
Abstract
Background
Brucellosis is a major bacterial zoonosis affecting domestic livestock and wild mammals, as well as humans around the globe. While conducting proteomics studies to better understand Brucella abortus virulence, we consolidated the proteomic data collected and compared it to publically available genomic data.
Results
The proteomic data was compiled from several independent comparative studies of Brucella abortus that used either outer membrane blebs, cytosols, or whole bacteria grown in media, as well as intracellular bacteria recovered at different times following macrophage infection. We identified a total of 621 bacterial proteins that were differentially expressed in a condition-specific manner. For 305 of these proteins we provide the first experimental evidence of their expression. Using a custom-built protein sequence database, we uncovered 7 annotation errors. We provide experimental evidence of expression of 5 genes that were originally annotated as non-expressed pseudogenes, as well as start site annotation errors for 2 other genes.
Conclusions
An essential element for ensuring correct functional studies is the correspondence between reported genome sequences and subsequent proteomics studies. In this study, we have used proteomics evidence to confirm expression of multiple proteins previously considered to be putative, as well as correct annotation errors in the genome of Brucella abortus strain 2308.
Similar content being viewed by others

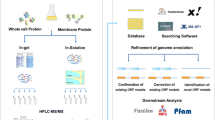

Background
Brucella species bacteria are gram negative alpha proteobacteria superbly adapted for survival in intracellular environments. They infect a wide range of mammals, including essentially all economically important domestic mammals, many wild species, and humans. Brucellosis is the largest bacterial zoonosis in the world [1–3]. In humans, untreated brucellosis is a long lasting disease characterized by recurrent fever episodes and clinical manifestations that include spondylitis, severe headaches, joint or abdominal pain, endocarditis, and meningoencephalitis. In severe non-treated cases brucellosis can cause death [1–3].
Seven terrestrial Brucella species have been defined: Brucella melitensis, Brucella abortus, Brucella suis, Brucella ovis, Brucella canis, Brucella neotomae and Brucella microti which infect goats, cattle, pigs, sheep, dogs, desert wood rats and common voles, respectively [1, 4]. Two Brucella species infecting marine mammals such as dolphins, whales, seals, sea lions and walrus have also been defined as Brucella ceti and Brucella pinnipedialis [5–7]. With the exception of B. suis biovar 3, the Brucella genome is encoded on two chromosomes, containing in total approximately 3,500 genes. Genome sequences from 32 different Brucella strains, representing all species, have been published either as complete genomes (10 strains) or as draft assemblies in NCBI (22 strains) [8–14]. The raw genome sequencing data of 78 other strains is also available in the Sequence Read Archive of NCBI. The genome sequences were very highly homologous, although regions of unique genetic material were also observed. It is possible that these regions are involved in establishing the distinct host preferences and biological behavior of the different Brucella species sequenced to date [15].
Unlike other pathogenic bacteria, Brucella virulence does not appear to be the result of relatively few virulence genes that can be transferred horizontally via plasmids, phages, or assembled in pathogenicity islands. Brucella also lack typical virulence factors such as exotoxins, flagella, capsules, and type III secretion systems. Rather, the pathogen's virulence appears to be an integrated aspect of its physiology. Therefore, to better understand Brucella virulence, we will need to better understand the Brucella proteome, including how it changes during the different stages of the intracellular and extracellular Brucella lifecycles, and how it interacts with host proteins and processes. Indeed, we have previously demonstrated that Brucella bacteria are capable of extensive, reversible, remodeling of their cell envelopes [16]. Furthermore, during the establishment of an intracellular infection, Brucella bacteria also appear able to carry out extensive, and reversible, modifications to their biosynthetic pathways and respiration in order to adapt to the changing microenvironments encountered in infected host cells [17]. This suggests that the Brucella proteome is considerably more dynamic than previously suspected, and that in depth proteomic analysis of the pathogen, as well as integration of these data with the available genomic information, will result in novel mechanistic and possibly therapeutic insights.
In this work we have generated a synthesis of the proteomic datasets we produced from multiple independent comparisons of Brucella strains either grown in media or retrieved from infected host cells. Some of this data is currently publicly available [[16, 17];http://proteomicsresource.org/Default.aspx] with the remainder becoming available as part of this work. These studies were originally designed to identify experimental condition-specific differences in the Brucella proteome. We compiled the experimental evidence for any Brucella protein detected and compared the proteomic data to the available genomic data. We provide the first direct experimental evidence for the expression of 305 Brucella proteins, but also identified experimental evidence for the expression of five genes previously annotated as pseudogenes, and of start site errors in two other genes.
Results and Discussion
First experimental evidence of the expression of 305 proteins in B. abortus 2308
Samples used for the proteomic analysis came from B. abortus either grown extracellularly in media or isolated from infected RAW264.7 macrophages. The extracellular samples included whole bacteria grown directly in tryptic soy broth, outer membrane preparations (blebs) [16] and cytosols. Intracellular samples consisted of viable B. abortus isolated at different time points post-infection from RAW264.7 macrophages [17] and of phagosomes isolated from infected murine phagocytic cells. We obtained 1704 peptides representing 621 different proteins, corresponding to approximately 20% of the predicted proteome. For 305 proteins, we are reporting the first experimental evidence of their expression in B. abortus 2308 (Table 1). We also report genome annotation errors for two proteins, expression of ORFs annotated pseudogenes for four proteins and one correction to the sequence of another previously annotated pseudogene which allows for its full length expression. Peptide sequences corresponding to these 312 proteins are listed in Additional File 1. The peptide coverage for the 305 newly demonstrated proteins varied from 1 to 20, with an average of three peptides per protein. In order to confirm the expression of proteins identified by a single peptide, we manually validated all MSMS spectra that had a sequence assignment score smaller than 45. Forty-four of the 305 proteins were described previously as hypothetical with no putative function. When subcellular localizations were predicted using three publicly available tools [18–20], 226 proteins were predicted to be cytosolic, ten were inner membrane proteins, 25 were periplasmic, three were outer membrane proteins and the localization of 48 proteins could not be predicted (Table 1). Experimental evidence for the expression of the other 309 of the 620 proteins has been demonstrated previously by our group [16, 17] and others [21–31]. It is important to note that we are reporting an analysis of the combined results of several independent experiments using the same bacterial strain and technology to acquire the data. However, each experiment was a separate comparative study designed to identify differentially expressed bacterial proteins under specific conditions per experiment. Proteins that were not sufficiently differentially expressed under the experimental conditions used would have not been identified. Thus, while our results can be used to confirm that the proteins reported were expressed, they may underestimate under what conditions they can become expressed.
Correction of five pseudogene annotations
In previous studies using B. abortus 2308, we used the genome databases available on NCBI for B. abortus, B. melitensis and B. suis for protein identification. More than once, we obtained peptides which matched proteins supposedly expressed only by the latter two species. Upon verification, those peptides were manually assigned to ORFs of previously annotated pseudogenes of B. abortus strain 2308 (NCBI taxonomy ID 359391). We therefore assembled a custom protein database which included the predicted translation sequence of all B. abortus 2308 ORFs annotated as pseudogenes. Using this database, we were able to confirm the protein expression of five of these ORFs (Figure 1): BAB1_1205, BAB1_1645, BAB1_1646, BAB1_1768 and BAB2_0216. The MSMS spectra of the 18 peptides representing these former pseudogenes were manually validated. We thus investigated the reasons for which these genes had been annotated as pseudogenes. The genomic sequence of the cytoplasmic protein with a conserved DUF 883 domain BAB1_1205 was found to be identical to BMEI0805, its B. melitensis counterpart. Apart from the short length of this protein, there was no apparent reason for its pseudogene annotation (Figure 1). For BAB1_1645 and BAB1_1646 (Figure 1), the nucleotidic sequence was 99% identical to their BMEI0397 and BMEI0396 counterparts, leading to two cytoplasmic B. abortus 2308 dihydroxyacetone kinases involved in glycerolipid metabolism that are 98% and 100% identical to the B. melitensis proteins, respectively. The case of BAB2_0216, which seems to be a 3-hydroxybutyryl-CoA dehydrogenase, was more complex and confusing, having a single nucleotide deletion when compared to B. melitensis. This deletion would lead to the silencing of the stop codon which creates two separate proteins in B. melitensis, BMEII1020 and BMEII1021. In B. abortus 2308, a fusion of the two genes would generate a much larger protein. However, the start codon in the corresponding ORF of vaccine B. abortus S19 (BAbS19_II02060) is different from BMEII1020, and even more different from the start codon and carboxyl terminal sequence of the counterparts in B. suis (BSUIS_B0227), B. ovis (BOV_A0203), B. canis (BCAN_B0224) and B. ceti (BCETI_6000534). As a consequence, the lengths of B. abortus and B. melitensis proteins differ considerably from those of other Brucella. Since the BAB2_0216 peptide that we found is located in the N-terminal section of the protein (Figure 1), we are able to confirm the expression of this originally annotated pseudogene, but were unable to confirm the expression of the full length protein.

B. abortus 2308 former pseudogenes. Peptide sequences identified by mass spectrometry are highlighted in grey. Corresponding B. melitensis 16 M locus tags are indicated between parentheses.
The sequence of the BAB1_1768 pseudogene was found to be misannotated in B. abortus 2308. The peptide sequence "TAGYGVGGAALGALAGGAIGGNGR" could not be found in the B. abortus 2308 nucleotide-derived proteome but matched the B. melitensis locus tag BMEI0287. In fact, except for 1 nucleotide, the corresponding 2308 genomic sequence is identical to that of BMEI0287 (Figure 2C). In B. abortus 2308, a single nucleotide insertion in BAB1_1768 modifies the reading frame, hence its original annotation as a pseudogene. The manually validated peptide matches B. abortus 2308 only when the additional nucleotide is removed, indicating that the sequence for locus BAB1_1768 should be corrected (Figure 1). Also to note is the earlier start site in B. abortus 2308, and all other species sequenced to date, when compared to B. melitensis 16 M. We believe that the B. abortus 2308 start site was correctly assigned in the publicly available genome given the clear presence of a ribosome binding site in position -8 of the B. abortus sequence.

Annotation errors in the B. abortus 2308 genome. (A, B) The original start codon annotation in the publicly available genome (NCBI taxonomy ID 359391) of the succinyl-CoA synthetase subunit beta (BAB1_1926, panel A) and of the KHG aldolase (BAB2_0083, panel B) are indicated by double asterisks whereas the corrected start site is indicated by a single asterisk (BAB1_1926 only). The peptides sequenced by mass spectrometry are highlighted in grey. The 5'-end of the CDS, as currently annotated, are underlined. The predicted sequence of the RBS found in proximity of the corrected start site of BAB1_1926 is boxed. Numbers next to the nucleotide sequence and the schematic gene representation indicate the position in the genome sequence (NC_007618 or NC_007624). (C) Genomic and amino acid sequences of BAB1_1768, as currently found in the publicly available genome, were aligned to their counterparts in B. melitensis 16 M (BMEI0287). The sequence of the peptide detected by mass spectrometry is highlighted in grey. Matching nucleotides are indicated by vertical bars and matching amino acids are indicated by asterisks. The predicted sequence of the RBS found in proximity of the B. abortus start site is boxed.
Correction of two start site annotations errors
Another type of annotation error identified in our studies was the erroneous assignment of gene translation start sites. For 2 proteins of B. abortus 2308, we report the expression of manually validated peptides corresponding to the sequence found upstream of their currently annotated start sites (Figure 2). The peptide sequence "MNIHEYQAK" was first found to match the cytoplasmic B. melitensis succinyl-CoA synthetase subunit beta protein (BMEI0138) and then assigned manually to BAB1_1926. Sequence comparison with other Brucella species and strains shows that the B. abortus 2308 protein start site is not shared with any of the subject sequences (Figure 2A). In fact, all homologues of this protein in other Brucella strains or species share the same start site, which is found 22 amino acids upstream of the B. abortus 2308 site. Moreover, a ribosome binding site can clearly be mapped to position -8 of the proposed new translation start site. We therefore believe this new start site to be accurate.
The second peptide, "TDLLPIMK", was found to match the cytoplasmic B. melitensis keto-hydroxyglutarate-aldolase (BMEII0009) and then assigned to BAB2_0083 in B. abortus 2308. This peptide overlaps the region upstream to the currently annotated translation start site and the first three amino acids based on the annotated translation start site (Figure 2B). Alignment of the current B. abortus 2308 protein sequence with its counterparts in other Brucella strains and species indicates that the 2308 protein sequence is falsely truncated. Other start sites lead to proteins having N-terminals longer by 11, 26 or 44 amino acids. Although we cannot clearly indicate the actual start site of BAB1_1926 or BAB2_0083, we can confirm that their N-terminals are longer than currently annotated. Based on the homology of the B. abortus 2308 genome being highest with that of other B. abortus strains, one can speculate that the start sites would be identical to those mapped in these strains.
Operons
Since genes that are part of an operon are usually co-transcribed, it is possible that these genes might also be co-translated [32]. Considering all proteins identified by our studies, we were able to almost fully reconstitute one of the two ribosomal RNA operons, with all but BAB1_1237 found. Additionally, the previously mentioned BAB1_1645 and BAB1_1646 genes are predicted to be part of an operon containing 6 genes, BAB1_1645 to BAB1_1650 http://www.microbesonline.org/operons/gnc359391.html. Four of these proteins were detected in our studies, although only BAB1_1645, -46 and -48 were found in the same experimental condition.
Conclusions
Mass spectrometry has proven to be a valuable tool to identify and correct genomic annotation errors in the study of microorganisms [33–37]. We performed a proteomics analysis of B. abortus 2308 proteins expressed upon extracellular and intracellular growth conditions to validate existing gene predictions at the protein level, to acquire useful information on B. abortus 2308 expressed proteins and to identify and correct inaccurately annotated ORFs. We were able to confirm the expression of over 300 previously unreported proteins and five pseudogenes, and corrected two wrongly assigned translation start sites. Taken together, these findings further demonstrate that computational genomic annotation errors can be corrected using proteomics. This will lead to improved databases and thus better protein identification and functional annotation.
Methods
Brucella abortus protein preparation for mass spectrometry analysis
Four types of B. abortus 2308 samples were prepared: outer membranes, cytosols, intracellular bacteria isolated from infected RAW264.7 macrophages and extracellular bacteria from overnight cultures. Outer membrane samples were prepared and processed for mass spectrometry analysis as previously described [16]. Cytoplasmic fractions were prepared as described previously [38]. Briefly, bacteria grown in tryptic soy broth (Difco) in 2-liter flasks on an orbital shaker and harvested by centrifugation in sealed cups at 7,000 × g for 20 min. The thick slurry of bacteria were suspended in 10 mM phosphate-buffered saline (pH 7.2) was passed twice through a French press (Pressure Cell 40 K, Aminco; SLM Instruments Inc., Urbana, Ill.) at an internal pressure of 35,000 lb/in2. The homogenate was digested with 50 mg of DNase II type V and RNase A per ml (Sigma) for 18 h at 37°C and fractionated by ultracentrifugation. The cell envelopes in the bottom of the tube removed and the cytoplasmic fractions in the supernatant, filtered, lyophilized and characterized as described previously [39]. Intracellular bacteria were isolated from RAW264.7 macrophages 3, 20 and 44 hours post-infection as previously described [17]. Proteins were extracted from intracellular and extracellular bacteria using the same method and digested for mass spectrometry as previously described [17].
Liquid Chromatography - Mass Spectrometry (LC-MS)
Peptide digests were analyzed by liquid chromatography coupled to mass spectrometry (LC-MS) as described [40]. Briefly, the samples were injected onto a reversed-phase column (Jupiter C18, Phenomenex, Torrance, CA) for HPLC separation. For LC-MS survey scans, the mass spectra were acquired over 400-1600 Da at a rate of 1 spectrum/second. Peptide sequencing was achieved by targeted and shotgun LC-MS/MS. For MS/MS scans, the mass range was 50-2000 Da, and each spectrum was acquired in 2 seconds. For LC-MS/MS, the duty cycle was one survey scan followed by one product ion scan (MS/MS).
Protein identification
Protein identification was done by submitting LC-MS/MS spectra to Mascot software (MatrixScience, Boston, MA) and searching against custom protein databases (see below). The parameters used for the Mascot search and protein homology clustering were previously detailed [16]. No multidimensional fingerprinting method was used. Annotation for each protein was performed using ExPASy Proteomics tools http://us.expasy.org/tools/#proteome, Kegg GenomeNet Database Service http://www.genome.jp/ and literature mining of orthologous genes and proteins.
Protein databases
The databases were composed of protein sequences obtained from the National Center for Biotechnology Information (NCBI) protein database (for B. abortus 2308, NC_007618 and NC_007624; for B. melitensis 16 M, NC_003317 and NC_003318; for Mus musculus, all protein sequences contained under taxonomy ID 10090) and of B. abortus 2308 "pseudoproteins" corresponding to the custom translation of pseudogenes. Genomic regions corresponding to the 316 entries annotated as pseudogenes in NCBI were directly translated and added to the database. Additionally, the ORF Finder tool from NCBI was used to determine other possible protein sequences corresponding to the pseudogenes. The ORF search was done by including 0 to 200 bp upstream or downstream from these regions. All resulting ORFs spanning the entire pseudogene sequence were kept. Ribosome binding sites were mapped when possible according to the sequence described in reference [41]. A total of 471 translated protein sequences were added to the NCBI databases.
Validation of mass spectrometry results
Sequences assigned to MS/MS spectra of peptides, which were mapped to pseudogenes or to genomic regions annotated as untranslated regions, were manually validated. For proteins identified by a single peptide, manual validation of the spectra was performed for peptide sequences having a Mascot score below 45.
Prediction of protein localization
The localization of newly demonstrated proteins was predicted using PSORTb version 2.0.4 http://www.psort.org/psortb/index.html, CELLO version 2.5 http://cello.life.nctu.edu.tw/ and PSLpred http://www.imtech.res.in/raghava/pslpred/index.html. For a localization to be assigned, a minimum of 2 of the 3 predictions had to match.
References
Moreno E, Moriyon I: The Genus Brucella. The Prokaryotes. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrant E. 2006, New York: Springer-Verlag, 315-456. full_text.
Mantur BG, Amarnath SK: Brucellosis in India - a review. J Biosci. 2008, 33: 539-547. 10.1007/s12038-008-0072-1.
Bouza E, Sanchez-Carrillo C, Hernangomez S, Gonzalez MJ: Laboratory-acquired brucellosis: a Spanish national survey. J Hosp Infect. 2005, 61: 80-83. 10.1016/j.jhin.2005.02.018.
Scholz HC, Hubalek Z, Sedlacek I: Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int J Syst Evol Microbiol. 2008, 58: 375-382. 10.1099/ijs.0.65356-0.
Ross HM, Foster G, Reid RJ, Jahans KL, Macmillan AP: Brucella species infection in sea-mammals. Vet Rec. Edited by: Ross H. 1994, 134: 359-10.1136/vr.134.14.359-b.
Ewalt DR, Payeur JB, Martin BM, Cummins DR, Miller WG: Characteristics of a Brucella species from a bottlenose dolphin (Tursiops truncatus). J Vet Diagn Invest. 1994, 6: 448-452.
Cloeckaert A, Verger JM, Grayon M: Classification of Brucella spp. isolated from marine mammals by DNA polymorphism at the omp2 locus. Microbes Infect. 2001, 3: 729-738. 10.1016/S1286-4579(01)01427-7.
Paulsen IT, Seshadri R, Nelson KE: The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci USA. 2002, 99: 13148-13153. 10.1073/pnas.192319099.
DelVecchio VG, Kapatral V, Redkar RJ: The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci USA. 2002, 99: 443-448. 10.1073/pnas.221575398.
Halling SM, Peterson-Burch BD, Bricker BJ: Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J Bacteriol. 2005, 187: 2715-2726. 10.1128/JB.187.8.2715-2726.2005.
Chain PS, Comerci DJ, Tolmasky ME: Whole-genome analyses of speciation events in pathogenic Brucellae. Infect Immun. 2005, 73: 8353-8361. 10.1128/IAI.73.12.8353-8361.2005.
Wattam AR, Williams KP, Snyder EE: Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J Bacteriol. 2009, 191: 3569-3579. 10.1128/JB.01767-08.
Audic S, Lescot M, Claverie JM, Scholz HC: Brucella microti: the genome sequence of an emerging pathogen. BMC Genomics. 2009, 10: 352-10.1186/1471-2164-10-352.
Crasta OR, Folkerts O, Fei Z, Mane SP, Evans C, Martino-Catt S, Bricker B, Yu G, Du L, Sobral BW: Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One. 2008, 3: e2193-10.1371/journal.pone.0002193.
Rajashekara G, Glasner JD, Glover DA, Splitter GA: Comparative whole-genome hybridization reveals genomic islands in Brucella species. J Bacteriol. 2004, 186: 5040-5051. 10.1128/JB.186.15.5040-5051.2004.
Lamontagne J, Butler H, Chaves-Olarte E: Extensive cell envelope modulation is associated with virulence in Brucella abortus. J Proteome Res. 2007, 6: 1519-1529. 10.1021/pr060636a.
Lamontagne J, Forest A, Marazzo E: Intracellular adaptation of Brucella abortus. J Proteome Res. 2009, 8: 1594-1609. 10.1021/pr800978p.
Yu CS, Lin CJ, Hwang JK: Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13: 1402-1406. 10.1110/ps.03479604.
Bhasin M, Garg A, Raghava GPS: PSLpred: prediction of subcellular localization of bacterial proteins. Bioinformatics. 2005, 21: 2522-2524. 10.1093/bioinformatics/bti309.
Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FSL: PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005, 21: 617-623. 10.1093/bioinformatics/bti057.
Connolly JP, Comerci D, Alefantis TG: Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics. 2006, 6: 3767-3780. 10.1002/pmic.200500730.
Klinke S, Zylberman V, Bonomi HR, Haase I, Guimarães BG, Braden BC, Bacher A, Fischer M, Goldbaum FA: Structural and kinetic properties of lumazine synthase isoenzymes in the order Rhizobiales. J Mol Biol. 2007, 26: 664-680. 10.1016/j.jmb.2007.08.021.
Zylberman V, Klinke S, Haase I, Bacher A, Fischer M, Goldbaum FA: Evolution of vitamin B2 biosynthesis: 6,7-dimethyl-8-ribityllumazine synthases of Brucella. J Bacteriol. 2006, 188: 6135-6142. 10.1128/JB.00207-06.
Robertson GT, Roop RM: The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol Microbiol. 1999, 34: 690-700. 10.1046/j.1365-2958.1999.01629.x.
Bellefontaine AF, Pierreux CE, Mertens P, Vandenhaute J, Letesson JJ, De Bolle X: Plasticity of a transcriptional regulation network among alpha-proteobacteria is supported by the identification of CtrA targets in Brucella abortus. Mol Microbiol. 2002, 43: 945-960. 10.1046/j.1365-2958.2002.02777.x.
Manterola L, Guzmán-Verri C, Chaves-Olarte E, Barquero-Calvo E, de Miguel MJ, Moriyón I, Grilló MJ, López-Goñi I, Moreno E: BvrR/BvrS-controlled outer membrane proteins Omp3a and Omp3b are not essential for Brucella abortus virulence. Infect Immun. 2007, 75: 4867-4874. 10.1128/IAI.00439-07.
Tibor A, Wansard V, Bielartz V, Delrue RM, Danese I, Michel P, Walravens K, Godfroid J, Letesson JJ: Effect of omp10 or omp19 deletion on Brucella abortus outer membrane properties and virulence in mice. Infect Immun. 2002, 70: 5540-5546. 10.1128/IAI.70.10.5540-5546.2002.
Essenberg RC, Sharma YK: Cloning of genes for proline and leucine biosynthesis from Brucella abortus by functional complementation in Escherichia coli. J Gen Microbiol. 1993, 139: 87-93.
Castañeda-Roldán EI, Ouahrani-Bettache S, Saldaña Z, Avelino F, Rendón MA, Dornand J, Girón JA: Characterization of SP41, a surface protein of Brucella associated with adherence and invasion of host epithelial cells. Cell Microbiol. 2006, 8: 1877-1887. 10.1111/j.1462-5822.2006.00754.x.
Valderas MW, Alcantara RB, Baumgartner JE, Bellaire BH, Robertson GT, Ng WL, Richardson JM, Winkler ME, Roop RM: Role of HdeA in acid resistance and virulence in Brucella abortus 2308. Vet Microbiol. 2005, 107: 307-312. 10.1016/j.vetmic.2005.01.018.
Essenberg RC: Cloning and characterization of the glucokinase gene of Brucella abortus 19 and identification of three other genes. J Bacteriol. 1995, 177: 6297-6300.
Wang R, Prince JT, Marcotte EM: Mass spectrometry of the M. smegmatis proteome: protein expression levels correlate with function, operons, and codon bias. Genome Res. 2005, 15: 1118-1126. 10.1101/gr.3994105.
Brunner E, Ahrens CH, Mohanty S, Baetschmann H, Loevenich S, Potthast F, Deutsch EW, Panse C, de Lichtenberg U, Rinner O, Lee H, Pedrioli PG, Malmstrom J, Koehler K, Schrimpf S, Krijgsveld J, Kregenow F, Heck AJ, Hafen E, Schlapbach R, Aebersold R: A high-quality catalog of the Drosophila melanogaster proteome. Nat Biotechnol. 2007, 25: 576-583. 10.1038/nbt1300.
Gupta N, Tanner S, Jaitly N, Adkins JN, Lipton M, Edwards R, Romine M, Osterman A, Bafna V, Smith RD, Pevzner PA: Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Res. 2007, 17: 1362-1377. 10.1101/gr.6427907.
Merrihew GE, Davis C, Ewing B, Williams G, Käll L, Frewen BE, Noble WS, Green P, Thomas JH, MacCoss MJ: Use of shotgun proteomics for the identification, confirmation, and correction of C. elegans gene annotations. Genome Res. 2008, 18: 1660-1669. 10.1101/gr.077644.108.
Mandel MJ, Stabb EV, Ruby EG: Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics. 2008, 9: 138-10.1186/1471-2164-9-138.
Deshayes C, Perrodou E, Gallien S, Euphrasie D, Schaeffer C, Van-Dorsselaer A, Poch O, Lecompte O, Reyrat JM: Interrupted coding sequences in Mycobacterium smegmatis: authentic mutations or sequencing errors. Genome Biol. 2007, 8: R20-10.1186/gb-2007-8-2-r20.
Aragón V, Díaz R, Moreno E, Moriyón I: Characterization of Brucella abortus and Brucella melitensis native haptens as outer membrane O-type polysaccharides independent from the smooth lipopolysaccharide. J Bacteriol. 1996, 178: 1070-
Moriyon I, Berman DT: Effects of nonionic, ionic, and dipolar ionic detergents and EDTA on the Brucella cell envelope. J Bacteriol. 1982, 152: 822-
Lucero NE, Jacob NO, Ayala SM, Escobar GI, Tuccillo P, Jacques I: Unusual clinical presentation of brucellosis caused by Brucella canis. J Med Microbiol. 2005, 54: 505-508. 10.1099/jmm.0.45928-0.
Dricot A, Rual JF, Lamesch P, Bertin N, Dupuy D, Hao T, Lambert C, Hallez R, Delroisse JM, Vandenhaute J, Lopez-Goñi I, Moriyon I, Garcia-Lobo JM, Sangari FJ, Macmillan AP, Cutler SJ, Whatmore AM, Bozak S, Sequerra R, Doucette-Stamm L, Vidal M, Hill DE, Letesson JJ, De Bolle X: Generation of the Brucella melitensis ORFeome version 1.1. Genome Res. 2004, 14: 2201-10.1101/gr.2456204.
Acknowledgements
This work was funded by the NIAID/NIH contract HHSN266200400056C.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JL designed and coordinated the study, analyzed the data and wrote the manuscript. MB participated in the data analysis and manuscript writing. AF performed the mass spectrometry experiments and peptide validations. ACM participated in the data analysis. NN performed the protein identification steps. FT participated in the protein identification steps. IM participated in the data analysis and manuscript writing. EM participated in the data analysis and manuscript writing. EP conceived of the study and participated in manuscript writing and study coordination. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2009_2894_MOESM1_ESM.PDF
Additional file 1: Proteins newly demonstrated in B. abortus 2308. Each entry is represented by a gene locus tag, description of the protein and the sequences of the peptides measured. Proteins are organized by predicted subcellular localization. (PDF 110 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lamontagne, J., Béland, M., Forest, A. et al. Proteomics-based confirmation of protein expression and correction of annotation errors in the Brucella abortus genome. BMC Genomics 11, 300 (2010). https://doi.org/10.1186/1471-2164-11-300
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-300