lkt produced by different M. haemolytica serotypes [40, 41]. Furthermore, the leukotoxin operon (lktCABD) of M. haemolytica is a complex mosaic structure derived by extensive inter- and intra-species horizontal DNA transfer and intragenic recombination events [43]. LktA protein sequences of B, O and A1 (COK_0274, COI_0481 and MHA_0254 respectively) have an overall dissimilarity of 12%, accounting for 17% amino acid substitution at the amino- terminal (1-378) and 21% substitution at the carboxyl- terminal (780-953) (Figure 3A). However, the amino terminus of LktA of A1 has 51% amino acid substitution in this region when compared to B and O, but shares 98% overall identity with O and 88% with B respectively. The region encompassing the first 35 amino acids of LktA, which is involved in pore formation, is 100% identical between the O and B isolates [44]. At the nucleotide level, there are a total of 415 variations across the multiple sequence alignment of the lktA genes from B, O and A1 genomes, which account for 113 amino acid sequence variations (Figure 3A, Additional file 5, Figure S5). The multiple alignment result also shows that lktA sequences from O and A1 are more similar to one another and most of the variations identified by three-way comparisons are due to the lktA sequence of the B genome. Among all the variations identified, 116 mutations found between ltkA genes of B and O genomes are considered as hcSNP by our SNP filtering methodology encompassing 21 non-synonymous mutations (Additional file 6, Table S6). Similarly, 135 (28 non-synonymous) and 3 (1 non-synonymous) mutations found between B versus A1 and O versus A1 respectively are considered as hcSNPs (Additional file 7, Table S7, and Additional file 8, Table S8).

Cumulative counts of nucleotide and amino acid variation of LktA (A) and LktB (B) genes across A1, B and O isolates.
The DNA sequence of Lkt translocation ATP-binding gene, lktB, appears to be more conserved between the B, O and A1 genomes (COK_0273, COI_0482 and MHA_0255 respectively) relative to the lktA gene. The multiple sequence alignment shows 82 nucleotide substitutions, but amounts to only five amino acid substitutions (Figure 3B, Additional file 5, Figure, S5). The lktB sequence from A1 and O genomes are more similar to each other, but both are more diverged from the lktB sequence of the B genome. Out of the 54 mutations identified between lktB of B and O that are considered as hcSNP, five are non-synonymous (Additional file 6, Table S6). Similarly, out of the 78 mutations found between B and A1, four are non-synonymous and only one non-synonymous mutation is shared between O and A1 (Additional file 7, Table S7, and Additional file 8, Table S8). Although Lkt polymorphism has been implicated in species specificity, we found that the Lkt isolated from individual M. haemolytica isolates bind to β2 integrins from various ruminant species [40, 41, 43]. We suspect that this degree of polymorphism in LktA is important to enhance the ability of M. haemolytica serotypes to adapt to its niche, the ruminant respiratory tract.
Although Lkt is an important virulence factor, our earlier experiments have shown that lktA-deletion mutants of M. haemolytica still cause mild lung lesions with reduced mortality when compared to the wild type bacteria [45]. Therefore, it is logical to hypothesize that this organism has an arsenal of accessory virulence factors, which aid in host colonization, help gain a competitive advantage to act synergistically and modulate host gene expression. These accessory factors possibly include CRISPR and a type III secretion system identified during this study along with the previously known factors such as lipopolysaccharides, O-sialoglycoprotein endopeptidase, capsular polysaccharides, iron-regulated outer-membrane proteins and adhesins [46].
Lipopolysaccharides
Lipopolysaccharide (LPS) is an integral part of the Gram-negative bacterial cell wall and is the chief endotoxin that contains pathogen-associated molecular patterns (PAMPs) [46]. LPS activates the macrophages through toll like receptors (TLRs) and elicits inflammatory cytokine production resulting in septicemia [46, 47]. M. haemolytica LPS also induces an inflammatory cytokine response leading to increased expression of β2-integrins in the host [48]. The structure of M. haemolytica A1 O-antigen consists of trisaccharide repeat of two D-galactose residues and one N-acetyl-D-galactosamine residue [49]. The Leloir pathway catalyzes the epimerization of UDP-galactose to UDP-glucose in most Gram-negative bacteria, which is an important step in LPS biosynthesis [50]. This enzymatic reaction is carried out by UDP-galactose 4-epimerase (GalE). GalE mutants of Salmonella enteric serovar Typhimurium, Neisseria gonorrhoeae, and Haemophilus influenzae, which have truncated LPS, are avirulent when compared to their wild type, while in M. haemolytica they may abrogate adhesion [51–55]. Both the B and O genomes contain 32 out of the 38 previously reported genes found in the A1 LPS biosynthetic pathway (Additional file 9, Table S9). The A1 LPS biosynthetic pathway enzymes missing in the B and O genomes include UDP-N-acetylglucosamine 2-epimerase (MHA_0521), glycosyltranferase (MHA_1849, MHA_1850), possible sialyltransferase (MHA_1852) and hypothetical proteins (MHA_1847, MHA_1851). Most of the LPS biosynthetic genes shared between these three genomes exhibit 99-100% sequence identity (Additional file 9, Table S9), suggesting that the pathway leading to carbohydrate addition during LPS synthesis is critical and resistant to mutations.
O-sialoglycoprotein endopeptidase
The adherence of pathogenic bacteria to mucosal epithelium is dependent on the expression of adhesive molecules or ligands called adhesions that allow attachment of the organisms to complementary molecules on mucosal surfaces or receptors. Pathogens from the family of Pasteurellaceae, employ various types of ligands, which enable them to adhere, colonize, and cause infection. These include pili, filamentous proteins (fimbriae), outer membrane proteins and capsular polysaccharides.
Although not cytotoxic, O-sialoglycoprotein endopeptidase helps M. haemolytica to colonize the upper respiratory tract of ruminants in a host-specific manner and serves as an important virulence factor. A1 and B are serotypically different, but the gene (MHA_1559 and COK_2067 respectively) encoding O-sialoglycoprotein endopeptidase has only one nucleotide substitution resulting in one amino acid substitution at position 70 (P → T) (Additional file 7, Table S7), while B (COK_2067) and O (COI_0128) show four substitutions, 70 (P → E), 191(T → E), 327 (S → G) and 340 (P → S) (Additional file 6, Table S6). Interestingly, the glycoprotease domain (amino acids, 96 - 116) remains conserved across all the three isolates.
Iron-regulated outer-membrane proteins
Pathogenic bacteria often use iron as an environmental signal for the regulation of virulence genes [56, 57]. In mammalian host, pathogens from the families of Neisseriaceae and Pasteurellaceae frequently deal with the lack of free iron for uptake, as most of it is stored in intracellular or extracellular (transferrin, lactoferrin, haemopexin and haptoglobin) bound forms [58, 59]. To overcome this problem, Gram-negative pathogenic bacteria have evolved an elaborate iron-regulatory system, to acquire this element from the host. These include the direct binding of iron-containing proteins to outer-membrane receptors and the secretion of siderophores or haemophores [60]. Although M. haemolytica has no known siderophores, these bacteria produce two highly conserved transferrin (Tf)-binding proteins that specifically bind the Tf of their particular host [61, 62]. Earlier experiments clearly indicate that iron is required for the proper growth and Lkt production in M. haemolytica, as pathogenic gonococcal mutants devoid of these proteins lose their virulence [63, 64]. The B and O genomes encode several iron acquisition and iron homeostasis proteins similar to serotype A1. The presence of an elaborate set of iron-acquisition genes, therefore reiterates the importance of iron in controlling the transcription and expression of Lkt and other virulence factors in modulating M. haemolytica pathogenesis. Expression of these proteins allows M. haemolytica to acquire iron from host the hemoglobin, hemopexin, and transferrin. B and O encode two hemoglobin receptors HmbR1 (COK_2539 and COI_1763) and HmbR2 (COK_1624 and COI_2258) that are 99-100% identical to HmbR1 (MHA_1639) and HmbR2 (MHA_2261) of A1. The previously described transferrin-binding proteins, TbpA (MHA_0196) found in A1, is shared by B (COK_1753) and O (COI_2333) with 98% DNA sequence identity, but the TbpB (MHA_0197) is only 50% identical to TbpB, found in the B and O genomes (COK_1752 and COI_2332 respectively).
Adhesins
Adhesins help in tethering the bacteria to the host cell surface in a manner similar to a grappling hook. Most of the adhesins are pili, and the type IV pilus locus pilABCD of O (COI_1201-COI_1998) and B (COK_1994-COK_1991) genomes are encoded in an opposite orientation, when compared to A1 (MHA_0662-MHA_0665). The PilC protein from A1 and B are 100% identical, indicating their bovine origin, whereas, strain O has a 29 amino acid deletion in its amino-terminus. PilC in N. meningitides is implicated in human epithelial cell-specific interaction and pilus biogenesis [65, 66]. The amino-terminal deletion of PilC in M. haemolytica strain O may serve as a modification, necessary for ovine epithelial cell-specific colonization. The biogenesis and function of type IV pili is controlled by a large number of genes, almost 40 of which has been identified in P. aeruginosa[67]. B and O genomes share a high degree of homology to a number of genes required for pili assembly that are involved in type II protein secretion and competence for DNA uptake, suggesting that these systems share a common hierarchy along with H. influenzae, P. aeruginosa, and Neisseria species [68].
Filamentous hemagglutinin (FHA) is a major cell surface-associated adhesin that attaches to the host ciliary epithelial cells and is a virulence determinant [69, 70]. Pfam http://pfam.sanger.ac.uk/ analysis indicates the possible involvement of internal FhaB domain of FHA protein in heme utilization. FHA (fhaB) from A1 (MHA_0866) shares 99% DNA sequence identity to the homologous regions of strains B (COK_0334) and O (Contig00015). The fhaB ortholog of M. haemolytica is also shared by Bordella pertussis, N. meningitidis, A. pleuropneumoniae, M. succiniciproducens and P. syringae. The fhaB genes of the B and O strains are adjacent to the fhaC ortholog, similar to the two-partner secretory pathway found in A1. The fhaB proteins in the B and O genomes lack the integrin-binding RGD motif, but are characterized by the presence of three bacterial intein-like (BIL) regions at their carboxyl- termini, similar to A1. BILs belong to the HINT (hedgehog/intein) superfamily of domains, which post-translationally self-process by protein splicing and self-cleavage, hence interferes with the host innate immune system [71].
The adhesin, serotype A1-specific antigen (Ssa1) is present in both the B and O genomes [72]. Surprisingly the amino acid sequence of Ssa1 from A1 (MHA_2492) is only 79% identical to B (COK_2411) considering their origin from a common host, but 95% identical to O (COI_0850). The Ssa1 protein also contains an amino-terminal peptidase S8 superfamily domain, which can be cleaved by serine peptidases and a carboxyl-terminal autotransporter superfamily domain.
The orthologs of H. influenzae and N. meningitides IgA-specific serine metallo-endopeptidase of A1 (MHA_0563 and MHA_2800), is 99% identical at amino acid level to B and O enzymes (COK_0634, COI2_430 and COK_1350, COI_2438). However, the MHA_2800 homologues of B and O (COK_1350, COI_2438) are only half the size and are devoid of amino acids ranging from 704-1503, including their carboxyl- terminal. This deletion removes the entire pertactin and autotransporter domains and almost 75% of the second peptidase S6 domain. These domains are not predicted to contain any active amino acids http://pfam.sanger.ac.uk/search/sequence. On the other hand MHA_0563 homologues, COK_0634 and COI_2430 contain all the three domains, i.e., S6 peptidase, AT-pertactin and carboxyl- terminal autotransporter. Iga1 hydrolyses the host mucosal antibody IgA and possibly IgG, and helps in colonization by immune-evasion [73].
The autotransporter/adhesion protein of A1 (MHA_2701), shares 75% identity with B (COK_1437) and O (COI_2393) genomes, whereas the AT family of autotransporter/adhesion MHA_1367 shares 96% (COK_2435) and 99% (COI_1943) identities respectively. These proteins are involved in promoting adhesion to the host mucosal surfaces and are closely related to autotransporter/adhesins of A. pleuropneumoniae, M. succiniciproducens and hep/hag family proteins of N. mucosa. The hep/hag domain is a seven-residue repeat that makes up the majority of the sequence of a family of bacterial haemagglutinins and invasins. The ORFs COK_2435 and COI_1943 show four and seven hep/hag repeats and one carboxyl-terminal YadA-like domain from Yersinia species. The hep/hag proteins also serve as immuno-dominant antigens in Burkholderia mallei and B. pseudomallei[74]. Therefore, hep/hag domains can be exploited for serodiagnosis in M. haemolytica along with hcSNP markers, for increased fidelity.
Clustered regularly interspaced short palindromic repeats
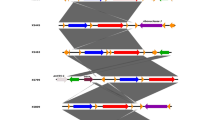
CRISPR loci consists of a family of DNA direct repeats separated by regularly sized non-repetitive spacer sequences that are found in most bacterial and archaeal genomes [75]. CRISPR regions provide acquired immunity against super infecting bacteriophages, possibly acting via RNA interference-like mechanism [76]. The differences in the number and type of the spacers between CRISPR repeats correlate with phage sensitivity. CRISPR regions are often hypervariable between the otherwise closely related strains [77]. In addition, there are many protein families known as CRISPR-associated sequences (Cas), which are encoded in the vicinity of CRISPR loci [78]. CRISPR/Cas gene regions can be quite large, with up to 20 different, tandem-arranged cas genes next to a CRISPR cluster or filling the region between two repeat clusters. Cas proteins are thought to be involved in the propagation and functioning of CRISPRs and some show similarity to helicases and repair proteins [78]. The CRISPR/Cas loci of B and O consist of four genes, COK_0798, COK_0799, COK_0800, COK_0801 and COI_0267, COI_0268, COI_0269, COI_0270 respectively. The Cas family, which is represented by CT1134 and CT1133 from Chlorobium tepidum, is also found in the B and O genomes (COK_0798, COK_0799 and COI_0267, COI_0268). This family belongs to the three-gene CRISPR/Cas subtypes found in Desulfovibrio vulgaris and is a member of the Dvulg subtype [78]. There are five hcSNPs in CT1134 and eleven in CT1133 loci between strains O and B, which can be used for strain typing. The locus, COK_0800 and COI_0269 are 100% identical to Thermotoga maritima Cas family, TM1801 [78]. The last gene in this cluster is a recB exonuclease (COK_0801 and COI_0270) which is 100% identical between these two isolates. Furthermore, we also identified CRISPR/Cas loci in the A1 genome (MHA_0343, MHA_0344, MHA_0345, and MHA_0346), previously reported as hypothetical proteins [14]. A schematic representation of the CRIPSR loci along with spacers from all the three isolates is shown in Figure 4. The presence of CRISPR regions in the M. haemolytica genome indicates its potential resistance to superinfection by phages. The CRISRP/Cas mechanism is analogous to the eukaryotic RNAi system [79, 80].

Schematic representation of CRISPR/Cas locus in B, O (A) and A1 (B). All the three isolates have a 10 bp spacer region between CT1133 and TM1801 genes. The A1 also locus has an additional 94 bp spacer between CT1134 and CT1133 genes, which is absent in B and O genomes.
Type III secretion system
We performed a novel computational analysis to search for previously undiscovered secretion systems in these genomes. In addition to genes encoding a type I secretion system similar to E. coli haemolysin, both the B and O genomes also contain genes that encode a previously undiscovered putative type III secretion system (T3SS) and twin arginine targeting (Tat) systems [81]. B and O encode proteins that show low to moderate homology to T3SS effector components of E. coli O157:H7, needle-like protein, SctF (49%), secretin SctC (35%), outer membrane protein SctW (40%), inner membrane proteins SctJ (37%), SctR (< 20%), SctS (> 20%), SctT (> 20%), SctU (41%), StcV (low), an ATPase SctN (46%) and an ATPase associated protein SctQ (low). They also share 40% identity to SctC and 47% to an ATPase SctN of B. pseudomallei. The genes encoding possible T3SS of B and O do not fall under a unique operon. The possible organization of T3SS genes is depicted in figure 5 along with type III secretion protein Ysc C homolog (COK_1782/COI_2362), which has a distinct secretin N-domain. Interestingly, we failed to identify T3SS operon in the A1 genome, due to either low sequence coverage or complete absence.

Schematic representation of possible type III secretion system (T3SS) in B/O genomes. COK_1772/COI_2352 to COK_1775/COI_2355- Putative arginine superfamily ATP binding cassette; COK_1776/COI_2356 & COK_1777/COI_2357- Cross over junction endonuclease Ruv A & B; COK_1778/COI_2358- RhtB family homoserine/threonine resistance; COK_1779/COI_2359- Acid phosphatase; COK_1780/COI_2360- Thiamine phosphate kinase; COK_1781/COI_2361- N utilization substance B; COK_1782/COI_2362- Type III secretion protein YscC homolog (competence protein E); COK_1783/COI_2363 to COK_1785/COI_2365- Hypothetical proteins, COK_1786/COI_2366- Putative pilus protein ComA.
The presence of a T3SS in M. haemolytica B and O could equip them with a unique virulence mechanism that enables them to inject bacterial effector proteins directly into the host cell cytoplasm. The T3SS bypasses the extracellular milieu and potentially facilitates bacterial pathogenesis by specifically interfering with host cell signal transduction/transcription and other cellular processes [82, 83]. Additional file 10, Table S10 describes the list of possible effector proteins showing high, moderate and low probability of being secreted through T3SS across all the three isolates. These effectors were predicted using a machine-learning method that uses the N-terminal region of proteins to predict secretion and does not rely on detecting homologous effector domains [26]. Almost 50% of the proteins predicted to be exported through T3SS are hypothetical and phage proteins. It is interesting to note that the Lkt acyl transferase and LktA are predicted to be highly exported through this machinery, although LktA has been shown to be secreted by type 1 system [84]. A family of metallo-endopeptidase S6, specific for cleaving IgA (COK_1350 and COI_2438) is also secreted with high probability. Similary, the list of moderately exported protein is also filled with a large number of hypothetical and phage proteins. There are quite a few virulence associated proteins, that are exported with moderate probability, and includes a S6 metalloendopeptidase homolog, (COK_0634 and COI_2430), iron scavenging transferrin binding protein (COK_1753 and COI_2333), siderophore esterase (COK_ 1515 and COI_0656), TonB (COK_1730, COI_2309), which is required to transduce cytoplasmic membrane energy to the outer membrane, Ton B dependent outer membrane receptor (COK_0223 and COI_0091), hemoglobin receptor (COK_2540 and COI_1762) and a neuraminidase (COK_1504 and COI_0667). Secretion of these proteins clearly indicates that M. haemolytica interacts with the host immune system, by bypassing mucosal defense and by scavenging iron to colonize.
One of the interesting effector protein from B, O and A1 (COK_1445, COI_1309 and MHA_0531) shows 20-40% identity to a transcription activator-like (TAL) type III effector, gene locus from Xanthomonas oryzae (AvrXa27), that activates the transcription of the host resistance gene Xa27, resulting in resistance to bacterial blight in rice [85]. TAL effectors target host general transcription factors and manipulate the host transcriptional machinery for virulence and/or avirulence [86]. To date, no such effector proteins have been identified from any Mannheimia species, which re-route the ruminant transcriptional/translational machinery to their advantage. Further experimental evidence directed towards isolating and characterizing these effector molecules from M. haemolytica will be necessary to confirm these computational inferences.
The twin arginine translocation (Tat) machinery exports folded proteins across the cytoplasmic membrane. Many Tat-secreted proteins are periplasmic enzymes that catalyze multiprotein oxido-reduction systems involved in respiration or anaerobic growth [86]. M. haemolytica Tat proteins ABC and E shares high homology with E. coli O157:H7 sec-independent translocase components (43%, 41%, 65% and 53% respectively). The presence of Tat pathway in M. haemolytica is interesting, because all proteins or complexes of proteins destined for Tat export must be covalently attached to one of these specialized amino-terminal twin-arginine signal peptides unlike the Sec pathway [87]. The Tat system is also an important virulence factor in aiding bacterial pathogens to infect plants and animals [88, 89]. The B and O genomes encode trimethylamine N-oxide (TMAO)-inducible operon (torABCE), which is orthologous to E. coli[90]. The TorA protein, trimethylamine N-oxide reductase relies heavily on the bacterial twin-arginine system for its export [87]. Various bacteria grow anaerobically using TMAO as an alternative terminal electron acceptor of a respiratory transport chain [91], but its presence in M. haemolytica is intriguing since this organism grows in an oxygen rich environment.
Phylogenetic analysis
Overall, the phylogenetic tree (Figure 6) constructed in this study is in congruence with the earlier results indicating that M. haemolytica B, O, A1, A. pleuropneumoniae, and H. ducreyi form a group that is divergent from the other members of the family, Pasteurellaceae[14]. Due to the limitations in using 16 S rRNA gene sequences for phylogenetic analysis, especially in the Pasteurellaceae species [14, 92], we used genes encoding various 30 S and 50 S ribosomal subunits. In this analysis, the M. haemolytica isolates are closely related to H. ducreyi followed by two different isolates of Actinobacillus. This is in agreement to the earlier analysis obtained using the housekeeping genes from A1 [14]. A. pleuropneumoniae and M. haemolytica occupy the upper respiratory tract of their hosts and operate using identical virulence mechanisms and transition from commensalism to pathogenesis. On the other hand, H. ducreyi is an opportunistic organism that infects human genitalia through breaks in the skin or epidermis and appears to be closely related to M. haemolytica based on its competence genes, but shares the lowest number of orthologs with B, O and A1 [14]. Based on these analyses, it is reasonable to expect that M. haemolytica, H. ducreyi and A. pleuropneumoniae share a common Pasteurellaceae ancestory when compared to M. succiniciproducens which had been clustered with Mannheimia species based on 16 S rRNA sequences [92]. M. succiniciproducens MBEL55E used in this analysis is a capnophilic ruminant rumen bacterium, thus it is expected to have a low similarity to the other members of M. haemolytica cluster. Furthermore, it lacks virulence-related genes such as the leukotoxin and capsule biosynthesis genes [93]. Considering these phylogenetic trees, it will be interesting to see how the taxonomic classification of other M. haemolytica isolates fall into, once their genome sequences become available. Furthermore, comparative genomic sequence analysis involving other RTX toxin secreting pathogens will help in analyzing their molecular drives in causing such divergence.

Phylogenetic tree derived from concatenated sequences of 28 conserved genes. Bootstrap values (100 replicates) are given at branch points as described in the methods section. Bar represent 0.01 substitutions per site.
Conclusions
In summary, we have performed a three-way comparison between the genomic sequences of three strains of M. haemolytica. We identified a number of genes that are unique to each strain, which are "high value targets" for future studies that attempt to correlate the variable gene pool with phenotype (strain virulence, species specificity, etc). We also identified a number of hcSNPs and focused on non-synonymous SNPs in known virulence genes. This data will be used to design new hcSNP arrays that will aid in studying variation across strains and will potentially aid in understanding gene regulation and the mode of action of various virulence factors. The additional virulence factors identified in this study include a previously unknown type III secretion system and CRISPR regions, which can modulate host immune responses. These additional virulence factors along with adhesins, which contain protease cleavage domain will be used to investigate bovine immune responses and will serve as effective candidates for vaccine development.
Availability and requirements
The B and O genomes can be accessed via NCBI database through the following URLs: http://www.ncbi.nlm.nih.gov/sites/entrez?db=genome&cmd=Retrieve&dopt=Overview&list_uids=6714 (B) and http://www.ncbi.nlm.nih.gov/sites/entrez?db=genome&cmd=Retrieve&dopt=Overview&list_uids=6715 (O) respectively.
Abbreviations
- SNPs:
-
Single nucleotide polymorphisms
- bp:
-
base pairs
- kb:
-
kilo bases
- Mb:
-
mega bases: ATP: Adenosine Tri Phosphate.
References
Mosier DA: Bacterial pneumonia. Vet Clin N Am Food Anim Pract. 1997, 13: 483-493.
Ackermann MR, Brogden KA: Response of the ruminant respiratory tract to Mannheimia (Pasteurella) haemolytica. Microbes Infect. 2000, 2: 1079-1088. 10.1016/S1286-4579(00)01262-4.
Miller MW: Pasteurellosis. Infectious Diseases of Wild Mammals. Edited by: Williams ES, Barker IK. 2001, Iowa: Iowa State University Press, 330-339. full_text.
Miller WM, Harkness JW, Richards MS, Pritchard DG: Epidemiological studies of calf respiratory disease in a large commercial veal unit. Res Vet Sci. 1980, 28: 267-274.
Frank GH: Pasteurellosis of cattle. Pasteurella and pasteurellosis. Edited by: Adlam C, Rutter JM. 1989, New York: Academic Press, 197-221.
NAHMS Sheep 2001: National animal health monitoring system Part I: Reference of sheep management in the United States. USDA: APHIS: VS: CEAH. 2002, Fort Collins, 51-55.
Rice JA, Medina LC, Hodgins DC, Shewen PE: Mannheimia haemolytica and bovine respiratory disease. Animal Health Research Reviews. 2008, 8: 117-128. 10.1017/S1466252307001375.
Bowland SL, Shewn PE: Bovine respiratory disease: commercial vaccines currently available in Canada. Can Vet J. 2000, 41: 33-48.
Angen Ø, Mutters R, Caugant DA, Olsen JE, Bisgaard M: Taxonomic relationships of the [Pasteurella] haemolytica complex as evaluated by DNA-DNA hybridizations and 16 S rRNA sequencing with proposal of Mannheimia haemolytica gen. nov., comb. nov., Mannheimia granulomatis comb. nov., Mannheimia glucosida sp. nov., Mannheimia ruminalis sp. nov. and Mannheimia varigena sp. nov. Int J Syst Bacteriol. 1999, 49: 67-86. 10.1099/00207713-49-1-67.
Pitt TL: Molecular typing in practice. J Hospital Infect. 1999, 43: 85-88. 10.1016/S0195-6701(99)90069-5.
Pennington TH: Molecular typing methods for Neisseria meningitidis. J Medical Micro. 1999, 48: 1055-1064. 10.1099/00222615-48-12-1055.
Ewers C, Lübke-Becker A, Wieler LH: Mannheimia haemolytica and the pathogenesis of enzootic bronchopneumonia. Berl Munch Tierarztl Wochenschr. 2004, 117: 97-115.
Lawrence PK, Kittichotirat W, Bumgarner RE, McDermott JE, Herndon DR, Knowles DP, Srikumaran S: Genome sequences of Mannheimia haemolytica serotype A2: ovine and bovine isolates. J Bacteriol. 2010, 192: 1167-1168. 10.1128/JB.01527-09.
Gioia J, Qin X, Jiang H, Clinkerbeard K, Lo R, Liu Y, Fox GE, Yerrapragada S, McLeod MP, McNeill TZ, Hemphill L, Sodergren E, Wang Q, Muzny DM, Homsi FJ, Weinstock GM, Highlander SK: The genome sequence of Mannheimia haemolytica A1: Insight into virulence, natural competence, and Pasteurellaceae phylogeny. J Bacteriol. 2006, 188: 7257-7266. 10.1128/JB.00675-06.
Villard L, Gauthier D, Maurin F, Borges E, Richard Y, Abadie G, Kodjo A: Serotypes A1 and A2 of Mannheimia haemolytica are susceptible to genotypic, capsular and phenotypic variations in contrast to T3 and T4 serotypes of Bibersteinia (Pasteurella) trehalosi. FEMS Microbiol Lett. 2008, 280: 42-49. 10.1111/j.1574-6968.2007.01035.x.
Highlander SK: Molecular genetic analysis of virulence in Mannheimia (Pasteurella) haemolytica. Front Biosci. 2001, 6: 1128-1150. 10.2741/Highland.
Zecchinon L, Fett T, Desmecht D: How Mannheimia haemolytica defeats h ost defence through a kiss of death mechanism. Vet Res. 2005, 2: 133-156. 10.1051/vetres:2004065.
Vetsigian K, Goldenfeld N: Global divergence of microbial genome sequences mediated by propagating fronts. Proc Natl Acad Sci USA. 2005, 102: 7332-7337. 10.1073/pnas.0502757102.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380.
Delcher AL, Bratke KA, Powers EC, Salzberg SL: Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007, 23: 673-679. 10.1093/bioinformatics/btm009.
Slater GS, Birney E: Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005, 6: 31-10.1186/1471-2105-6-31.
Lowe TM, Eddy SR:TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 1999, 25: 955-964. 10.1093/nar/25.5.955.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O: The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008, 9: 75-10.1186/1471-2164-9-75.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003, 4: 41-10.1186/1471-2105-4-41.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Samudrala R, Heffron F, McDermott JE: Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009, 4: e1000375-10.1371/journal.ppat.1000375.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and Clustal × version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Felsenstein J: Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981, 17: 368-376. 10.1007/BF01734359.
Gilbert DG: Free Software in Molecular Biology for Macintosh and MS Windows Computers. Bioinformatics Methods and Protocols. Edited by: Misener S, Krawetz SA. 1999, New Jersey: Humana Press, [http://iubio.bio.indiana.edu/soft/molbio/Listings.html]
Hacker J, Carniel E: Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Reports. 2001, 2: 376-381.
Fraser CM, Eisen JA, Nelson KE, Paulsen IT, Salzberg SL: Th e value of complete microbial genome sequencing (you get what you pay for). J Bacteriol. 2002, 184: 6403-6405. 10.1128/JB.184.23.6403-6405.2002.
Baluyut CS, Simonson RR, Bemrick WJ, Maheswaran SK: Interaction of Pasteurella haemolytica with bovine neutrophils: identification and partial chara cterization of a cytotoxin. Am J Vet Res. 1981, 42: 1920-1926.
Kisiela DI, Aulik NA, Atapattu DN, Czuprynski CJ: N-terminal region of Mannheimia haemolytica leukotoxin serves as a mitochondrial targeting signal in mammalian cells. Cell Microbiol. 2010, 12 (7): 976-87. 10.1111/j.1462-5822.2010.01445.x.
Strathdee CA, Li RY: Cloning, nucleotide sequence, and characterization of genes encoding the secretion function of the Pasteurella haemolytica leukotoxin determinant. J Bacteriol. 1989, 171: 916-928.
Kolodrubetz D, Dailey T, Ebersole J, Kraig E: Cloning and expression of the leukotoxin gene from Actinobacillus actinomycetemcomitans. Infect Immun. 1980, 57: 1465-1469.
Devenish J, Rosendal S, Johnson R, Hubler S: Immunoserological comparison of 104-kilodalton proteins associated with hemolysis and cytolysis in Actinobacillus pleuropneumoniae, Actinobacillus suis, Pasteurella haemolytica, and Escherichia coli. Infect Immun. 1989, 57: 3210-3213.
Shewen PE, Wilkie BN: Evidence for the Pasteurella haemolytica cytotoxin as a product of actively growing bacteria. Am J Vet Res. 1985, 46: 1212-1214.
Jeyaseelan S, Hsuan SL, Kannan MS, Walcheck B, Wang JF, Kehrli ME, Lally ET, Sieck GC, Maheswaran SK: Lymphocyte function-associated antigen 1 is a receptor for Pasteurella haemolytica leukotoxin in bovine leukocytes. Infect Immun. 2000, 68: 72-79. 10.1128/IAI.68.1.72-79.2000.
Lawrence PK, Dassanayake RP, Knowles DP, Srikumaran S: Transfection of non-susceptible cells with Ovis aries recombinant lymphocyte function-associated antigen 1 renders susceptibility to Mannheimia haemolytica leukotoxin. Vet Microbiol. 2007, 125: 91-99. 10.1016/j.vetmic.2007.05.006.
Lawrence PK, Nelson WR, Liu W, Knowles DP, Foreyt WJ, Srikumaran S: Beta(2) integrin Mac-1 is a receptor for Mannheimia haemolytica leukotoxin on bovine and ovine leukocytes. Vet Immunol Immunopathol. 2008, 122: 285-294. 10.1016/j.vetimm.2007.12.005.
Lawrence PK, Dassanayake RP: Ovis aries CR4 is involved in Mannheimia haemolytica leukotoxin-induced cytotoxicity. Vet Immunol Immunopathol. 2009,
Dassanayake RP, Liu W, Davis WC, Foreyt WJ, Srikumaran SJ: Bighorn sheep beta2-integrin LFA-1 serves as a receptor for Mannheimia haemolytica leukotoxin. J Wildl Dis. 2008, 44: 743-747.
Davies RL, Campbell S, Whittam ST: Mosaic Structure and Molecular Evolution of the Leukotoxin Operon (lktCABD) in Mannheimia (Pasteurella) haemolytica, Mannheimia glucosida, and Pasteurella trehalosi. J Bacteriol. 2002, 184: 266-277. 10.1128/JB.184.1.266-277.2002.
Forestier C, Welch RA: Identification of RTX toxin target cell specificity domains by use of hybrid genes. Infect Immun. 1991, 59: 4212-4220.
Dassanayake RP, Shanthalingam S, Herndon CN, Lawrence PK, Cassirera EF, Potter KA, Foreyt WJ, Clinkenbeard KD, Srikumaran S: Mannheimia haemolytica serotype A1 exhibits differential pathogenicity in two related species, Ovis canadensis and Ovis aries. Vet Microbiol. 2009, 133: 366-371. 10.1016/j.vetmic.2008.07.015.
Raetz CRH, Whitfield C: Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002, 71: 635-700. 10.1146/annurev.biochem.71.110601.135414.
Imler JL, Hoffmann JA: Toll receptors in innate immunity. Trends in Cell Biol. 2001, 11: 304-311. 10.1016/S0962-8924(01)02004-9.
Yoo HS, Maheswaran SK, Lin G, Townsend EL, Ames TR: Induction of inflammatory cytokines in bovine alveolar macrophages following stimulation with Pasteurella haemolytica lipopolysaccharide. Infect Immun. 1995, 63: 381-388.
Richards JC, Leitch RA: Elucidation of the structure of the Pasteurella haemolytica serotype T10 lipopolysaccharide O-antigen by N.M.R. spectroscopy. Carbohydr Res. 1989, 186: 275-286. 10.1016/0008-6215(89)84041-8.
Frey PA: The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996, 10: 461-470.
Hone D, Morona R, Attridge S, Hackett J: Construction of defined galE mutants of Salmonella for use as vaccines. J Infect Dis. 1987, 156: 167-174.
Maskell DJ, Szabo MJ, Butler PD, Williams AE, Moxon ER: Molecular analysis of a complex locus from Haemophilus influenza involved in phase-variable lipopolysaccharide biosynthesis. Mol Microbiol. 1991, 5: 1013-1022. 10.1111/j.1365-2958.1991.tb01874.x.
Metzger M, Bellemann P, Bugert P Geider K: Genetics of galactose metabolism of Erwinia amylovora and its influence on polysaccharide synthesis and virulence of the fire blight pathogen. J Bacteriol. 1994, 176: 450-459.
Robertson BD, Frosch M, van Putten JPM: The role of gale in the biosynthesis and function of gonococcal lipopolysaccharide. Mol Microbiol. 1993, 8: 891-901. 10.1111/j.1365-2958.1993.tb01635.x.
Whiteley LO, Maheswaran SK, Weiss DJ, Ames TR, Kannan MS: Pasteurella haemolytica A1 and bovine respiratory disease: pathogenesis. J Vet Internal Med. 1992, 6: 11-22. 10.1111/j.1939-1676.1992.tb00980.x.
Mekalanos JJ: Environmental signals controlling expression of virulence determinants in bacteria. J Bacteriol. 1992, 174: 1-7.
McIver KS, Heath AS, Scott JR: Regulation of virulence by environmental signals in group A streptococci: influence of osmolarity, temperature, gas exchange, and iron limitation on emm transcription. Infect Immun. 1995, 63: 4540-4542.
Litwin CM, Calderwood SB: Role of iron in regulation of virulence genes. Clin Microbiol Rev. 1993, 6: 137-149.
Braun V, Gaisser S, Herrmann C, Kampfenkel K, Killmann H, Traub I: Energy-coupled transport across the outer membrane of Escherichia coli: ExbB binds ExbD and TonB in vitro, and leucine 132 in the periplasmic region and aspartate 25 in the transmembrane region are important for ExbD activity. J Bacteriol. 1996, 178: 2836-2845.
Stojiljkovic I, Kumar V, Srinivasan N: Non-iron metalloporphyrins: potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol Microbiol. 1999, 3: 429-442. 10.1046/j.1365-2958.1999.01175.x.
Fuller CA, Yu R, Irwin AW, Schryvers AB: Biochemical evidence for a conserved interaction between bacterial transferrin binding protein A and transferrin binding protein B. Microb Pathog. 1998, 24: 75-78. 10.1006/mpat.1997.0174.
Morton DJ, Williams P: Utilization of transferrin-bound iron by Haemophilus species of human and porcine origins. FEMS Microbiol Lett. 1989, 53: 123-127. 10.1111/j.1574-6968.1989.tb03609.x.
Gentry MJ, Confer AW, Weinberg ED, Homer JT: Cytotoxin (leukotoxin) production by Pasteurella haemolytica: requirement for an iron-containing compound. Am J Vet Res. 1986, 47: 1919-1923.
Cornelissen CN, Kelley M, Hobbs MM, Anderson JE, Cannon JG: The transferrin receptor expressed by gonococcal strain FA1090 is required for the experimental infection of human male volunteers. Mol Microbiol. 1998, 27: 611-616. 10.1046/j.1365-2958.1998.00710.x.
Jonsson AB, Nyberg G, Normark S: Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J. 1991, 10: 477-488.
Rudel T, Scheurerpflug I, Meyer TF: Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature. 1995, 373: 357-359. 10.1038/373357a0.
Mattick JS: Type IV pili and twitching motility. Annu Rev Microbiol. 2002, 56: 289-314. 10.1146/annurev.micro.56.012302.160938.
Bakaletz LO, Baker BD, Jurcisek JA, Harrison A, Novotny LA, Bookwalter JE, Mungur R, unson RS: Demonstration of type IV pilus expression and a twitching phenotype by Haemophilus influenzae. Infect Immun. 2005, 73: 1635-1643. 10.1128/IAI.73.3.1635-1643.2005.
Coutte L, Alonso S, Reveneau N, Willery E, Quatannens B, Locht C, Jacob-Dubuisson F: Role of adhesin release for mucosal colonization by a bacterial pathogen. J Exp Med. 2003, 197: 735-742. 10.1084/jem.20021153.
Davies RL, Donachie W: Intra-specific diversity and host specificity within Pasteurella haemolytica based on variation of capsular polysaccharide, lipopolysaccharide and outer-membrane proteins. Microbiol. 1996, 142: 1895-1907. 10.1099/13500872-142-7-1895.
Bachash MD, Dassa B, Peleg O, Pineiro SA, E Pietrokovski S: Bacterial intein-like domains of predatory bacteria: a new domain type characterized in Bdellovibrio bacteriovorus. Functional Integrative Genomics. 2009, 9: 153-166. 10.1007/s10142-008-0106-7.
Lo RY, Strathdee CA, Shewen PE, Cooney BJ: Molecular studies of Ssa1, a serotype-specific antigen of Pasteurella haemolytica A1. Infect Immun. 1991, 59: 3398-3406.
St. Geme JW: The pathogenesis of nontypable Haemophilus influenzae otitis media. Vaccine. 2000, 19 (Suppl. 1): 41-50. 10.1016/S0264-410X(00)00277-2.
Tiyawisutsri R, Holden MT, Tumapa S, Rengpipat S, Clarke SR, Foster SJ, Nierman WC, Day NP, Peacock SJ: Burkholderia Hep_Hag autotransporter (BuHA) proteins elicit a strong antibody response during experimental glanders but not human melioidosis. BMC Microbiol. 2007, 7: 19-10.1186/1471-2180-7-19.
Mojica FJ, Ferrer C, Juez G, Rodriguez-Valera F: Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol. 1995, 17: 85-93. 10.1111/j.1365-2958.1995.mmi_17010085.x.
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV: A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006, 1: 7-10.1186/1745-6150-1-7.
Pourcel C, Salvignol G, Vergnaud G: CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiol. 2005, 151: 653-663. 10.1099/mic.0.27437-0.
Haft DH, Selengut J, Mongodin EF, Nelson KE: A Guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005, 1: e60-10.1371/journal.pcbi.0010060.
Sorek R, Kunin V, Hugenholtz P: CRISPR-a widespread system that provides acquired resistance against phages in bacteria and archaea. Nature Rev Microbiol. 2008, 6: 181-186. 10.1038/nrmicro1793.
Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP: RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009, 139: 945-956. 10.1016/j.cell.2009.07.040.
Felmlee T, Pellett S, Welch RA: Nucleotide sequence of an Escherichia coli chromosomal hemolysin. J Bacteriol. 1985, 163: 94-105.
Gotoh H, Okada N, Kim YG, Shiraishi K, Hirami N, Haneda T, Kurita A, Kikuchi Y, Danbara H: Extracellular secretion of the virulence plasmid-encoded ADP-ribosyltransferase SpvB in Salmonella. Microbial Pathog. 2003, 34: 227-238. 10.1016/S0882-4010(03)00034-2.
Coburn B, Sekirov I, Finlay BB: Type III secretion systems and disease. Clin Microbiol Rev. 2007, 20: 535-549. 10.1128/CMR.00013-07.
Young R, Moulds TL, Struck DK: Secretion of the Pasteurella leukotoxin by Escherichia coli. FEMS Microb Lett. 1989, 60: 169-173. 10.1111/j.1574-6968.1989.tb03439.x.
Keyu G, Dongsheng T, Chengxiang Q, Zhongchao Y: Transcription activator-like type III effector AvrXa27 depends on OsTFIIAγ5 for the activation of Xa27 transcription in rice that triggers disease resistance to Xanthomonas oryzae pv. Oryzae. Mol Plant Pathol. 2009, 10: 829-835. 10.1111/j.1364-3703.2009.00567.x.
Sargent F, Berks BC, Palmer T: Pathfinders and trailblazers: a prokaryotic targeting system for transport of folded proteins. FEMS Microbiology Letters. 2005, 254: 198-207. 10.1111/j.1574-6968.2005.00049.x.
Berks BC: A common export pathway for proteins binding complex redox cofactors?. Mol Microbiol. 1996, 22: 393-404. 10.1046/j.1365-2958.1996.00114.x.
Ochsner UA, Snyder A, Vasil AI, Vasil ML: Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc Natl Acad Sci USA. 2002, 99: 8312-8317. 10.1073/pnas.082238299.
Ding Z, Christie PJ: Agrobacterium tumefaciens twin-arginine-dependent translocation is important for virulence, flagellation, and chemotaxis but not type IV secretion. J Bacteriol. 2003, 185: 760-771. 10.1128/JB.185.3.760-771.2003.
Bogsch E, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T: An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998, 273: 18003-18006. 10.1074/jbc.273.29.18003.
Barrett EL, Kwan HS: Bacterial reduction of trimethylamine oxide. Annu Rev Microbiol. 1985, 39: 131-149. 10.1146/annurev.mi.39.100185.001023.
Christensen H, Kuhnert P, Olsen JE, Bisgaard M: Comparative phylogenies of the housekeeping genes atpD, infB and rpoB and the 16 S rRNA gene within the Pasteurellaceae. Int J Syst Evol Microbiol. 2004, 54: 1601-1609. 10.1099/ijs.0.03018-0.
Hong SH, Kim JS, Lee SY, In YH, Choi SS, Jeong-Keun : The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nature Biotech. 2004, 22: 1275-1281. 10.1038/nbt1010.
Acknowledgements
We are thankful to Roche Applied Science for funding these sequencing projects. We also thank Drs. William Foreyt, Washington State University and Anthony Confer, Oklahoma State University for providing us with M. haemolytica serotype A2 isolates from domestic sheep and cattle, respectively. We thank Matt Parsek of the Department of Microbiology at the University of Washington for allowing us to use unpublished Pseudomonas aeruginosa sequence data generated in collaboration with him in order to estimate the errors in our sequence assemblies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
PKL and REB conceived and formulated the MS outline. PKL wrote the major part of the manuscript. PKL, REB and WK analyzed the data; WK produced all the figures and tables. JEM used computational software to analyze T3SS and effectors. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lawrence, P.K., Kittichotirat, W., McDermott, J.E. et al. A three-way comparative genomic analysis of Mannheimia haemolytica isolates. BMC Genomics 11, 535 (2010). https://doi.org/10.1186/1471-2164-11-535
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-535