
Abstract
Background
Saliva of adult female mosquitoes help sugar and blood feeding by providing enzymes and polypeptides that help sugar digestion, control microbial growth and counteract their vertebrate host hemostasis and inflammation. Mosquito saliva also potentiates the transmission of vector borne pathogens, including arboviruses. Culex tarsalis is a bird feeding mosquito vector of West Nile Virus closely related to C. quinquefasciatus, a mosquito relatively recently adapted to feed on humans, and the only mosquito of the genus Culex to have its sialotranscriptome so far described.
Results
A total of 1,753 clones randomly selected from an adult female C. tarsalis salivary glands (SG) cDNA library were sequenced and used to assemble a database that yielded 809 clusters of related sequences, 675 of which were singletons. Primer extension experiments were performed in selected clones to further extend sequence coverage, allowing for the identification of 283 protein sequences, 80 of which code for putative secreted proteins.
Conclusion
Comparison of the C. tarsalis sialotranscriptome with that of C. quinquefasciatus reveals accelerated evolution of salivary proteins as compared to housekeeping proteins. The average amino acid identity among salivary proteins is 70.1%, while that for housekeeping proteins is 91.2% (P < 0.05), and the codon volatility of secreted proteins is significantly higher than those of housekeeping proteins. Several protein families previously found exclusive of mosquitoes, including only in the Aedes genus have been identified in C. tarsalis. Interestingly, a protein family so far unique to C. quinquefasciatus, with 30 genes, is also found in C. tarsalis, indicating it was not a specific C. quinquefasciatus acquisition in its evolution to optimize mammal blood feeding.
Similar content being viewed by others


Background
Most adult female mosquitoes are hematophagous, in addition to taking sugar meals. Saliva helps blood feeding by interfering with host reactions that could disrupt the blood flow, assists sugar meals with glycosidases, and contain antimicrobials that may control microbial growth in their meals [1]. With the advent of tissue transcriptomics, we can postulate that these functions are mediated by large numbers (70-100) of polypeptides, many of which are expressed solely in the adult female salivary glands [2]. Unique protein families have been found in Anopheles, Aedes or Culex mosquitoes, as well as a group of common proteins or enzymes [3–5]. Functional characterization of these proteins uncovers scavengers of biogenic amines [6, 7] or leukotrienes [8], inhibitors of blood clotting [9–11], bradykinin formation [12, 13], platelet aggregation [14, 15] and vasodilators [16, 17]. Other molecularly uncharacterized activities include inhibitors of mast cell TNF production [18] and inhibition of T cell activation [19, 20]. It is apparent that the complexity of the salivary components affecting host hemostasis and inflammation mirrors the complexity of host hemostasis and inflammation itself, which must be disarmed for successful blood feeding. Indeed mosquitoes lacking salivation by salivary duct ablation feed less and take more dangerous time of exposure to their hosts [21, 22].
Perhaps due to the potent pharmacological activities of saliva, or the immune reactions to it, mosquito saliva also plays a role in pathogen transmission, including in arboviral transmission [23–25]. Accordingly, determination of the salivary composition of vector mosquitoes not only discovers new potentially pharmacologically active molecules, but also can also help generating vaccine targets for disruption of arboviral transmission. These proteins may also be of epidemiological significance as selective human or animal markers of vector exposure [26–29].
We have previously described the sialotranscriptome of C. quinquefasciatus[5], where several unique protein families were discovered, many of which are products of gene duplications. Indications of horizontal gene transfer from bacteria to mosquitoes were also pointed out as participating in the generation of mosquito sialomes [3, 4]. To this date, no other sialotranscriptome from a member of the Culex genus has been described. We currently portray the sialotranscriptome of Culex tarsalis, a North American bird feeding mosquito [30], and, like C. pipiens, a good vector of West Nile virus [31].
Results and Discussion
Characteristics of the assembled salivary EST set
A total of 1,753 cDNA clones were used to assemble a database (Additional file 1, Table S1) that yielded 809 clusters of related sequences, 675 of which contained only one expressed sequence tag (EST). The 809 clusters were compared, using the programs BlastX, BlastN, or RPSBLAST [32], to the nonredundant protein database of the National Center of Biological Information (NCBI), to a gene ontology database [33] to the conserved domains database of the NCBI [34], and to a custom prepared subset of the NCBI nucleotide database containing either mitochondrial or rRNA sequences.
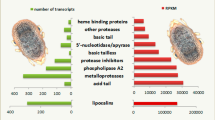
Three categories of expressed genes derived from the manual annotation of the contigs (Fig 1). The putatively secreted (S) category contained 44% of the sequences, the housekeeping (H) category had 36.3% and 19.7% of the ESTs could not be classified and belong to the Unknown (U) class. The transcripts of the unknown class could represent novel proteins or derive from the less conserved 3' or 5' untranslated regions of genes, as was indicated for the sialotranscriptome of An. gambiae[3]. This distribution is typical of previous mosquito sialotranscriptomes [4, 5, 35–37].

Distribution of the transcripts from the salivary gland cDNA library of Culex tarsalis according to functional class.
Housekeeping (H) genes
The 637 ESTs attributed to H genes expressed in the salivary glands of C. tarsalis were further characterized into 19 subgroups according to function (Table 1 and Additional file 1, Table S1). Transcripts associated with the protein synthesis machinery represented 44% of all transcripts associated with a housekeeping function, an expected result for the secretory nature of the organ. Nineteen percent of the transcripts were classified as either "Unknown conserved" or "Conserved secreted proteins". These represent highly conserved proteins of unknown function, presumably associated with cellular function, but still uncharacterized. These sets may help functional identification of the "Conserved hypothetical" proteins as previously reviewed in [38]. Transporters accounted for 7% and included several members of the vacuolar ATPase machinery, important for mosquito salivary secretion [39]. Energy metabolism and protein modification related enzymes accounted for nearly 5% each of the transcripts of the H class.
Possibly secreted (S) class of expressed genes
A total of 771 ESTs represent putative C. tarsalis salivary components (Tables 2 and Additional file 1, Table S1). These include ubiquitous known gene families, e.g, families seen in other organs and organisms, as well as protein families unique to hematophagous Diptera, or to Culicines, or to Culex genus alone. Table 2 also indicates our degree of knowledge, or ignorance, regarding these protein families, 15 of which we have no hint for their function, more than half of the 24 classes of proteins represented in Table 2. Many of these putatively secreted protein families of unknown function are multigenic, such as the CWRP family found previously only in C. quinquefasciatus[5], which contributes to 58% of the ESTs associated with the S class in C. tarsalis. The D7/OBP-like families contribute to 9% of all transcripts associated with secreted products, which is less than previously seen in Anopheles or Aedes sialotranscriptomes, probably due to the dilution effect following abundant expression of the CWRP family.
The salivary secretome of Culex tarsalis
From the sequenced cDNAs, a total of 283 novel C. tarsalis protein sequences were derived, 80 of which code for putative secreted products (Additional file 2, Table S2). Because cDNA sequences coding for many of these proteins were found as singletons, this secretome is to be considered preliminary and incomplete, but many parallels with previous mosquito sialotranscriptomes can be drawn, as follows:
Proteins with presumed or experimentally validated function
The D7/Odorant binding protein-like family
The D7 proteins constitute a unique multi gene family found expressed in the salivary glands of blood sucking Diptera [40], belonging to the superfamily of Odorant Binding Proteins (OBP) [41]. Long and short versions exist, the long versions containing two and the short versions containing one OBP domain. Three genes codes for long D7 proteins and 5 genes code for short D7 proteins in An. gambiae[3], while in Ae. aegypti 2 genes code for long and 3 genes code for short D7 proteins [4]. Short versions of anopheline D7 mosquitoes were shown to bind biogenic amines such as serotonin and histamine [6], thus counteracting hemostasis and inflammation. More recently, the amino terminal OBP domain of a D7 long form of Ae. aegypti was shown to bind peptidic leukotrienes with high affinity. The crystal structures of a short D7 protein from An. gambiae and a long D7 protein from Ae. aegypti revealed that the D7 OBP domains have 7 alpha helices, 2 more than the canonical OBP family [7]. In addition to these inflammatory agonist binding functions, a short D7 protein from An. stephensi, named hamadarin, was shown to inhibit bradykinin formation by inhibiting the FXII/Kallikrein pathway [12].
Eight members of the D7 protein family were found in the C. tarsalis sialotranscriptome, in addition to two canonical OBP proteins. Alignment of these proteins with their close blast matches found in the NR protein database, and construction of a bootstrapped phylogram (Fig 2) shows several clades with strong bootstrap support, and allows the following inferences: The eight C. tarsalis proteins are the probable results of 4 genes. Ctar-34, Ctar-35 and Ctar-36 are possible alleles of a single gene related to other long C. quinquefasciatus D7 proteins shown in Clade I, while Ctar-37, Ctar-38 and Ctar-40 are probably alleles of a second C. tarsalis gene coding for yet another long D7 protein, and are associated within Clade II. Ctar-173, results from a gene coding for a third long D7 protein grouping in Clade V with other mosquito long D7 proteins, including, Anopheles and Aedes proteins. Ctar-371 is the only C. tarsalis sequence for a short D7 protein, grouping in clade IV with other Culex pipiens D7 proteins. Notice also Clades III and Clade VI, which exclusively contain long D7 Anopheline or Phlebotomine proteins, respectively.

Phylogram of the D7 protein family of mosquitoes. The numbers on the tree nodes represent the percent bootstrap support in 10,000 trials (only values above 50% are shown). The bar at the bottom indicates amino acid divergence. The Culex tarsalis sequences are named Ctar-XXX where the XXX represents the cluster number that originated it. The remaining sequences are named in a five letter followed by number format where the 3 first letters represent the 3 first letters of the genus, followed by the first 2 letters of the species binomial name. The number represents the NCBI gi| access. For more details, see text.
Ctar-34, Ctar-35, Ctar-37 and Ctar-38 possess the signature [ED]- [EQ]-x(7)-C-x(12,17)-W-x(2)-W-x(7,9)- [TS]-x-C- [YF]-x- [KR]-C-x(8,22)-Q-x(22,32)-C-x(2)- [VLI] found in lipid binding D7 domain of Ae. aegypti[8]. The serotonin binding motif found on short anopheline D7 proteins as well as the carboxydomain of the long D7 protein of A. aegypti[6, 7] was not found on any C. tarsalis D7 protein (nor in any D7 protein of C. quinquefasciatus - results not shown), indicating this motif may have evolved beyond recognition, or that other proteins in Culex may undertake this task (see below under CWRP heading).
Ctar-195 and Ctar-525 represent 2 canonical OBP protein sequences, transcripts of which were found expressed in the salivary glands of C. tarsalis. Homologs of these canonical OBP proteins were also previously found expressed in C. quinquefasciatus and An. gambiae sialotranscriptomes. These proteins tend to be much more conserved in sequence to mosquito and even Drosophila proteins and it is possible that they may play an endogenous or housekeeping function.
The 30 kDa antigen/GE rich/Aegyptin family
This protein family is typical of the salivary glands of adult female mosquitoes, and was first identified as a salivary antigen in Ae. aegypti[42], and later found in salivary transcriptomes and proteomes of both culicine and anopheline mosquitoes [4, 5, 35–37, 43, 44], where it was named GE rich protein. Distantly related proteins also exist in black flies and sand flies. More recently, proteins of this family from Aedes and Anopheles were shown to prevent platelet aggregation by collagen [14, 45], indicating conservation of function after the split of the culicidae into the culicines and anophelines, approximately 150 MYA [46].
The sialotranscriptome of C. tarsalis allowed the identification of 8 protein sequences from the 30 kDa antigen/Aegyptin family (Additional file 2, Table S2), all represented by 1-10 ESTs found in the library. Blast searches against the NR database retrieved closely related sequences. Those that were more than 95% identical and of the same species were excluded to avoid inclusion of alleles or very closely related genes. Clustal alignment of this subset was used to produce a bootstrapped phylogram (Fig 3) that allows the following inferences: The 8 proteins from C. tarsalis most probably derive from 3 genes, Ctar-49, Ctar-50 and Ctar-51 being probable alleles of a gene closely related to a C. quinquefasciatus salivary gene shown in Clade I. Clade I includes Aedes aegypti salivary proteins that are short versions of the canonical 30 kDa protein, as are all Culex proteins in this clade. A second C. tarsalis gene possibly codes for Ctar-103, Ctar-104 and Ctar-105, and a third gene for Ctar-55 and Ctar-57. These two gene products groups with strong bootstrap support to their C. quinquefasciatus homologs, and these in turn group relatively weakly (48% bootstrap support) in Clade II as shown in Fig 3. Fig 3 also shows the uniquely Aedes containing proteins in clade III, represented by pairs of homologs between Ae. aegypti and Ae. albopictus, and the uniquely anopheline clade IV, with one sequence each of 5 different mosquito species. This phylogram suggests the single gene status of this family in the Anopheles genus, and the multi gene character in Culicines. At least 3 genes for canonical 30 kDa Ag/Aegyptin exists in the Aedes genus, and at least one more gene for the shorter protein shown in Clade I. Culex also have the shorter version gene, plus at least 2 genes of the canonical type.

Phylogram of the 30 kD/GE rich/Aegyptin protein family of mosquitoes. The numbers on the tree nodes represent the percent bootstrap support in 10,000 trials. The bar at the bottom indicates 20% amino acid divergence. The Culex tarsalis sequences are named Ctar-XXX where the XXX represents the cluster number that originated it. The remaining sequences are named in a five letter followed by number format where the 3 first letters represent the 3 first letters of the genus, followed by the first 2 letters of the species binomial name. The number represents the NCBI gi| access. For more details, see text.
Mucins and Peritrofins
Serine and threonine rich proteins are a common finding in sialotranscriptomes. These proteins are generally modified post-translationally and their mature forms have N-acetyl galactosamine residues, typical of mucins [47]. They probably have a function to lubricate the food canals and may also have antimicrobial function. Ctar-246 and Ctar-429 encode related protein sequences that might reflect splice variants of the same gene. These 2 truncated protein sequences have over 60 predicted glycosylation sites, and are similar to a previously described salivary mucin of C. quinquefasciatus. Ctar-261 is related to Aedes and Culex mucins that, in the case of the Aedes protein, were associated with induction by viral infection suggesting an immune function for this protein with 13 putative glycosylation sites. Ctar-581 encodes a 5' truncated protein sequence similar to salivary mucins of C. quinquefasciatus and Aedes albopictus.
Enzymes
Additional file 1, Table S1 indicate the presence of transcripts coding for enzymes possibly associated with the sugar meal and blood meal. Related to the sugar meal are several transcripts coding for glycosidases similar to proteins annotated as maltase and amylase. Additional file 2, Table S2 provides one full length sequence of a salivary alpha-glucosidase similar to other Culicine salivary maltases, plus 4 other truncated sequences coding for different sugar hydrolases. Regarding blood meal-related enzymes, 4 EST's coding for fragments of adenosine deaminase were found, as well as for an endonuclease. In C. quinquefasciatus a salivary endonuclease has been previously characterized and associated with helping forming the feeding hematoma [48]. However, the C. tarsalis enzyme is most closely related to sand flies salivary endonucleases and only distantly related to the salivary endonuclease of C. quinquefasciatus, although it is closely related to a non-salivary C. quinquefasciatus enzyme, indicating this C. tarsalis enzyme may be playing a housekeeping function.
Remarkably absent from the C. tarsalis sialotranscriptome are ESTs coding for members of the 5' nucleotidase, which functions as the salivary apyrase of mosquitoes. Apyrase hydrolyzes ATP and ADP to AMP and orthophosphate, destroying these important agonists of inflammation and platelet aggregation [2, 49]. It has been previously noticed that C. quinquefasciatus has little salivary apyrase activity when compared to Aedes and Anopheles mosquitoes, and this observation was postulated to be a consequence of the lack of platelets in birds, the most common host of Culex[50], while Aedes and Anopheles are mostly mammalian feeders [51]. This may explain the absence of 5'-nucleotidase/apyrase coding transcripts in C. tarsalis, although an increased sequencing effort could produce 5'-nucleotidases that might be secreted. As an example of the abundance of this type of transcript in mosquito sialotranscriptomes, only one transcript out of 503 clones coded for apyrase in the C. quinquefasciatus sialotranscriptome [5], while 99 out of 4,066 were found in a similarly made library from An. gambiae, and 66 out of 4,232 ESTs were found in the Ae. aegypti sialotranscriptome [4]. From these observed frequencies, the expected number of EST's for C. tarsalis and C. quinquefasciatus are 27 and 8, respectively, producing a Chi square of 54.6 and a P < 0.001, indicating the low expression of apyrase transcripts in Culex.
Three ESTs coding for a phosphatase produce the predicted truncated sequence encoded by Ctar-194, which is similar to a salivary alkaline phosphatase previously described in Ae. aegypti. The function of this enzyme in feeding, if any, is unknown.
Transcripts coding for at least 6 different serine proteases were found in C. tarsalis sialotranscriptome (Additional files 1 and 2, Tables S1 and S2). These enzymes may function in immunity as prophenoloxidase activators, or in digesting skin matrix components, such as in an elastase function, or hydrolysing host blood clotting enzymes such as fibrinogen/fibrin, or activating plasminogen.
Immunity related products
Antimicrobial peptides, lysozyme, and pathogen pattern recognition polypeptides are commonly found in the sialotranscriptome of blood sucking arthropods. Additional file 2, Table S2 shows the full length sequence of C. tarsalis salivary lysozyme, which is 91% identical to the C. quinquefasciatus homolog, and 75% identical to the salivary homolog of Ae. albopictus. Truncated ORF's of a C-type lectin and a Gram-negative binding protein were also found. They both match previously described salivary proteins of Aedes and Culex.
Secreted proteins with unknown function
Promiscuous Antigen 5 (AG-5) family
This is a ubiquitous protein family found in animals and plants [52], and in all sialotranscriptomes of blood sucking Diptera analyzed so far. The function of these proteins in mosquito saliva is not known, but in blood sucking Brachycera two proteins of this family have been functionally characterized. Remarkably, in a tabanid fly, a member of the AG-5 family acquired a typical RGD domain surrounded by Cys residues and acts as a main platelet aggregation [53], and in the stable fly a salivary AG-5 protein binds immunoglobulins and may function as an inhibitor of the classical complement pathway [54]. We present evidence, in the form of truncated transcripts, for the expression of at least two members of the family in C. tarsalis salivary glands; Ctar-151, assembled from 3 ESTs, matches with 94% identity the salivary secreted antigen-5 precursor AG5-3 from Culex quinquefasciatus while Ctar-438 matches with 91% identity another C. quinquefasciatus protein of the same family.
Insect specific Cys-Rich polypeptide family
Two transcripts were found in the C. tarsalis sialotranscriptome coding for basic (pI = 8.8) peptides of mature MW of 9.7 kDa containing 12 Cys residues. This peptide family was previously found in the salivary transcriptome of C. quinquefasciatus, but close relatives of the same size exist in Drosophila, Bombyx, Tribolium and Apis. The ubiquity of this protein family in insects, its size and pI suggests an antimicrobial role.
Hematophagous Diptera specific 41.0 kDa family
The first 41.0 kDa family member was characterized in the sialotranscriptome of Ae. aegypti, and later found in C. quinquefasciatus and in Ae. albopictus[4, 5, 36, 55]. Although not present in the sialotranscriptomes of members of the anopheline Cellia subgenus An. funestus, An. stephensi and An. gambiae (including scaning of the deducted proteome), it was recently found in An. darlingi, a member of the Nyssorhynchus subgenus [56], characterizing this family as uniquely Culicidae. Additional file 2, Table S2 provides evidence of a member of this protein family in C. tarsalis, encoded by Ctar-541, producing a predicted protein of mature MW of 43 kDa, being 69% identical to its C. quinquefasciatus homolog. Psiblast of Ctar-541 against the NR protein database retrieves on its first blastp cycle only mosquito salivary proteins, as expected. On the second cycle, it retrieves with lower significance (e value > 0.005) mostly bacterial proteins, but also salivary proteins from Simulium vittatum and Culicoides sonorensis. Further iterations of Psiblast retrieves with high significance bacterial proteins of the methyl-accepting chemotaxis receptor family (MCP), which may suggest this bacterial family to be originated from horizontal gene transfer to an ancestral blood feeding Nematocera. The gene structure of this protein in C. quinquefasciatus and Ae. aegypti is similar, containing 2 exons with a short intron of ca. 60 nt. Inclusion of Simulium and Culicoides sequences in this family indicates an ancient origin before the Nematocera split [57].
Mosquito specific 23.4 kda protein
Ctar-345 codes for a protein related to mosquito proteins so far only found expressed in mosquito adult salivary glands. Psiblast of Ctar-345 against the NR database does not retrieve any additional protein with an e value better than 0.02.
Mosquito specific hyp37 family
This member of this mosquito salivary protein family was identified in An. stephensi[35], and later found also An. gambiae[3], but not previously in other mosquito transcriptomes, including C. quinquefasciatus and Ae. aegypti. Ctar-769, represented by a single EST in our database, codes for a member of this family as indicated by blastp comparisons to the NR protein database, where it retrieves only Culex and Anopheles proteins. Psiblast of Ctar-769 against the NR database does not increase finding matches beyond additional mosquito proteins, including proteins deducted from the uncovering of the C. quinquefasciatus genome with and without signal peptide, and additional proteins from An. gambiae lacking a signal peptide indicative of secretion. Interestingly, the Anopheles gambiae gene found expressed in the salivary glands was shown to reside as a single exon in chromosome arm 2R [3]. The An. gambiae gene was also found to be selectively expressed in the adult female salivary glands suggesting a role in blood feeding [3].
Culicine specific 30.5 kDa family
Members of the 30.5 kDa protein were previously discovered in Ae. aegypti, Ae. albopictus and C. quinquefasciatus sialotranscriptomes. Additional related proteins were also deducted from the genomes of Ae. aegypti and C. quinquefasciatus, but not An. gambiae. The sialotranscriptome of C. tarsalis provides additional evidence for this multi copy family exclusive of Culicines. Clustal alignment of these protein sequences (Fig 4A) shows many conserved amino acids and a conserved Cys framework on the second half of the protein. The bootstrapped phylogram shows 3 robust clades. Clade I contains a sub clade of 2 Ae. aegypti and 2 Ae. albopictus proteins, and a second sub clade of a C. quinquefasciatus and Ctar-129. Clade II has 4 C. tarsalis sequences, possibly alleles of a single gene or closely related genes, and one C. quinquefasciatus sequence. Clade III shows a possible gene expansion in C. quinquefasciatus containing 4 gene products, clustering with one Ae. aegypti sequence. Overall the phylogram indicates that Aedes aegypti has at least 3 genes coding for this protein family, while C. quinquefasciatus has at least 6, 3 of which possibly arrived by further gene duplications within clade III. Culex tarsalis and Ae. albopictus, have at least two genes expressing these proteins in their adult female salivary glands. Psiblast of members of this protein family against the NR protein database converges after 6 iterations, retrieving solely Aedes and Culex proteins (not shown). The function of this protein family is unknown, but transcripts for this family were found enriched in the salivary glands of adult female Ae. aegypti[4] suggesting a blood feeding role.

The 30.5 kDa family of culicine proteins. A) Clustal alignment. The Culex tarsalis proteins are identified by their Ctar-prefix. The remaining sequences are named with the first three letters from the genus name followed by two letters from the species name and by their NCBI protein accession number. The symbols above the alignment indicate: (*) identical sites; (:) conserved sites; less conserved sites. B) Neighbor Joining bootstrapped phylogram of the alignment in A. The Culex tarsalis proteins are marked with a square symbol. The numbers on the branches represent the percent bootstrap support. The bar in the bottom represents 20% amino acid divergence. For more details, see text.
Culicine specific 7.8 kDa family
Ctar-761 codes for a mature peptide of predicted MW = 11.7 kDa and pI = 8.8. It is similar to previously described salivary peptides found in sialotranscriptomes of Ae. aegypti and Ae. albopictus, but not Culex. Psiblast using an inclusion e value of 0.1 (relatively high to compensate for the small size of the sequences) against the NR protein database additionally identifies a salivary peptide previously found in C. quinquefasciatus, annotated as 9.7 kDa salivary peptide. Inclusion of this peptide with the previous to generate the reverse position search model [32] additionally identifies other salivary peptides from C. quinquefasciatus, with convergence of the search and identification of the 6 non redundant peptides with an e value of 8 e-16 or smaller. Alignment of these 6 peptides with Ctar-761 shows that the Aedes sequences are more compact than those from Culex, containing a conserved framework of 6 cysteines, while those of Culex have 2 additional Cys residues (Fig 5a). Only 3 other residues are identical in all sequences, indicating a high evolutionary rate of this peptide family, if they indeed share a common ancestor. The bootstrapped phylogram (Fig 5B) shows 2 robust clades, clade I containing only Aedes sequences in two subclades, each containing a pair of Ae. aegypti or Ae. albopictus sequences. Clade II has 2 sequences from C. quinquefasciatus. Ctar-761 does not cluster with either of the clades, indicating again the possible fast evolutionary rate of this peptide family.

The 7.8 kDa family of culicine peptides. A) Clustal alignment. The Culex tarsalis proteins are identified by the square symbol and their Ctar-prefix. The remaining sequences are named with the first three letters from the genus name followed by two letters from the species name and by their NCBI protein accession number. Notice conserved cysteine framework in black background and identical amino acids in yellow or gray background. B) Neighbor Joining bootstrapped phylogram of the alignment in A. The numbers on the branches represent the percent bootstrap support. The bar in the bottom represents 10% amino acid divergence. For more details, see text.
Culicine specific 4.2 kDa peptide family
Ctar-146, Ctar-147 and Ctar-208 encode peptides with similarities to C. quinquefasciatus 4.2 kDa salivary peptide, which has similarity solely to another salivary peptide previously found in Ae. albopictus.
Culex genus specific cysteine and tryptophan rich protein family (CWRP)
This protein family was previously discovered in the C. quinquefasciatus sialotranscriptome [5], where 10 proteins were deduced from the EST sequences. These have a signal peptide indicative of secretion, 4 conserved cysteins, 3 conserved tryptophans and one conserved asparagine and leucine residues. Inspection of the C. quinquefasciatus genome indicates a total of 30 genes coding for this protein family, most (28) members being single exonic. Expression of these genes in C. quinquefasciatus salivary glands was confirmed by Edman degradation of electrophoretically separated salivary homogenate [5]. The C. tarsalis sialotranscriptome led to identification of 6 full length members of this protein family, plus 14 other fragments. Alignment of the members of this family (Fig 6) shows a conserved cysteine framework in most members, two conserved tryptophan residues (in addition to 4 other less conserved tryptophan residues), as well at the conserved asparagine and leucine residues. This protein family is by far the most transcribed in C. tarsalis adult female salivary glands, accounting for 25% of the transcripts that were sequenced. Interestingly, the most transcribed member in C. tarsalis, Ctar-1, is a truncated member of the family by relatively recent acquisition of a stop codon just after the second conserve Cysteine. This abundant expression suggests that this family may be responsible for host serotonin or histamine antagonism, as is performed by the abundantly expressed D7 proteins of Aedes or Culex. This possibility is further supported by the relatively low expression of the D7 protein family in C. tarsalis and their absent serotonin binding motifs which are found both in Aedes and Anopheles D7 proteins (see above description of the D7 proteins). Psiblast of members of the CWRP protein family retrieves, interestingly, sugar binding proteins including bacterial glycosidases, lectins such as ricin and hemolysins, the crystal structure of which have a trefoil structure [58]. The single exon structure of this protein family in C. quinquefasciatus suggests either acquition of this gene family by horizontal transfer, and/or gene duplication occurring by retroposition of an RNA template.

The cysteine tryptophane rich proteins (CWRP) of Culex. The number above the alignment indicates the conserved cysteine framework. The bar indicates the conserved Q-X-W motif found in lectin families. Also shown are the conserved tryptophane, asparagine and leucine residues. The C. tarsalis proteins are indicated by the prefix Ctar-. The C. quinquefasciatus proteins were obtained from the Vectorbase proteome prediction.
Culex genus specific 9.7 peptide family
Ctar-520 and Ctar-29 encode truncated peptides matching the previously described putative 9.7 kDa salivary peptide of C. quinquefasciatus, which has no other matches to known proteins.
Culex genus specific GQP repeat protein family
Ctar-31, Ctar-32 and Ctar-33 produce matches to a low complexity protein previously described in the C. quinquefasciatus sialome characterized by a poly histidine repeat in the mature aminoterminal region, and a series of GQP/GQG repeats. The polyhistidine domain may confer a bacteriostatic role for this protein if it chelates Zn ions, a bacterial growth factor, and a domain existing in various antimicrobial peptides [59–61].
Comparison of protein sequence identities between C. tarsalis and C. quinquefasciatus gene products
Over 170 deduced protein sequences coding for putative housekeeping (H) products are presented in Additional file 2, Table S2. These proteins allow comparison of the evolutionary rate of the S proteins compared to the H proteins, using the C. quinquefasciatus proteome as a reference set, as done before for comparing An. stephensi salivary proteins to those of An. gambiae[35]. For this comparison, we used only protein sequences from C. tarsalis that had at least 100 AA of alignment by the blastp tool to a C. quinquefasciatus protein, and excluded from this set possible alleles or closely related gene duplications by removing the smaller sequence(s) that had 80% or more similarity to another one within the set. We thus compared 50 putative secreted C. tarsalis proteins with their C. quinquefasciatus proteome, obtaining an average of 70.1% protein identity, while 169 putative housekeeping proteins from C. tarsalis were 91.2% identical to C. quinquefasciatus predicted proteins (see Additional file 2, Table S2 worksheets - P < 0.001 Mann-Whitney rank sum test). This significant difference further supports the concept that the evolution of mosquito salivary secreted proteins occur at a faster pace than housekeeping proteins, as indicated before for anopheline proteins [35, 37, 56].
It has been suggested that codon volatility (the proportion of the point-mutation neighbors of a codon that encode different amino acids) could be a measure of selection for fast evolution of proteins, as could occur in pathogens in a constant avoidance of antibody recognition [62]. Although this intuitive idea has created strong opposition [63–65], it is supported by published models [66, 67]. We accordingly decided to measure the average codon volatility for the 205 sequences coding for housekeeping and 80 sequences coding for putative secreted proteins shown in Additional file 2, Table S2. The average codon volatility for the H class genes was 0.7609 + 0.001 while the S class had an average volatility of 0.7746 + 0.002 (Average + SE), a highly significant result (P = 6.9 × 10-8, double tailed t test). Whatever the discussion regarding the value of this index, it indicates that a single point mutation on an S class gene has a significantly higher chance of producing a non-synonymous amino acid substitution than in an H class gene.
Conclusions
From a conservative perspective, the sialotranscriptome of C. tarsalis confirms the presence of ubiquitous salivary mosquito protein families, such as the D7, 41 kDa, hyp37, 30 kDa antigen/Aegyptin, 23.4 kDa, mucins, cysteine rich peptide, antigen 5, amylase/maltase, and the immunity related proteins lysozyme, pathogen recognition molecules, and serine proteases possibly associated with immunity or with host matrix/fibrin degradation. Adenosine deaminase has also been found, but not the 5'nucleotidase/apyrase that has been abundantly found in mammalian feeding mosquitoes, but possibly absent or reduced in the bird feeding C. tarsalis. From another stand point, the C. tarsalis sialotranscriptome confirmed the presence of proteins so far known exclusively in culicine mosquitoes, such as endonucleases and the 62 kDa, 30.5 kDa, 7.8 kDa and 4.2 kDa families, all without a known function. Additionally, further confirmation of unique proteins of the Culex genus were found for members of the highly expanded CWRP, 9.7 kDa and the GPQ repeat families.
It has been previously indicated that many unique mosquito protein families appear to be related to bacterial proteins, (when using the Psiblast tool against the NR database) suggesting their acquisition by horizontal transfer. Interestingly, the genes coding for such proteins, now available for An. gambiae, C. quinquefasciatus and Ae. aegypti are often single exonic, as is the case of bacterial genes. In the case of C. tarsalis, this appears to be the case with the 41 kDa (which have a single short intron) and the CWRP (mostly uniexonic in C. quinquefasciatus) families. However, other possibilities for single exonic genes are their acquisition as gene duplication resulting from retrotransposition of an mRNA precursor deriving from an endogenous, multi exonic gene. At any rate, the frequency of the relatively unusual processes of horizontal transfer and/or retrotransposition in the acquisition of new genes associated with blood feeding appear to occur at a high rate in the formation of mosquito sialomes.
Since our transcriptome was based on ~2,000 ESTs from a non-normalized library, the question arises as to what extent novel putative secreted proteins can be discovered with a more extensive sequencing, or the use of a normalized library. As indicated in a recent review [68], it has been our experience that, perhaps due to the relatively low complexity of the salivary gland proteome (when compared to organs like the mammalian liver or brain, for example), 1000-2000 sequenced clones are enough to display the majority of the sialome. Indeed, ~2,000 clones sequenced from an Ae. aegypti salivary gland cDNA library [4] discovered virtually all those found in another effort that obtained ~20,000 sequences [69]. A similar situation was encountered with the An. gambiae sialome, where ~4,000 sequences identified basically all those in a large sequencing effort, also ~20,000 sequences done by the Pasteur Institute [3]. Sequencing of normalized libraries, or more intensive sequencing of existing libraries, perhaps with newer generation of pyrosequencing methodology, may increase the number of salivary secreted peptides, but possibly to no more than 10% of the number found with random sequencing of ~2,000 clones. It should be indicated, however, that these additional low abundance transcripts may account for pharmacologically potent peptides.
Finally, the fast divergence of salivary proteins allows the possibility of using C. tarsalis proteins as specific markers of vector exposure, as is being attempted for ticks [70–72], mosquitoes [26–29] and sand flies [73].
Methods
Mosquitoes and cDNA library construction
Culex tarsalis, strain Bakersfield (Bakersfield, California) was supplied by Dr. W.K. Riesen University of California-Davis. Colonized mosquitoes were maintained on mouse blood (for egg laying) and given 10% sucrose ad libitum. Larvae were reared and adults maintained under controlled conditions of temperature (27°C), humidity (70% RH), and light (16:8 L:D diurnal cycle). PolyA+ RNA was extracted from 80 dissected pairs of salivary glands using the Micro-FastTrack mRNA isolation kit (Invitrogen, Carlsbad, CA), which was then used to make a non-normalized PCR-based cDNA library using the SMART™ cDNA library construction kit (BD Biosciences-Clontech, Palo Alto, CA) as described before [55].
cDNA sequencing
The salivary gland cDNA library was plated on LB/MgSO4 plates containing X gal/IPTG to an average of 250 plaques per 150 mm Petri plate. Recombinant (white) plaques were randomly selected and transferred to 96 well MICROTEST TM U bottom plates (BD BioSciences, Franklin Lakes, NJ), containing 100 μl of SM buffer (0.1 M NaCl; 0.01 M MgSO4; 7 H2 O; 0.035 M Tris HCl (pH 7.5); 0.01% gelatin) per well. The plates were covered and placed on a gyrating shaker for 30 min at room temperature. The phage suspension was either immediately used for PCR or stored at 4°C for future use.
To amplify the cDNA using a PCR reaction, 4 μl of the phage sample was used as a template. The primers were sequences from the λ TriplEx2 vector and named pTEx2 5seq (5/TCC GAG ATC TGG ACG AGC 3/) and pTEx2 3LD (5/ATA CGA CTC ACT ATA GGG CGA ATT GGC 3/), positioned at the 5/end and the 3/end of the cDNA insert, respectively. The reaction was carried out in 96 well flexible PCR plates (Fisher Scientific, Pittsburgh, PA) using TaKaRa EX Taq polymerase (TAKARA Mirus Bio, Madison, WI) on a GeneAmp® PCR system 9700 (Perkin Elmer Corp., Foster City, CA). The PCR conditions were: one hold of 95°C for 3 min; 25 cycles of 95°C for 1 min, 61°C for 30 sec; 72°C for 5 min. The amplified products were analysed on a 1.5% agarose/EtBr gel. cDNA library clones were PCR amplified, and the ones showing single band were selected for sequencing. Approximately 200-250 ng of each PCR product was transferred to Thermo Fast 96 well PCR plates (ABgene Corp., Epsom, Surray, UK) and frozen at -20°C before cycle sequencing. Samples were shipped on dry ice to the Rocky Mountain Laboratories Genomics Unit with primer and template combined together in an ABI 96-well Optical Reaction Plate (P/N 4306737) following the manufacturers recommended concentrations. Sequencing reactions were setup as recommended by Applied Biosystems BigDye® Terminator v3.1 Cycle Sequencing Kit by adding 1 μl ABI BigDye® Terminator Ready Reaction Mix v3.1 (P/N 4336921), 3 μl 5× ABI Sequencing Buffer (P/N 4336699), and 2 μl of water for a final volume of 10 μl. Cycle sequencing was performed at 96°C for 10 seconds, 50°C for 5 seconds, 60°C for 4 minutes for 27 cycles on either a Bio-Rad Tetrad 2 (Bio-Rad Laboratories, Hercules, CA) or ABI 9700 (Applied Biosystems, Inc., Foster City, CA) thermal cycler. Fluorescently-labeled extension products were purified following Applied Biosystems BigDye® XTerminator™ Purification protocol and subsequently processed on an ABI 3730xL DNA Analyzer (Applied Biosystems, Inc., Foster City, CA). The AB1 file generated for each sample from the 3730xL DNA Analyzer was provided to researchers in Rockville, MD through a secure network drive for all subsequent downstream sequencing analysis. In addition to the sequencing of the cDNA clones, primer extension experiments were performed in selected clones to further extend sequence coverage.
Bioinformatic Tools and Procedures
Expressed sequence tags (EST) were trimmed of primer and vector sequences. The BLAST tool [32], CAP3 assembler [74] and ClustalW [75] software were used to compare, assemble, and align sequences, respectively. For assembly of ESTs, a pipeline using blast and the CAP3 assembler was used, by first blasting all sequences using blastn with a word size of 36 and feeding the results as a fasta file to the CAP3 assembler[76]. The CAP3 parameters were set at default values, with an overlap length cut off of 40 and percent identity of overlap at 80%. Phylogenetic analysis and statistical neighbor-joining (NJ) bootstrap tests of the phylogenies were done with the Mega package [77]. For functional annotation of the transcripts we used the tool BlastX [32] to compare the nucleotide sequences to the non-redundant (NR) protein database of the National Center for Biotechnology Information (NCBI, National Library of Medicine, NIH,) and to the Gene Ontology (GO) database [33]. The tool, reverse position specific Blast (RPSBLAST) [32]was used to search for conserved protein domains in the Pfam [78], SMART [79], Kog [80], and conserved domains databases (CDD) [81]. We have also compared the transcripts with other subsets of mitochondrial and rRNA nucleotide sequences downloaded from NCBI and to several organism proteomes downloaded from NCBI, ENSEMBL, or VectorBase. Segments of the three-frame translations of the EST (because the libraries were unidirectional, 6-frame translations were not used), starting with a methionine found in the first 300 predicted amino acids (AA), or the predicted protein translation in the case of complete coding sequences, were submitted to the SignalP server [82] to help identify translation products that could be secreted. O-glycosylation sites on the proteins were predicted with the program NetOGlyc [83]. Functional annotation of the transcripts was based on all the comparisons above. Following inspection of all these results, transcripts were classified as either Secretory (S), Housekeeping (H) or of Unknown (U) function, with further subdivisions based on function and/or protein families. Codon volatility was calculated as previously described [62].
Abbreviations
- aa:
-
amino acid
- AMP:
-
antimicrobial peptide
- AG5:
-
antigen 5 family
- EST:
-
expressed sequence tag
- H class:
-
housekeeping
- kbase:
-
kilobase
- NR:
-
nonredundant
- nt:
-
nucleotide
- OBP:
-
odorant binding protein
- S class:
-
secreted
- SG:
-
salivary gland
- SMART:
-
switching mechanism at 5/end of RNA transcript
- U class:
-
unknown function.
References
Clements AN: The physiology of mosquitoes. Pergamon Press Oxford. 1963, 393-
Ribeiro JM, Francischetti IM: Role of arthropod saliva in blood feeding: sialome and post-sialome perspectives. Annu Rev Entomol. 2003, 48: 73-88. 10.1146/annurev.ento.48.060402.102812.
Arca B, Lombardo F, Valenzuela JG, Francischetti IM, Marinotti O, Coluzzi M, Ribeiro JM: An updated catalogue of salivary gland transcripts in the adult female mosquito, Anopheles gambiae. J Exp Biol. 2005, 208 (Pt 20): 3971-3986. 10.1242/jeb.01849.
Ribeiro JM, Arca B, Lombardo F, Calvo E, Phan VM, Chandra PK, Wikel SK: An annotated catalogue of salivary gland transcripts in the adult female mosquito, Aedes aegypti. BMC genomics. 2007, 8 (1): 6-10.1186/1471-2164-8-6.
Ribeiro JM, Charlab R, Pham VM, Garfield M, Valenzuela JG: An insight into the salivary transcriptome and proteome of the adult female mosquito Culex pipiens quinquefasciatus. Insect Biochem Mol Biol. 2004, 34 (6): 543-563. 10.1016/j.ibmb.2004.02.008.
Calvo E, Mans BJ, Andersen JF, Ribeiro JM: Function and evolution of a mosquito salivary protein family. J Biol Chem. 2006, 281 (4): 1935-1942. 10.1074/jbc.M510359200.
Mans BJ, Calvo E, Ribeiro JM, Andersen JF: The crystal structure of D7r4, a salivary biogenic amine-binding protein from the malaria mosquito Anopheles gambiae. J Biol Chem. 2007, 282 (50): 36626-36633. 10.1074/jbc.M706410200.
Calvo E, Mans BJ, Ribeiro JM, Andersen JF: Multifunctionality and mechanism of ligand binding in a mosquito antiinflammatory protein. Proc Natl Acad Sci USA. 2009, 106 (10): 3728-3733. 10.1073/pnas.0813190106.
Valenzuela JG, Francischetti IM, Ribeiro JM: Purification, cloning, and synthesis of a novel salivary anti-thrombin from the mosquito Anopheles albimanus. Biochemistry. 1999, 38 (34): 11209-11215. 10.1021/bi990761i.
Francischetti IM, Valenzuela JG, Ribeiro JM: Anophelin: kinetics and mechanism of thrombin inhibition. Biochemistry. 1999, 38 (50): 16678-16685. 10.1021/bi991231p.
Stark KR, James AA: Isolation and characterization of the gene encoding a novel factor Xa-directed anticoagulant from the yellow fever mosquito, Aedes aegypti. J Biol Chem. 1998, 273 (33): 20802-20809. 10.1074/jbc.273.33.20802.
Isawa H, Yuda M, Orito Y, Chinzei Y: A mosquito salivary protein inhibits activation of the plasma contact system by binding to factor XII and high molecular weight kininogen. J Biol Chem. 2002, 13: 13-
Isawa H, Orito Y, Iwanaga S, Jingushi N, Morita A, Chinzei Y, Yuda M: Identification and characterization of a new kallikrein-kinin system inhibitor from the salivary glands of the malaria vector mosquito Anopheles stephens i. Insect biochemistry and molecular biology. 2007, 37 (5): 466-477. 10.1016/j.ibmb.2007.02.002.
Calvo E, Tokumasu F, Marinotti O, Villeval JL, Ribeiro JM, Francischetti IM: Aegyptin, a novel mosquito salivary gland protein, specifically binds to collagen and prevents its interaction with platelet glycoprotein VI, integrin alpha2beta1, and von Willebrand factor. J Biol Chem. 2007, 282 (37): 26928-26938. 10.1074/jbc.M705669200.
Champagne DE, Smartt CT, Ribeiro JM, James AA: The salivary gland-specific apyrase of the mosquito Aedes aegypti is a member of the 5'-nucleotidase family. Proc Natl Acad Sci USA. 1995, 92 (3): 694-698. 10.1073/pnas.92.3.694.
Ribeiro JMC, Nussenzveig RH: The salivary catechol oxidase/peroxidase activities of the mosquito, Anopheles albimanus. J Exp Biol. 1993, 179: 273-287.
Champagne D, Ribeiro JMC: Sialokinins I and II: Two salivary tachykinins from the Yellow Fever mosquito, Aedes aegypti. Proc Natl Acad Sci (USA). 1994, 91: 138-142. 10.1073/pnas.91.1.138.
Bissonnette EY, Rossignol PA, Befus AD: Extracts of mosquito salivary gland inhibit tumour necrosis factor alpha release from mast cells. Parasite Immunol. 1993, 15: 27-33. 10.1111/j.1365-3024.1993.tb00569.x.
Wasserman HA, Singh S, Champagne DE: Saliva of the Yellow Fever mosquito, Aedes aegypti, modulates murine lymphocyte function. Parasite immunology. 2004, 26 (6-7): 295-306. 10.1111/j.0141-9838.2004.00712.x.
Wanasen N, Nussenzveig RH, Champagne DE, Soong L, Higgs S: Differential modulation of murine host immune response by salivary gland extracts from the mosquitoes Aedes aegypti and Culex quinquefasciatus. Medical and veterinary entomology. 2004, 18 (2): 191-199. 10.1111/j.1365-2915.2004.00498.x.
Mellink JJ, Boven Kamp Van Den W: Functional aspects of mosquito salivation in blood feeding in Aedes aegypti. Mosquito News. 1981, 41: 115-119.
Ribeiro JMC, Rossignol PA, Spielman A: Role of mosquito saliva in blood vessel location. J Exp Biol. 1984, 108: 1-7.
Edwards JF, Higgs S, Beaty BJ: Mosquito feeding-induced enhancement of Cache Valley Virus (Bunyaviridae) infection in mice. J Med Entomol. 1998, 35 (3): 261-265.
Labuda M, Kozuch O: Amplification of arbovirus transmission by mosquito intradermal probing and interrupted feeding. Acta virologica. 1989, 33 (1): 63-67.
Schneider BS, Higgs S: The enhancement of arbovirus transmission and disease by mosquito saliva is associated with modulation of the host immune response. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2008, 102 (5): 400-408. 10.1016/j.trstmh.2008.01.024.
Remoue F, Cisse B, Ba F, Sokhna C, Herve JP, Boulanger D, Simondon F: Evaluation of the antibody response to Anopheles salivary antigens as a potential marker of risk of malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2006, 100 (4): 363-370. 10.1016/j.trstmh.2005.06.032.
Orlandi-Pradines E, Almeras L, Denis de Senneville L, Barbe S, Remoue F, Villard C, Cornelie S, Penhoat K, Pascual A, Bourgouin C: Antibody response against saliva antigens of Anopheles gambiae and Aedes aegypti in travellers in tropical Africa. Microbes and infection/Institut Pasteur. 2007, 9 (12-13): 1454-1462.
Cornelie S, Remoue F, Doucoure S, Ndiaye T, Sauvage FX, Boulanger D, Simondon F: An insight into immunogenic salivary proteins of Anopheles gambiae in African children. Malaria journal. 2007, 6: 75-10.1186/1475-2875-6-75.
Poinsignon A, Cornelie S, Mestres-Simon M, Lanfrancotti A, Rossignol M, Boulanger D, Cisse B, Sokhna C, Arca B, Simondon F: Novel peptide marker corresponding to salivary protein gSG6 potentially identifies exposure to Anopheles bites. PLoS ONE. 2008, 3 (6): e2472-10.1371/journal.pone.0002472.
Dow RP, Reeves WC, Bellamy RE: Field tests of avian host preference of Culex tarsalis Coq. Am J Trop Med Hyg. 1957, 6 (2): 294-303.
Turell MJ, Dohm DJ, Sardelis MR, Oguinn ML, Andreadis TG, Blow JA: An update on the potential of north American mosquitoes (Diptera: Culicidae) to transmit West Nile Virus. Journal of medical entomology. 2005, 42 (1): 57-62. 10.1603/0022-2585(2005)042[0057:AUOTPO]2.0.CO;2.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25 (1): 25-29. 10.1038/75556.
Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH: CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002, 30 (1): 281-283. 10.1093/nar/30.1.281.
Valenzuela JG, Francischetti IM, Pham VM, Garfield MK, Ribeiro JM: Exploring the salivary gland transcriptome and proteome of the Anopheles stephensi mosquito. Insect Biochem Mol Biol. 2003, 33 (7): 717-732. 10.1016/S0965-1748(03)00067-5.
Arca B, Lombardo F, Francischetti IM, Pham VM, Mestres-Simon M, Andersen JF, Ribeiro JM: An insight into the sialome of the adult female mosquito Aedes albopictus. Insect biochemistry and molecular biology. 2007, 37 (2): 107-127. 10.1016/j.ibmb.2006.10.007.
Calvo E, Dao A, Pham VM, Ribeiro JM: An insight into the sialome of Anopheles funestus reveals an emerging pattern in anopheline salivary protein families. Insect biochemistry and molecular biology. 2007, 37 (2): 164-175. 10.1016/j.ibmb.2006.11.005.
Galperin MY, Koonin EV: 'Conserved hypothetical' proteins: prioritization of targets for experimental study. Nucleic acids research. 2004, 32 (18): 5452-5463. 10.1093/nar/gkh885.
Novak MG, Ribeiro JMC, Hildebrand JG: 5-Hydroxytriptamine in the salivaryt glands of adult female Aedes aegypti and its role in regulation of salivation. J Exp Biol. 1995, 198: 167-174.
Valenzuela JG, Charlab R, Gonzalez EC, Miranda-Santos IKF, Marinotti O, Francischetti IM, Ribeiro JMC: The D7 family of salivary proteins in blood sucking Diptera. Insect Mol Biol. 2002, 11 (2): 149-155. 10.1046/j.1365-2583.2002.00319.x.
Hekmat-Scafe DS, Dorit RL, Carlson JR: Molecular evolution of odorant-binding protein genes OS-E and OS-F in Drosophila. Genetics. 2000, 155 (1): 117-127.
Simons FE, Peng Z: Mosquito allergy: recombinant mosquito salivary antigens for new diagnostic tests. Int Arch Allergy Immunol. 2001, 124 (1-3): 403-405. 10.1159/000053771.
Cazares-Raga FE, Gonzalez-Lazaro M, Montero-Solis C, Gonzalez-Ceron L, Zamudio F, Martinez-Barnetche J, Torres-Monzon JA, Ovilla-Munoz M, Aguilar-Fuentes J, Rodriguez MH: GP35 ANOAL, an abundant acidic glycoprotein of female Anopheles albimanus saliva. Insect molecular biology. 2007, 16 (2): 187-198. 10.1111/j.1365-2583.2006.00712.x.
Jariyapan N, Choochote W, Jitpakdi A, Harnnoi T, Siriyasatein P, Wilkinson MC, Bates PA: A glycine- and glutamate-rich protein is female salivary gland-specific and abundant in the malaria vector Anopheles dirus B (Diptera: Culicidae). Journal of medical entomology. 2006, 43 (5): 867-874. 10.1603/0022-2585(2006)43[867:AGAGPI]2.0.CO;2.
Yoshida S, Sudo T, Niimi M, Tao L, Sun B, Kambayashi J, Watanabe H, Luo E, Matsuoka H: Inhibition of collagen-induced platelet aggregation by anopheline antiplatelet protein, a saliva protein from a malaria vector mosquito. Blood. 2008, 111 (4): 2007-2014. 10.1182/blood-2007-06-097824.
Krzywinski J, Grushko OG, Besansky NJ: Analysis of the complete mitochondrial DNA from Anopheles funestus: an improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Molecular phylogenetics and evolution. 2006, 39 (2): 417-423. 10.1016/j.ympev.2006.01.006.
Hang HC, Bertozzi CR: The chemistry and biology of mucin-type O-linked glycosylation. Bioorganic & medicinal chemistry. 2005, 13 (17): 5021-5034.
Calvo E, Ribeiro JM: A novel secreted endonuclease from Culex quinquefasciatus salivary glands. J Exp Biol. 2006, 209 (Pt 14): 2651-2659. 10.1242/jeb.02267.
Ribeiro JMC: Role of arthropod saliva in blood feeding. Ann Rev Entomol. 1987, 32: 463-478. 10.1146/annurev.en.32.010187.002335.
Ribeiro JM: Blood-feeding in mosquitoes: probing time and salivary gland anti-haemostatic activities in representatives of three genera (Aedes, Anopheles, Culex). Med Vet Entomol. 2000, 14 (2): 142-148. 10.1046/j.1365-2915.2000.00227.x.
Horsfall WR: Mosquitoes: Their bionomics and relation to disease transmission. 1955, New York: The Ronald Press Co
Megraw T, Kaufman TC, Kovalick GE: Sequence and expression of Drosophila Antigen 5-related 2, a new member of the CAP gene family. Gene. 1998, 222 (2): 297-304. 10.1016/S0378-1119(98)00489-2.
Xu X, Yang H, Ma D, Wu J, Wang Y, Song Y, Wang X, Lu Y, Yang J, Lai R: Toward an understanding of the molecular mechanism for successful blood feeding by coupling proteomics analysis with pharmacological testing of horsefly salivary glands. Mol Cell Proteomics. 2008, 7 (3): 582-590.
Ameri M, Wang X, Wilkerson MJ, Kanost MR, Broce AB: An immunoglobulin binding protein (antigen 5) of the stable fly (Diptera: Muscidae) salivary gland stimulates bovine immune responses. Journal of medical entomology. 2008, 45 (1): 94-101. 10.1603/0022-2585(2008)45[94:AIBPAO]2.0.CO;2.
Valenzuela JG, Pham VM, Garfield MK, Francischetti IM, Ribeiro JMC: Toward a description of the sialome of the adult female mosquito Aedes aegypti. Insect Biochem Mol Biol. 2002, 32: 1101-1122. 10.1016/S0965-1748(02)00047-4.
Calvo E, Pham VM, Marinotti O, Andersen JF, Ribeiro JM: The salivary gland transcriptome of the neotropical malaria vector Anopheles darlingi reveals accelerated evolution of genes relevant to hematophagy. BMC genomics. 2009, 10: 57-10.1186/1471-2164-10-57.
Grimaldi D, Engel M: Evolution of the insects. 2005, New York: Cambridge University Press
Hatakeyama T, Unno H, Kouzuma Y, Uchida T, Eto S, Hidemura H, Kato N, Yonekura M, Kusunoki M: C-type lectin-like carbohydrate recognition of the hemolytic lectin CEL-III containing ricin-type-trefoil folds. The Journal of biological chemistry. 2007, 282 (52): 37826-37835. 10.1074/jbc.M705604200.
Bulet P, Stocklin R, Menin L: Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. 2004, 198: 169-184. 10.1111/j.0105-2896.2004.0124.x.
Lai R, Takeuchi H, Lomas LO, Jonczy J, Rigden DJ, Rees HH, Turner PC: A new type of antimicrobial protein with multiple histidines from the hard tick, Amblyomma hebraeum. Faseb J. 2004, 18 (12): 1447-1449.
Loomans HJ, Hahn BL, Li QQ, Phadnis SH, Sohnle PG: Histidine-based zinc-binding sequences and the antimicrobial activity of calprotectin. The Journal of infectious diseases. 1998, 177 (3): 812-814. 10.1086/517816.
Friedman R, Hughes AL: Codon volatility as an indicator of positive selection: data from eukaryotic genome comparisons. Molecular biology and evolution. 2005, 22 (3): 542-546. 10.1093/molbev/msi038.
Pillai SK, Kosakovsky Pond SL, Woelk CH, Richman DD, Smith DM: Codon volatility does not reflect selective pressure on the HIV-1 genome. Virology. 2005, 336 (2): 137-143. 10.1016/j.virol.2005.03.014.
Chen Y, Emerson JJ, Martin TM: Evolutionary genomics: codon volatility does not detect selection. Nature. 2005, 433 (7023): E6-7. 10.1038/nature03223. discussion E7-8
Sharp PM: Gene "volatility" is most unlikely to reveal adaptation. Molecular biology and evolution. 2005, 22 (4): 807-809. 10.1093/molbev/msi073.
Archetti M: Genetic robustness at the codon level as a measure of selection. Gene. 2009, 443 (1-2): 64-69. 10.1016/j.gene.2009.05.009.
Plotkin JB, Dushoff J, Desai MM, Fraser HB: Codon usage and selection on proteins. Journal of molecular evolution. 2006, 63 (5): 635-653. 10.1007/s00239-005-0233-x.
Ribeiro JMC, Arca B: From sialomes to the sialoverse: An insight into the salivary potion of blood feeding insects. Adv Insect Physiol. 2009, 37: 59-118. full_text.
Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi Z, Megy K, Grabherr M: Genome sequence of Aedes aegypti, a major arbovirus vector. Science (New York, NY). 2007, 316 (5832): 1718-1723.
Schwartz BS, Ribeiro JM, Goldstein MD: Anti-tick antibodies: an epidemiologic tool in Lyme disease research. Am JEpidemiol. 1990, 132 (1): 58-66.
Schwartz BS, Nadelman RB, Fish D, Childs JE, Forseter G, Wormser GP: Entomologic and demographic correlates of anti-tick saliva antibody in a prospective study of tick bite subjects in Westchester County, New York. Am JTrop Med Hyg. 1993, 48 (1): 50-57.
Sanders ML, Jaworski DC, Sanchez JL, DeFraites RF, Glass GE, Scott AL, Raha S, Ritchie BC, Needham GR, Schwartz BS: Antibody to a cDNA-derived calreticulin protein from Amblyomma americanum as a biomarker of tick exposure in humans. Am JTrop Med Hyg. 1998, 59 (2): 279-285.
Barral A, Honda E, Caldas A, Costa J, Vinhas V, Rowton ED, Valenzuela JG, Charlab R, Barral-Netto M, Ribeiro JM: Human immune response to sand fly salivary gland antigens: a useful epidemiological marker?. Am J Trop Med Hyg. 2000, 62 (6): 740-745.
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9 (9): 868-877. 10.1101/gr.9.9.868.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25 (24): 4876-4882. 10.1093/nar/25.24.4876.
Guo Y, Ribeiro JM, Anderson JM, Bour S: dCAS: a desktop application for cDNA sequence annotation. Bioinformatics (Oxford, England). 2009, 25 (9): 1195-1196. 10.1093/bioinformatics/btp129.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5 (2): 150-163. 10.1093/bib/5.2.150.
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2000, 28 (1): 263-266. 10.1093/nar/28.1.263.
Schultz J, Copley RR, Doerks T, Ponting CP, Bork P: SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000, 28 (1): 231-234. 10.1093/nar/28.1.231.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN: The COG database: an updated version includes eukaryotes. BMC bioinformatics. 2003, 4 (1): 41-10.1186/1471-2105-4-41.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005, D39-45. 33 Database
Nielsen H, Engelbrecht J, Brunak S, von Heijne G: Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10 (1): 1-6. 10.1093/protein/10.1.1.
Julenius K, Molgaard A, Gupta R, Brunak S: Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005, 15 (2): 153-164. 10.1093/glycob/cwh151.
Acknowledgements
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, and NIH R01 AI46435 grant to KEO. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the government of the United States of America. We are grateful to NIAID intramural editor Brenda Rae Marshall for assistance.
Because E.C., A.J.V, K.D.B, V.M.P, and J.M.C.R. are government employees and this is a government work, the work is in the public domain in the United States. Notwithstanding any other agreements, the NIH reserves the right to provide the work to PubMedCentral for display and use by the public, and PubMedCentral may tag or modify the work consistent with its customary practices. You can establish rights outside of the U.S. subject to a government use license.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
EC helped with library manufacture, sequencing, data analysis, and contributed to the manuscript. AJF and KDB. helped with sequencing the cDNA. VMP participated in sequencing the library. JMCR performed data analysis, and contributed to the manuscript. IS-V and KEO provided biological specimens and contributed to the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Calvo, E., Sanchez-Vargas, I., Favreau, A.J. et al. An insight into the sialotranscriptome of the West Nile mosquito vector, Culex tarsalis. BMC Genomics 11, 51 (2010). https://doi.org/10.1186/1471-2164-11-51
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-51