
Abstract
Background
Using a combination of pyrosequencing and conventional Sanger sequencing, the complete genome sequence of the recently described novel Brucella species, Brucella microti, was determined. B. microti is a member of the genus Brucella within the Alphaproteobacteria, which consists of medically important highly pathogenic facultative intracellular bacteria. In contrast to all other Brucella species, B. microti is a fast growing and biochemically very active microorganism with a phenotype more similar to that of Ochrobactrum, a facultative human pathogen. The atypical phenotype of B. microti prompted us to look for genomic differences compared to other Brucella species and to look for similarities with Ochrobactrum.
Results
The genome is composed of two circular chromosomes of 2,117,050 and 1,220,319 base pairs. Unexpectedly, we found that the genome sequence of B. microti is almost identical to that of Brucella suis 1330 with an overall sequence identity of 99.84% in aligned regions. The most significant structural difference between the two genomes is a bacteriophage-related 11,742 base pairs insert only present in B. microti. However, this insert is unlikely to have any phenotypical consequence. Only four protein coding genes are shared between B. microti and Ochrobactrum anthropi but impaired in other sequenced Brucella. The most noticeable difference between B. microti and other Brucella species was found in the sequence of the 23S ribosomal RNA gene. This unusual variation could have pleiotropic effects and explain the fast growth of B. microti.
Conclusion
Contrary to expectations from the phenotypic analysis, the genome sequence of B. microti is highly similar to that of known Brucella species, and is remotely related to the one of O. anthropi. How the few differences in gene content between B. microti and B. suis 1330 could result in vastly different phenotypes remains to be elucidated. This unexpected finding will complicate the task of identifying virulence determinants in the Brucella genus. The genome sequence of B. microti will serve as a model for differential expression analysis and complementation studies. Our results also raise some concerns about the importance given to phenotypical traits in the definition of bacterial species.
Similar content being viewed by others


Background
The genus Brucella comprises important mammal and human pathogens. Low infectious doses (10 to 100 bacteria, [1]), transmission through aerosols, and a difficult treatment of the disease by antibiotics, have led Brucella to be classified as potential bioterrorism agents. The genus Brucella [2] belongs to the family Brucellaceae within the order Rhizobiales of the Alphaproteobacteria. Ochrobactrum, a soil living facultative human pathogen, is the most closely related genus [3] with a 16S rRNA gene sequence [Genbank: U70978] more than 98% identical to that of Brucella spp.
Characterized Brucella comprise six classical Brucella species, B. melitensis, B. abortus, B. ovis, B. canis, B. suis, and B. neotomae, two species of marine mammal origin, namely B. pinnipedialis and B. ceti, and the recently described species B. microti and B. inopinata. B. microti was initially isolated from systemically diseased common voles (Microtus arvalis) in the Czech Republic [4]. More recently, it has also been isolated from red foxes in Lower Austria [5] and even directly from soil in the same geographical area [6]. Importantly, B. microti is thus the only Brucella species with a known reservoir outside of its mammalian host. Recent and frequent isolations of B. microti from different animals from different geographical regions indicate that B. microti could constitute an emerging pathogen. All Brucella species are genetically highly related, exhibiting identical 16S rRNA and rec A gene sequences [7]. In fact, DNA-DNA hybridization studies, today's gold standard for bacterial species delineation, suggest that all Brucella spp. should be unified into a single species (B. melitensis) and not be regarded as different species [8]. The high genetic relatedness of all Brucella species was further confirmed by whole genome sequencing [9–11].
Although Brucella spp. are facultative intracellular bacteria adapted to specific mammalian hosts, the whole genome sequence of B. suis revealed a clear similarity to that of soil bacteria associated with plants, such as Agrobacterium and Rhizobium [11]. It was therefore speculated that the mammal host-adapted Brucella species evolved from a plant-associated soil living ancestor. The recent isolation of B. microti directly from soil supported this hypothesis. In addition to its persistence in soils, B. microti is unique in sharing other phenotypic traits with its closest phylogenetic relative Ochrobactrum. In particular, B. microti is biochemically highly active, at odds with other Brucella species, but a common feature among Ochrobactrum species. We therefore speculated that B. microti might represent an intermediary stage in Brucella evolution, closer to the Brucella/Ochrobactrum common ancestor. To substantiate this hypothesis, the whole genome sequence of B. microti was determined and compared to that of other Brucella and to the genome sequence of O. anthropi. Differences in gene content that could explain B. microti phenotypic peculiarities were carefully studied.
Results
The genome sequence of Brucella microti CCM 4915T was determined (25× coverage) by shotgun analysis using the GS-FLX pyrosequencing technology and direct Sanger sequencing of remaining gaps. The genome is composed of two circular chromosomes of lengths 2,117,050 bp (base-pairs) and 1,220,319 bp. We predicted the presence of 3,291 protein coding genes, 55 tRNAs and 9 ribosomal RNAs. A comparison with the other Brucella genomes revealed the presence of 60 pseudogenes.
Comparison of genome structures
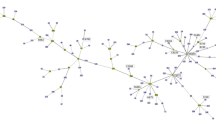
Dotplots (Figure 1) of the chromosomes of B. microti against the 8 Brucella genome sequences available at the time of writing and the genome sequence of O. anthropi show that: i) the overall genome structures of Brucella are remarkably conserved, ii) Brucella genomes are markedly different from that of O. anthropi and iii) based on this genome structure only, B. microti is more similar to B. suis 1330, B. canis and B. melitensis 16M than to the other genomes with which it has at least one major difference. Consistently, phylogenetic reconstruction based on a set of 1,486 orthologous genes clearly placed B. microti closer to B. suis 1330 than to any other Brucella (Figure 2). We thus based our subsequent analyses on the detailed comparison of B. microti and B. suis 1330.

Dotplots of 7 Brucella and Ochrobactrum anthropi genomes against the two chromosomes of B. microti. B. microti chromosomes are in abscissa of each plot and the corresponding chromosomes of target genomes are in ordinate. In chromosome 2 plots, the 12 kbp region specific to B. microti is circled. Plots for B. abortus S19 and B. abortus 9–941 are not shown because of their similarity to the plot for B. melitensis biovar abortus 2308. In the case of O. anthropi, the dotplots of the two chromosomes of B. microti against the 2 large chromosomes of O. anthropi are shown. O. anthropi plasmids are not shown as they have no similarity with B. microti chromosomes.

Phylogenetic representation of the alignment of 1,486 groups of orthologous genes from 8 available Brucella genome sequences and that of O. anthropi. The long branch leading to O. anthropi has been shortened. Even though B. suis and B. microti are not found within the same clade, they both exhibit a slower evolution rate than most other Brucella species (as shown by their short branch length) resulting in a high overall similarity at the genome sequence level.
Global alignment of the chromosomes
The global alignment of B. suis 1330 and B. microti chromosomes revealed their almost perfect co-linearity. On chromosome 1, we identified 270 indels (insertions or deletions) of one or more base-pairs in the alignment (163 insertions in B. microti and 107 in B. suis 1330). On chromosome 2, we identified 135 indels (75 insertions in B. microti and 60 insertions in B. suis 1330). Insertions in B. microti range in size from 1 bp (61 occurrences) to 845 bp on chromosome 1, and from 1 bp (32 occurrences) to 11,742 bp on chromosome 2 (the second largest insertion being 1071 bp long). Insertions in B. suis 1330 range in size from 1 bp (57 occurrences) to 844 bp on chromosome 1 and from 1 bp (29 occurrences) to 510 bp on chromosome 2. The global alignment of the chromosomes is given as Additional Files 1 and 2 and the list of indels between B. microti and B. suis 1330 is reported in Additional file 3.
Out of a total of 3,312,414 aligned positions, we observed 5213 SNPs, corresponding to 0.16% nucleotide difference between the two species. In comparison, alignment of B. suis 1330 with B. melitensis 16M revealed a total of 7307 SNP for a total of 3,237,820 aligned nucleotides (0.23%).
The 12 kbp insertion
The largest difference between the genome of B. suis 1330 and that of B. microti is a 12 kbp insertion (11,742 bp), that is unique to B. microti (Figure 1). This insertion is located in chromosome 2 (position 1,038,869 to 1,050,617), between a gene for tRNALeu and gene BMI_II1054, ortholog of BRA1053 in B. suis 1330 genome. A significant, but partial match to a phage integrase (BMI_II1048) was detected at an extremity of this island (best hit in the genome of Hyphomonas neptunium, YP_760435.1, 59% identity). This putative integrase is flanked by an ORF (BMI_II1049) showing similarity with phage excisionases. Altogether, these findings suggest that the 12 kbp genome insertion originates from a yet unidentified phage. Moreover, the target for this insertion was a GGCACCA motif, found both at the end of the tRNA, and identically conserved at the end of the insert. Accordingly, most of the other ORFs within this island exhibit remote similarities with phage ORFs in protein sequence databases. However, these phage-derived genes now appear defective, as the sequence homology only partially cover the ORFs. Interestingly, a predicted gene in this region (BMI_II1051) has a best match against O. anthropi (Oan_0220). Finally, this B. microti specific genomic island bears no similarity with the 26.5 kbp island recently reported in the genome of B. ovis [12]. The origin of this insertion remains unclear since its tentative detection by a recently developed specific PCR [6] with Brucella phage DNA (Tb, Wb, F1 and F25) as template was negative. Therefore, it seems unlikely that the insertion has derived from the most commonly known Brucella phages.
Insertion sequences
Insertion sequences of the IS711/IS6501 family are commonly found in Brucella, but other IS elements are also present scattered through the genomes. In B. microti, we identified 13 copies of IS711 elements, 2 IS2020 elements, 2 IS1953 elements, 2 ISBm1 elements, one of them disrupted by the insertion of an IS711 element, one ISBm2 element and one ISBm3 element. The main difference with respect to B. suis 1330 is the number of IS711 insertion sequences. IS711 Insertion sequences which are not present in both B. microti and B. suis 1330 are listed in Table 1. Seven are present in B. microti only and one in B. suis 1330 only. Interestingly, those changes are all found on the large chromosome. The insertion site between BMI_I1295 and cob L has already been reported in B. microti [7], and the insertion site between tRNAMet and omp 28 (BMI_I1490/BR1475) was previously thought to be specific of Brucella isolated from marine mammals [13]. One of those insertions impairs a hypothetical protein in B. suis 1330 (BR0722) and in two other cases, the insertion is located in the 5' upstream-region of a gene (thr S/BMI_I1076 and BMI_I1903) but does not seem to disrupt a putative promoter [14]. None of the insertions thus appear to disrupt existing operon structures or inactivate putative promoters.
Tandem Repeat Analysis
The MLVA-15 typing systems [15], based on multilocus VNTR (Variable Number of Tandem Repeats) and extended later into MLVA-16 [16] used a set of primer pairs (Listed in Table 2, [15]), selected for their variability in a set of three Brucella genomes. We searched for these primers in the genome of B. microti and reported their positions and the theoretical lengths of the corresponding PCR products in Additional file 4. Of the 80 primer pairs reported in [15], 78 had a perfect match to the B. microti genome sequence, and two exhibited one mismatch in one of the primers (Bruce07 and Bruce24). The genomic positions leading to VNTR amplicons of different sizes between B. microti and B. suis 1330, as well as other indels, are listed in Additional file 3. On the 16 MLVA-16 primer pairs, 14 yield PCR fragments of different sizes. This explains the differences in the MLVA profile of B. microti with respect to other Brucella
Ribosomal RNA
Brucella contains three copies of the ribosomal RNA operon. The sequences of 5S and 16S ribosomal RNA genes are nearly identical among all species. In contrast, we noticed that the 23S ribosomal RNA gene sequence of B. microti differs markedly from that of the other Brucella (Additional file 5), and that the differences map into the intervening sequence (IVS I) localized in the helix 9, cleaved by the RNase III during the maturation of the 23S rRNA. Interestingly, the B. microti IVS region differs from that of O. anthropi, but is very similar to that of another fast growing Brucella, B. inopinata sp. nov. strain B01 [17], whose genome sequence is in the form of 55 contigs on the Patric Web site [18]. Curiously, the similarity between B. microti and B. inopinata B01 is limited to this particular 23S rRNA region, whereas the rest of their genome sequences are notably different, for instance showing different theoretical PCR fragment size for 2 of the 3 MLVA-16 primer pairs that we were able to locate on the preliminary data (we looked only for exact matches to the primers). Additionally, a tree built from a fragment of Brucella genomes (corresponding to the 10,000 first nucleotides of B. microti chromosome 1 sequence) showed that B. inopinata B01 likely diverged before B. microti from the other Brucella (Additional file 6). The alignment of the Brucella IVS I regions is presented in Figure 3 and an analysis of the secondary structure of the IVS regions in Figure 4. In this figure, the black boxes correspond to previously identified conserved motifs [19]. The nucleotides around the putative cleavage site (red box) are well conserved in all Brucella, except for B. microti where the CUG consensus cleavage motif is split. In contrast to this IVS I region which is shared between B. microti and B. inopinata B01, a second region, between positions 446 and 523 in the alignment (Figure 3 and Additional file 5), is perfectly identical between O. anthropi and B. microti, and distinct from B. inopinata B01 which is identical to other Brucella in this region.

Alignment of the region around intervening sequence (IVS I) in selected Brucella and O. anthropi. Fragments of the alignment where all sequences are identical are not shown. The whole alignment with numbering is given in Additional file 5. Sequence fragments shared by B. microti and Brucella sp. B01 are in blue. Sequence fragments shared by B. microti and O. anthropi are in green. Other regions are in red. Fragments of the alignment highlighted in yellow correspond to the terminal nucleotides of the secondary structures represented in Figure 4.

Predicted secondary structure of the intervening sequence (IVS I). The predicted secondary structure of the intervening sequences (IVS I) of the 23S ribosomal RNA in B. microti and Brucella sp. B01 (left), B. suis (middle) and O. anthropi (right). Arrows with a dark head represent conserved cleavage sites. Arrows with a thin head represent unconserved cleavage sites. In O. anthropi, only the lower part of the cleavage motif is present.
Gene content analysis and comparison with other Brucella and Ochrobactrum anthropi
Genes differentially annotated or presenting notable differences between B. microti and B. suis 1330 are listed in Table 2 (for the genes discussed in the text) and Additional file 7 (for a complete version). We reported the status of each of these genes in the other Brucella and O. anthropi, a s well as the nature of the change at the sequence level that turns the intact gene in B. microti or B. suis 1330 into a pseudogene in the other organisms. In agreement with the observation (Figure 2) that the branch length leading to B. microti is shorter than those leading to other Brucella, we note (Additional file 7) that the number of pseudogenes in B. microti is half that of B. suis 1330.
Genes specific to B. microti and O. anthropi and impaired in all other Brucella
In term of metabolic capabilities, B. microti (Table 1 in [7]) was experimentally found much more similar to O. anthropi than to other Brucella. We thus examined the gene content of B. microti to identify genes shared with O. anthropi but impaired in all other sequenced Brucella. Only 4 such genes were found. Phylogenetic trees for those genes are shown in Additional file 8(A–D).
BMI_I149 has an ortholog in O. anthropi (Oant_0158) and is impaired in other Brucella. In B. suis 1330, the corresponding pseudogene is BR0146. Homologues for this gene are found in many Rhizobiales such as Bartonella or Mezorhizobium (Additional file 8A). The enzyme encoded by this gene, malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) [EC:1.1.1.40], is involved in pyruvate metabolism. The reaction is: (S)- malate + NADP+ = pyruvate + CO2 + NADPH. This gene has a paralog BMI_I1020, the orthologs of which are intact in all Brucella and Ochrobactrum (Additional file 8A).
BMI_I1566, corresponding to pseudogene BR1552 in B. suis 1330, is an aspartyl/asparaginyl beta-hydroxylase which is intact in B. microti and O. anthropi (Oant_1613) and impaired in other Brucella (Additional file 8B). The product of this gene [EC:1.14.11.16] catalyzes the reaction: peptide-L-aspartate + 2-oxoglutarate + O2 = peptide-3-hydroxy-L-aspartate + succinate + CO2. The succinate (or succinic acid), product of this reaction, is one of the compounds used in the physiological reactions that differentiate B. microti from other Brucella [7]. Succinate is involved in many metabolic pathways such as the citrate cycle (TCA cycle).
BMI_I1599, corresponding to pseudogene BR1586 in B. suis 1330 is intact in B. microti, impaired in other Brucella, and intact in O. anthropi (Oant_1582) (Additional file 8C). This gene is predicted to encode an extracellular solute-binding protein belonging to an ABC-type transport system probably involved in dipeptide transport. However, in the case of B. microti, the nearby dpp C gene encoding the permease component of this transport system is impaired (pseudogene BMI_I1597), with probable consequences on the whole transport system.
BMI_II978, encoding a transcriptional regulator of the MarR family is present in B. microti and O. anthropi (Oant_1375) and contains an internal STOP codon in other Brucella (Additional file 8D). It corresponds to pseudogene BRA0985 in B. suis 1330. This family of transcriptional regulators mediates the response to multiple environmental stresses and the resistance to multiple antibiotics.
Finally, the B. microti hydrolase BMI_I2199/BR2178, is similar to the one in O. anthropi, while its homologues in other Brucella species are much shorter, lacking the last 40 amino-acids, which is likely to have functional consequences. Its homologues in B. canis and B. suis ATCC 23365 are annotated as acetyltransferases, part of the acetoin cleaving system.
Genes intact in B. microti and O. anthropi and impaired in almost all other Brucella
Some genes are intact in B. microti and O. anthropi but are impaired in almost all Brucella, with some exceptions. For instance, the ATP-dependent helicase HtrB (BMI_I135), pseudogene BR0132 in B. suis 1330, presents a frameshift in all Brucella except B. ovis (BOV_0127) and is intact in O. anthropi. This is also the case for BMI_I947 corresponding to pseudogene BR0949 in B. suis 1330. This gene exhibits an internal STOP codon in all Brucella except B. abortus S19 (BabS19_I09030) and is present in O. anthropi (Oant_2240). This gene encodes a transport protein sometimes annotated as outer membrane protein E. The gene encoding sarcosine dehydrogenase (BMI_I1332, EC:1.5.99.1), corresponding to pseudogene BR1320 in B. suis 1330, is intact in B. microti and O. anthropi but impaired in other Brucella except Brucella suis ATCC 23445. This enzyme catalyzes the following reaction: sarcosine + acceptor + H2O = glycine + formaldehyde + reduced acceptor. The flagellar motor switch gene, fli G (BMI_II122, pseudogene BRA0122 in B. suis 1330), which is apparently intact in B. ovis, B. microti and O. anthropi, is impaired in other Brucella.
Other potentially significant differences in gene content between B. microti and other Brucella
Glutamate metabolism
Quite a number of genes involved in glutamate metabolism are missing from certain Brucella species.
The gene of glutamate decarboxylase beta (gad B, BMI_II334) contains an in-frame STOP codon in B. suis 1330 and appears to be damaged in many Brucella (except for B. ovis and B. suis ATCC 23445). The product of this gene catalyzes the decarboxylation of glutamate into gamma-aminobutyric acid (GABA) and CO2. The nearby glutamate/gamma-aminobutyrate antiporter (gad C, BMI_II335) is also impaired in B. suis 1330, presenting an internal frameshift as in B. ovis, B. canis and B. suis ATCC 23445. Taken together, these two genes are only intact in B. microti (O. anthropi does not have any of those genes). These two genes are involved in the generation of a proton motive force in Lactobacillus strains [20], but also in acid resistance mechanisms [21–23].
Also involved in glutamate and other amino-acids metabolisms, the B. suis 1330 gene (BRA1118) encoding a N-acetylglucosamine kinase presents a frameshift and is thus impaired in B. microti (BMI_II1124). This enzyme [EC:2.7.1.59] catalyzes the following reaction: ATP + N-acetyl-D-glucosamine = ADP + N-acetyl-D-glucosamine 6-phosphate.
The proline/dehydrogenase/delta-1-pyrroline-5-carboxylate dehydrogenase (BMI_II715) is a pseudogene in B. suis 1330 (BRA0722). This enzyme [EC:1.5.1.12] catalyzes the reaction: (S)-1-pyrroline-5-carboxylate + NAD(P)+ + 2 H2O = L-glutamate +NAD(P)H + H+ and is involved in glutamate, arginine and proline metabolism.
Membrane proteins presenting significant differences between B. microti and other Brucella
We also observed a number of changes in genes encoding membrane proteins. The outer membrane protein encoding gene BMI_I75 exhibits large differences in size, being larger in B. microti than in its counterparts in other Brucella. A small conductance mechanosensitive ion channel protein encoding gene (BMI_I1045) has frameshifts in B. suis 1330 (pseudogene BR1042), B. ovis and B. canis and is intact in B. microti and O. anthropi. A second small conductance mechanosensitive ion channel protein encoding gene (BMI_I1334) is impaired in B. suis 1330 (pseudogene BR1322), B. canis and B. ovis
An outer membrane autotransporter, encoded by BMI_II170, is paradoxically most similar to the protein Meso_3532 in Mesorhizobium sp. BNC1 than to its homologues in all Brucella and Ochrobactrum.
The B. microti membrane protein Bme3, encoded by BMI_II422, corresponds to pseudogene BRA0425 in B. suis 1330. Genes at this locus have been reported to be involved in polysaccharide synthesis [24].
The ortholog of gene BMI_II547, encoding a cell wall surface hemagglutinin in B. microti, presents a premature STOP codon in B. suis 1330 (BRA0553). Remarkably, a fragment of this gene, position 534037 to 534117 in B. microti chromosome 2 sequence, is found multiple times in Brucella genomes. Its abundance (blastn search [25], no filter, E-value < 10-30) ranges between 38 times in B. ovis to 13 times in B. microti and B. suis ATCC 23445 and 6 to 7 times in the other Brucella studied in this work. This repeated element could reveal useful for typing purpose.
B. microti gene BMI_I1862 is quite different in length with respect to its counterparts in other Brucella. The B. microti version has a 126 nucleotides insert. This gene encodes a protein with a Yada-like C-terminal domain characteristic of a family of surface exposed bacterial proteins. O. anthropi has no homolog for this gene.
Gene clusters encoding the components of the flagellum
All genes involved in flagella assembly and present in O. anthropi are apparently intact in B. microti, in contrast to the situation in other Brucella where at least one gene is impaired (Table 3). However, there is a significant difference in flagella gene organization between Ochrobactrum spp. and Brucella spp. In O. anthropi, the flagella genes are essentially found in two clusters. One contains flh B, fli G, fli N, fli M, mot A, flg F, fli I, flg B, flg C, fli E, flg G, flg A, flg I, flg H, fli L, fli P and the other contains fli R, flh A, fli Q, flg D, flb T, fla F, flg L, flg K, flg E, fli K, mot C, mot B, fli F, fli C. In Brucella, a 15 kb insert containing genes not related to flagella interrupts the first group between genes fli I and flg B (Additional file 9).
Survival in the soil
Paulsen et al. [11] noticed that the B. suis operon (BRA0636-BRA0647), encoding an homoprotocatechuate pathway, is widely distributed among diverse soil microorganisms and may contribute to the survival of the bacteria outside of its host. Interestingly, 3 of these genes present a frameshift in B. microti, suggesting that they do not significantly contribute to its survival in the soil. In contrast, an other operon cited in the same context (BRA1155-BRA1162) [11], encoding a beta-ketoadipate pathway is intact in both B. suis 1330 and B. microti.
Discussion
Voges-Proskauer reaction
In terms of metabolic capabilities, B. microti (Table 1 in [7]) was found much more similar to O. anthropi than to other Brucella. One of the tests for which B. microti was positive is the Voges-Proskauer reaction. The Voges-Proskauer reaction demonstrates the possibility for an organism to produce acetoin and 2,3-butanediol and is an established biochemical test for distinguishing wide classes of bacteria. The pathway leading to the production of acetoin is described for instance in [26]. It involves the transformation of pyruvate into alpha-acetolactate by the acetolactate synthase, then the conversion of alpha-acetolactate to acetoin by the alpha-acetolactate decarboxylase followed by the conversion of acetoin to 2,3-butanediol. Brucella species possess genes for the acetolactate synthase 3 (BMI_I1399, large subunit and BMI_I1400, small subunit) and the acetolactate synthase 2 (large subunit only BMI_II939). The conversion of acetoin to 2,3-butanediol is performed by the homolog of als O (VC1591) in Vibrio cholera which is BMI_I1134. A gene apparently missing from this pathway is the homolog of als D (VC1589) in V. cholera, the alpha-acetolactate decarboxylase. However, as reported for Bacillus subtilis [27], this reaction can occur spontaneously at low pH, in absence of als D. This suggests that known Brucella species have all the enzymes necessary to produce acetoin. Their negative testing for the Voges-Proskauer reaction might thus be due to an indirect cause, such as the lack of a sufficient supply of pyruvate. Like O. anthropi, but in contrast with other Brucella, B. microti possesses two paralogs of malate dehydrogenase (BMI_I149 and BMI_I1020) which catalyzes a reaction producing pyruvate. It is thus tempting to speculate that this enzyme duplication might be linked to the positivity of the Voges-Proskauer reaction in B. microti.
Proton motive force and acid-resistance mechanism
Of notable interest is the presence of the gene tandem BMI_II334 and BMI_II335 encoding a glutamate decarboxylase beta GadB and a glutamate/gamma-aminobutyrate antiporter GadC, respectively. These proteins might give B. microti the potential to generate a proton motive force from the decarboxylation of glutamate. This capacity might have been lost in other Brucella where either of these genes are found impaired. In Lactococcus lactis [21], Shigella flexneri [22] and Escherichia coli [23] GadB and GadC were shown to participate in a glutamate-dependent acid resistance mechanism. Acid resistance mechanisms allow enteric pathogens to overcome acid stress in the gastrointestinal tract of their host [28]. In the case of B. microti, this system may help the bacteria to survive in acid soils. More importantly, it might also play a role in intracellular survival, as within hosts macrophages, Brucella species reside in a low pH environment, where the importance of the gad B, gad C, hde A gene cluster as an acid resistance locus as already been suggested [29, 30].
Motility
Due to their lifestyle, many intracellular bacteria have lost their capacity to produce functional flagella [31]. Despite the presence of numerous flagella genes [32], Burkholderia mallei is non-motile whereas Burkholderia pseudomallei and Burkholderia thailendensis are motile. The lack of motility of B. mallei was traced back to a 65 kb insertion within the fli P gene as well as a frameshift in the flagellar motor gene mot B [32]. Similarly, Yersinia pseudotuberculosis and Yersinia enterocolitica are motile [33] whereas Yersinia pestis KIM is non-motile in spite of the presence of a nearly complete set of flagella genes, but with a truncated gene for transcription factor FlhD. In each of the above cases, the host adapted species are non-motile (B. mallei/Y. pestis) whereas the others are motile. Although all available Brucella genomes possess genes for the flagellum complex, Brucella are non-motile and display no flagellum under standard conditions. However, under specific conditions during early exponential growth phase, B. melitensis 16M has been reported to express some of the key genes of the flagellar apparatus and assemble a sheathed flagellum which is required for virulence in a mouse infection model [34, 35].
The motor switch gene fli G is the gene most often found impaired in Brucella (Table 3), with exceptions in B. ovis and B. microti. This rotor protein is essential for the assembly and the function of the flagellar motor [36]. In B. ovis, fli M, encoding the flagellar C ring protein is impaired (as in B. abortus S19, B. abortus 9–941 and B. melitensis biovar Abortus 2308), explaining the lack of flagella in B. ovis. In contrast with these Brucella species, B. microti does not present a readily apparent defect among the proteins constituting the flagellar assembly complex. Its lack of a visible flagellum could thus be due to more subtle causes. In this context, we noticed that one of the two flagella gene clusters of O. anthropi is split in Brucella genomes (Additional file 9). Such a modification might have disrupted the coordinated expression of those flagella genes. However, given the integrity of its individual flagellar genes, B. microti might express a sheathed flagellum under specific conditions, as observed for B. melitensis 16M [34, 35].
The 12 kbp insertion
The specific 12 kbp genomic island of probable phage origin is of obvious interest for identification purpose, and has already been used for the recovery of B. microti from soil samples [6]. Blast (tblastn) searches of nucleotide sequences within this island against the Genbank database found best matches in O. anthropi, Nitrobacter hamburgensis X14 and other Rhizobiales. This suggests that this element originated from a phage commonly infecting Rhizobiales. In a recent study [37], evidence of horizontal gene transfers in Brucella genomes were reported. Those SARs (Shared Anomalous Regions) consist of regions 2 to 19 kb long, sometimes flanked on one side by a tRNA and on the other side by a fragment of that tRNA, as found in the B. microti genomic island. Although some important genes (e.g. Type IV secretion or LPS [lipopolysaccharide] synthesis genes) apparently entered the Brucella genomes through this mechanism, the B. microti island appears devoid of functional genes. Paradoxically, the most striking genome structure difference between B. microti and other Brucella is probably of no phenotypic consequence.
23S ribosomal rna
The cleavage of the 23S rRNA IVSs by RNase III results in a specific fragmentation pattern. In B. microti, the 5' fragment is predicted to be 127 bp long, the IVS I (helix 9) 153 bp long and the 3' fragment 2.6 kbp long. The IVS is composed of palindrome sequences and repeated motifs forming stable secondary structures (stem-loop) (Figure 4) [38]. The complementary ends of the IVS are highly conserved between Brucella species and correspond to inverted DNA repeats characteristic of mobile genetic elements [38]. In B. microti, the 23S maturation leading to the IVS removal may not occur because of sequence variations at the cleavage site. In Salmonella typhimurium, RNase III- mutants are viable, suggesting that the removal of the intervening sequence is not required for 23S function [39, 40]. The fragmentation of 23S rRNA during post-transcriptional processing of precursor rRNA has been reported for Brucella [41], however, no information is available concerning B. microti. Concerning the role of the 23S rRNA fragmentation, results in Salmonella [42] indicate that the degree of fragmentation correlates with the amount of 23S rRNA degradation in stationary phase, allowing for a post-transcriptional control of ribosome production. Knowing that two fast-growing Brucella isolates, B. microti and B. inopinata B01, share a similar change in their 23S rRNA structure, it is tempting to speculate that this change, impeding IVS removal, could have an impact on their growth rates. It is surprising that two phylogenetically distant species of Brucella, as revealed by sequence analysis of VNTR regions and phylogenetic analysis (Additional file 6), have exactly the same IVS sequence. This finding pleads either for an ancestral nature of this IVS or for a recent exchange of 23S rRNA sequences between B. inopinata B01 and B. microti. However, part of the 23S rRNA gene sequence of B. microti was found identical to that of O. anthropi, this region not being shared with B. inopinata B01. This mosaic structure of the 23S rRNA gene sequence of B. microti, partly identical to that of B. inopinata B01 and partly identical to that of O. anthropi confirms the existence of horizontal gene transfers in Brucella.
Brucella virulence genes
Major virulence factors of Brucella that have been characterized include the Type IV secretion system [43, 44], LPS [45], Omp25 [46], and the BvrS-BvrR two component system [47]. All of these virulence gene sequences were found to be identical in B. microti and B. suis 1330. Interestingly, two ORFs involved in LPS biosynthesis as well as Omp25 are found in one of the nine genomic islands (GI-2) which were revealed by whole-genome micro-array analysis in B. melitensis 16M [48, 49].
Phylogeny of the genus Brucella
The newly determined genome sequence of B. microti allows us to revisit the phylogeny of the Brucella genus. Our results (Figure 2) are in agreement with previous works [37, 50] which regroup on one hand B. suis 1330, B. suis ATCC 23445 and B. canis, and on the other hand B. abortus S19, B. abortus 9–941 and B. melitensis biovar Abortus 2308 together with B. melitensis 16M. B. microti and B. ovis separated earlier from those groups. It was claimed [50] that the B. ovis lineage was "basal" to the rest of the Brucella lineage, dating the divergence of most Brucella species from their common ancestor 86,000 to 296,000 years ago. Our analyses now indicate that the B. microti lineage is at least as "basal" as B. ovis, and anticipating on the completion of its genome sequence, the divergence of B. inopinata B01 will probably appear even more ancestral.
Conclusion
Unexpectedly in the light of its numerous phenotypic peculiarities, B. microti was found to have a genome sequence very close to that of previously characterized Brucella species. With respect to its closest relative, B. suis 1330, the genome sequence of B. microti was found 99.84% identical in perfectly aligned regions, and no less than 99% identical taking into account insertion-deletions. Although we identified at least 4 genes impaired in all studied Brucella but intact in B. microti and O. anthropi, it is unlikely that these differences alone could explain the numerous Ochrobactrum-like phenotypic traits exhibited by B. microti, as well as its increased virulence. Additionally, we have identified an unexpected alteration of the 23S rRNA gene sequence of B. microti, also shared by an other fast growing novel Brucella species B. inopinata sp. nov. strain B01. This sequence variation could have a pleiotropic effect by increasing the number of ribosomes per bacterial cell and thus enhance the overall translation activity. Finally, the phenotypic characteristics of B. microti might also be due to genome variations in non-coding (regulatory) regions influencing the expression level of numerous genes. Our study appears to be the first encountering a new limitation of the comparative genomic approach in the elucidation of phenotypic traits: usually even close (e.g. virulent vs. non virulent) strains display too many differences in their genomes to allow the straightforward identification of the relevant genes. Here, we experienced the opposite problem, being left with too few gene differences to explain a large number of phenotypic variations. A differential analysis of the transcriptome of B. microti vs. that of its closest genomic relative B. suis 1330 as well as complementation studies should help reveal how their quasi-identical gene content could result in two microorganisms exhibiting so many differences in their metabolic behaviors, life-styles, and virulence.
Methods
Strain information and Accession Numbers
Brucella microti CCM 4915T genome sequence is deposited in the Genbank database under accession numbers CP001578 for the large chromosome and CP001579 for the small chromosome.
Shotgun sequencing and finishing
An initial shotgun sequencing using GS-FLX produced 414,552 reads of average size 213 bp. Assembly with the Newbler program resulted in 90 contigs above 500 bp. Newbler assembler contigs were converted into artificial Sanger reads. Based on the extensive similarity between the genomes of Brucella, we were able to determine tentative primer pairs that were tested by PCR and then used for sequencing by Sanger technology, allowing us to bridge the gaps between contigs. The resulting sequences were subsequently added to the assembly using the Phred/Phrap/Consed software packages [51–53].
Genomic comparisons
The genomic sequences reported in this article were compared to the available genomic sequences of B. melitensis 16M [NC_003317 (large chromosome) and NC_003318 (small)] [9], B. suis 1330 [NC_004310 (large) and NC_004311 (small)] [11], B. abortus 9–941 [NC_006932 (large) and NC_006933 (small)] [10], B. melitensis biovar Abortus 2308 [NC_007618 (large) and NC_007624 (small)], B. ovis [NC_009505 (large) and NC_009504(small)] [12], B. suis ATCC 23445 [NC_010169 (large) and NC_010167 (small)], B. canis ATCC 23365 [NC_010103 (large) and NC_010104 (small)] and Brucella abortus S19 [NC_010742 (large) and NC_010740 (small)]. In addition we used the genomic sequence of Ochrobactrum anthropi A TCC 49188 [NC_009671 (93,589 bp), NC_009672 (57,138 bp), NC_009668 (1,895,911 bp), NC_009667 (2,887,297 bp), NC_009670 (101,491 bp), NC_009669 (170,351 bp)]. Dotplots of the genome sequences were performed using programs from the MUMMER package [54].
Alignment of genomic sequences
Alignment of chromosomes of B. suis 1330 and B. microti were performed using software from the LAGAN Toolkit [55]. An in-house program to superimpose annotation on the alignment was used to have a finer view of the position of the differences with respect to the annotated genomes, and to aid in the annotation of B. microti. The alignment files are available as Additional Files 1 and 2. A list of indels computed from those alignments is presented as Additional file 3.
Genome annotation
Due to the close similarity between the genomes of B. suis 1330 and B. microti, a large use was made of the full alignment of sequences that is presented in Additional Files 1 and 2 as well as similarity searches against the complete genomes of other Brucella. Insertion sequences that were not present in the genome of B. suis 1330 were identified with the help of the Biotoul IS-Finder [56, 57].
Identification of candidate pseudogenes
Candidate pseudogenes either in B. microti or B. suis 1330 were identified as follows. First, the most similar and intact version of the homologous ORF was selected among one of the available Brucella genomes. This sequence was then used to query (using tblastn [25], no filter, E-value < 10-50) the remaining Brucella and Ochrobactrum genomes to detect the eventual absence or disruption of the homologous gene (premature STOP codon, frameshift). 170 cases of premature terminations or frameshifts were found and are listed in Additional file 7. Fifty of them correspond to ORFs altered in B. microti and intact in at least one other Brucella, while 120 correspond to genes presumably functional in B. microti and altered in its closest relative B. suis 1330.
Brucella phylogeny
The proteins of the B. microti chromosomes were grouped with those of 8 other Brucella and O. anthropi in a file containing 33,053 proteins. A blastp [25] search (Evalue < 10-5) of this set against itself yielded a table of blast results which was used to cluster proteins using a Markov chain clustering algorithm [58]. We selected clusters containing a single protein from each of the initial organism. This procedure resulted in 1,486 clusters of genes present in each Brucella and in O. anthropi. The 1,486 Brucella core proteins were first aligned individually using MUSCLE (v3.7)[59]. Poorly aligned regions were discarded by GBLOCKS (v0.91b) [60] using the Phylogeny.fr platform [61]. The resulting alignments were used as a guide to align the corresponding DNA sequences on a codon basis. After cleaning up the nucleotide alignments for poorly aligned regions, the 1,486 multiple alignments were concatenated in a single alignment of 431,655 codons. The phylogenetic tree was reconstructed using the maximum likelihood method implemented in the PhyML program (v3.0 aLRT) [62]. The default nucleotide substitution model (HKY85) was selected assuming an estimated proportion of invariant sites and 4 gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data as well as the transition/transversion ratio. Reliability for internal branches was assessed using the aLRT test (SH-Like). Graphical representation and edition of the phylogenetic tree were performed with TreeDyn (v198.3) [63] and MEGA3 [64].
Phylogeny of 4 genes unique to B. microti and O. anthropi
Only 4 genes common to B. microti and O. anthropi but impaired in all other Brucella were identified. These genes are a priori the most likely to contribute to the Ochrobactrum-like phenotypic traits of B. microti. The corresponding protein sequences were searched against the nr database with the blastp program (E-value < 10-5) using the Phylogeny.fr platform [61]. The homologous sequences were aligned with MUSCLE (v3.7) configured for highest accuracy (MUSCLE with default settings). After alignment, ambiguous regions (i.e. containing gaps and/or poorly aligned) were removed with GBLOCKS (v0.91b) using the following parameters: minimum length of a block after gap cleaning of 10, no gap positions were allowed in the final alignment, all segments with contiguous non-conserved positions bigger than 8 were rejected and minimum number of sequences for a flank position: 85%. The phylogenetic tree was reconstructed using the maximum likelihood method implemented in the PhyML program (v3.0 aLRT). The default substitution model (WAG) was selected assuming an estimated proportion of invariant sites and 4 gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data. Reliability for internal branch was assessed using the aLRT test (SH-Like). Graphical representation and edition of the phylogenetic tree were performed with TreeDyn (v198.3).
References
Valderas MW, Barrow WW: Establishment of a method for evaluating intracellular antibiotic efficacy in Brucella abortus-infected Mono Mac 6 monocytes. J Antimicrob Chemother. 2007, 61: 128-134. 10.1093/jac/dkm433.
Meyer KF, Shaw EB: A comparison of the morphologic, cultural, and biochemical characteristics of B. abortus and B. melitensis. J Infect Dis. 1920, 27: 173-184.
Velasco J, Romero C, López-Goñi I, Leiva J, Díaz R, Moriyón I: Evaluation of the relatedness of Brucella spp. and Ochrobactrum anthropi and description of Ochrobactrum intermedium sp. nov., a new species with a closer relationship to Brucella spp. Int J Syst Bacteriol. 1998, 48 (Pt 3): 759-768.
Hubálek Z, Scholz HC, Sedlácek I, Melzer F, Sanogo YO, Nesvadbová J: Brucellosis of the common vole (Microtus arvalis). Vector Borne Zoonotic Dis. 2007, 7: 679-87. 10.1089/vbz.2007.0143.
Scholz HC, Hofer E, Vergnaud G, Le Flèche P, Whatmore AM, Dahouk SA, Pfeffer M, Krüger M, Cloeckaert A, Tomaso H: Isolation of Brucella microti from Mandibular Lymph Nodes of Red Foxes, Vulpes vulpes, in Lower Austria. Vector Borne Zoonotic Dis. 2009, 9: 153-156. 10.1089/vbz.2008.0036.
Scholz HC, Hubalek Z, Nesvadbova J, Tomaso H, Vergnaud G, Le Flèche P, Whatmore AM, Al Dahouk S, Krüger M, Lodri C, Pfeffer M: Isolation of Brucella microti from soil. Emerg Infect Dis. 2008, 14: 1316-7. 10.3201/eid1408.080286.
Scholz HC, Hubalek Z, Sedlácek I, Vergnaud G, Tomaso H, Al Dahouk S, Melzer F, Kämpfer P, Neubauer H, Cloeckaert A, Maquart M, Zygmunt MS, Whatmore AM, Falsen E, Bahn P, Göllner C, Pfeffer M, Ayodeji B, Busse H, Nöckler K: Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int J Syst Evol Microbiol. 2008, 58: 375-82. 10.1099/ijs.0.65356-0.
Verger J, Grimont F, Grimont PAD, Grayon M: Brucella, a Monospecific Genus as Shown by Deoxyribonucleic Acid Hybridization. Int J Syst Bacteriol. 1985, 35: 292-295.
DelVecchio VG, Kapatral V, Redkar RJ, Patra G, Mujer C, Los T, Ivanova N, Anderson I, Bhattacharyya A, Lykidis A, Reznik G, Jablonski L, Larsen N, D'Souza M, Bernal A, Mazur M, Goltsman E, Selkov E, Elzer PH, Hagius S, O'Callaghan D, Letesson J, Haselkorn R, Kyrpides N, Overbeek R: The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci USA. 2002, 99: 443-8. 10.1073/pnas.221575398.
Halling SM, Peterson-Burch BD, Bricker BJ, Zuerner RL, Qing Z, Li L, Kapur V, Alt DP, Olsen SC: Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J Bacteriol. 2005, 187: 2715-26. 10.1128/JB.187.8.2715-2726.2005.
Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ, Umayam L, Brinkac LM, Beanan MJ, Daugherty SC, Deboy RT, Durkin AS, Kolonay JF, Madupu R, Nelson WC, Ayodeji B, Kraul M, Shetty J, Malek J, Van Aken SE, Riedmuller S, Tettelin H, Gill SR, White O, Salzberg SL, Hoover DL, Lindler LE, Halling SM, Boyle SM, et al: The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci USA. 2002, 99: 13148-53. 10.1073/pnas.192319099.
Tsolis RM, Seshadri R, Santos RL, Sangari FJ, Lobo JMG, de Jong MF, Ren Q, Myers G, Brinkac LM, Nelson WC, DeBoy RT, Angiuoli S, Khouri H, Dimitrov G, Robinson JR, Mulligan S, Walker RL, Elzer PE, Hassan KA, Paulsen IT: Genome Degradation in Brucella ovis Corresponds with Narrowing of Its Host Range and Tissue Tropism. PLoS ONE. 2009, 4: e5519-10.1371/journal.pone.0005519.
Cloeckaert A, Grayon M, Grepinet O: An IS711 Element Downstream of the bp26 Gene Is a Specific Marker of Brucella spp. Isolated from Marine Mammals. Clin Diagn Lab Immunol. 2000, 7: 835-839.
SoftBerry – BPROM – Prediction of bacterial promoters. [http://linux1.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb]
Le Flèche P, Jacques I, Grayon M, Al Dahouk S, Bouchon P, Denoeud F, Nöckler K, Neubauer H, Guilloteau LA, Vergnaud G: Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 2006, 6: 9-10.1186/1471-2180-6-9.
Al Dahouk S, Le Flèche P, Nöckler K, Jacques I, Grayon M, Scholz HC, Tomaso H, Vergnaud G, Neubauer H: Evaluation of Brucella MLVA typing for human brucellosis. J Microbiol Methods. 2007, 69: 137-45. 10.1016/j.mimet.2006.12.015.
Scholz HC, Nöckler K, Göllner C, Bahn P, Vergnaud G, Tomaso H, Al Dahouk S, Kämpfer P, Cloeckaert A, Marquart M, Zygmunt MS, Whatmore AM, Pfeffer M, Huber B, De BK: Brucella inopinata sp. nov., isolated from a breast implant infection. Int J Syst Evol Microbiol. 2009
PATRIC: PathoSystems Ressource Integration Center. [http://patric.vbi.vt.edu/]
Evguenieva-Hackenberg E, Klug G: RNase III processing of intervening sequences found in helix 9 of 23S rRNA in the alpha subclass of Proteobacteria. J Bacteriol. 2000, 182: 4719-29. 10.1128/JB.182.17.4719-4729.2000.
Higuchi T, Hayashi H, Abe K: Exchange of glutamate and gamma-aminobutyrate in a Lactobacillus strain. J Bacteriol. 1997, 179: 3362-3364.
Sanders JW, Leenhouts K, Burghoorn J, Brands JR, Venema G, Kok J: A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol Microbiol. 1998, 27: 299-310. 10.1046/j.1365-2958.1998.00676.x.
Waterman SR, Small PL: Identification of sigma S-dependent genes associated with the stationary-phase acid-resistance phenotype of Shigella flexneri. Mol Microbiol. 1996, 21: 925-40. 10.1046/j.1365-2958.1996.00058.x.
Hersh BM, Farooq FT, Barstad DN, Blankenhorn DL, Slonczewski JL: A glutamate-dependent acid resistance gene in Escherichia coli. J Bacteriol. 1996, 178: 3978-81.
Vizcaíno N, Cloeckaert A, Zygmunt MS, Fernández-Lago L: Molecular Characterization of a Brucella Species Large DNA Fragment Deleted in Brucella abortus Strains: Evidence for a Locus Involved in the Synthesis of a Polysaccharide. Infect Immun. 1999, 67: 2700-2712.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-402. 10.1093/nar/25.17.3389.
Kovacikova G, Lin W, Skorupski K: Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol Microbiol. 2005, 57: 420-33. 10.1111/j.1365-2958.2005.04700.x.
Renna MC, Najimudin N, Winik LR, Zahler SA: Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J Bacteriol. 1993, 175: 3863-75.
Richard HT, Foster JW: Acid resistance in Escherichia coli. Adv Appl Microbiol. 2003, 52: 167-86. full_text.
Ficht TA: Intracellular survival of Brucella: defining the link with persistence. Vet Microbiol. 2003, 92: 213-223. 10.1016/S0378-1135(02)00367-X.
Roop RM, Gee JM, Robertson GT, Richardson JM, Ng W, Winkler ME: Brucella stationary-phase gene expression and virulence. Annu Rev Microbiol. 2003, 57: 57-76. 10.1146/annurev.micro.57.030502.090803.
Toft C, Fares MA: The Evolution of the Flagellar Assembly Pathway in Endosymbiotic Bacterial Genomes. Mol Biol Evol. 2008, 25: 2069-2076. 10.1093/molbev/msn153.
Nierman WC, DeShazer D, Kim HS, Tettelin H, Nelson KE, Feldblyum T, Ulrich RL, Ronning CM, Brinkac LM, Daugherty SC, Davidsen TD, Deboy RT, Dimitrov G, Dodson RJ, Durkin AS, Gwinn ML, Haft DH, Khouri H, Kolonay JF, Madupu R, Mohammoud Y, Nelson WC, Radune D, Romero CM, Sarria S, Selengut J, Shamblin C, Sullivan SA, White O, Yu Y, et al: Structural flexibility in the Burkholderia mallei genome. Proceedings of the National Academy of Sciences of the United States of America. 2004, 101: 14246-14251. 10.1073/pnas.0403306101.
Bliska JB, Ryndak MB, Grabenstein JP: Type III Secretion Systems in Yersinia pestis and Yersinia pseudotuberculosis. Bacterial Genomes and Infectious Diseases. Edited by: Chan VL, Sherman PM, Bourke B. 2006, Totowa, New Jersey: Humana Press, 213-226.
Fretin D, Fauconnier A, Köhler S, Halling S, Léonard S, Nijskens C, Ferooz J, Lestrate P, Delrue R, Danese I, Vandenhaute J, Tibor A, DeBolle X, Letesson J: The sheathed flagellum of Brucella melitensis is involved in persistence in a murine model of infection. Cell Microbiol. 2005, 7: 687-698. 10.1111/j.1462-5822.2005.00502.x.
Leonard S, Ferooz J, Haine V, Danese I, Fretin D, Tibor A, de Walque S, De Bolle X, Letesson J: FtcR Is a New Master Regulator of the Flagellar System of Brucella melitensis 16M with Homologs in Rhizobiaceae. J Bacteriol. 2007, 189: 131-141. 10.1128/JB.00712-06.
Lloyd SA, Blair DF: Charged residues of the rotor protein FliG essential for torque generation in the flagellar motor of Escherichia coli. Journal of Molecular Biology. 1997, 266: 733-744. 10.1006/jmbi.1996.0836.
Wattam AR, Williams KP, Snyder EE, Almeida NF, Shukla M, Dickerman AW, Crasta OR, Kenyon R, Lu J, Shallom JM, Yoo H, Ficht TA, Tsolis RM, Munk C, Tapia R, Han CS, Detter JC, Bruce D, Brettin TS, Sobral BW, Boyle SM, Setubal JC: Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J Bacteriol. 2009, 191: 3569-79. 10.1128/JB.01767-08.
Evguenieva-Hackenberg E: Bacterial ribosomal RNA in pieces. Mol Microbiol. 2005, 57: 318-25. 10.1111/j.1365-2958.2005.04662.x.
Mattatall NR, Sanderson KE: RNase III deficient Salmonella typhimurium LT2 contains intervening sequences (IVSs) in its 23S rRNA. FEMS Microbiol Lett. 1998, 159: 179-85. 10.1111/j.1574-6968.1998.tb12858.x.
Nicholson AW: Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol Rev. 1999, 23: 371-90. 10.1111/j.1574-6976.1999.tb00405.x.
Hsu D, Zee YC, Ingraham J, Shih LM: Diversity of cleavage patterns of Salmonella 23S rRNA. J Gen Microbiol. 1992, 138: 199-203.
Hsu D, Shih LM, Zee YC: Degradation of rRNA in Salmonella strains: a novel mechanism to regulate the concentrations of rRNA and ribosomes. J Bacteriol. 1994, 176: 4761-5.
Boschiroli ML, Ouahrani-Bettache S, Foulongne V, Michaux-Charachon S, Bourg G, Allardet-Servent A, Cazevieille C, Lavigne J, Liautard JP, Ramuz M, O'Callaghan D: Type IV secretion and Brucella virulence. Veterinary Microbiology. 2002, 90: 341-348. 10.1016/S0378-1135(02)00219-5.
Rouot B, Alvarez-Martinez M, Marius C, Menanteau P, Guilloteau L, Boigegrain R, Zumbihl R, O'Callaghan D, Domke N, Baron C: Production of the Type IV Secretion System Differs among Brucella Species as Revealed with VirB5- and VirB8-Specific Antisera. Infect Immun. 2003, 71: 1075-1082. 10.1128/IAI.71.3.1075-1082.2003.
Cardoso P, Macedo G, Azevedo V, Oliveira S: Brucella spp noncanonical LPS: structure, biosynthesis, and interaction with host immune system. Microbial Cell Factories. 2006, 5: 13-10.1186/1475-2859-5-13.
Edmonds MD, Cloeckaert A, Elzer PH: Brucella species lacking the major outer membrane protein Omp25 are attenuated in mice and protect against Brucella melitensis and Brucella ovis. Vet Microbiol. 2002, 88: 205-221. 10.1016/S0378-1135(02)00110-4.
López-Goñi I, Guzmán-Verri C, Manterola L, Sola-Landa A, Moriyón I, Moreno E: Regulation of Brucella virulence by the two-component system BvrR/BvrS. Vet Microbiol. 2002, 90: 329-339. 10.1016/S0378-1135(02)00218-3.
Rajashekara G, Glasner JD, Glover DA, Splitter GA: Comparative Whole-Genome Hybridization Reveals Genomic Islands in Brucella Species. J Bacteriol. 2004, 186: 5040-5051. 10.1128/JB.186.15.5040-5051.2004.
Rajashekara G, Covert J, Petersen E, Eskra L, Splitter G: Genomic Island 2 of Brucella melitensis Is a Major Virulence Determinant: Functional Analyses of Genomic Islands. J Bacteriol. 2008, 190: 6243-6252. 10.1128/JB.00520-08.
Foster JT, Beckstrom-Sternberg SM, Pearson T, Beckstrom-Sternberg JS, Chain PSG, Roberto FF, Hnath J, Brettin T, Keim P: Whole Genome-Based Phylogeny and Divergence of the Genus Brucella. J Bacteriol. 2009, 191: 2864-70. 10.1128/JB.01581-08.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8: 195-202.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-85.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-94.
Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S: Versatile and open software for comparing large genomes. Genome Biology. 2004, 5: R12-10.1186/gb-2004-5-2-r12.
Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S: LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13: 721-31. 10.1101/gr.926603.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M: ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34: D32-6. 10.1093/nar/gkj014.
IS Finder. [http://www-is.biotoul.fr/]
Enright AJ, Van Dongen S, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30: 1575-84. 10.1093/nar/30.7.1575.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-7. 10.1093/nar/gkh340.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, 17: 540-52.
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J, Guindon S, Lefort V, Lescot M, Claverie J, Gascuel O: Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36: W465-9. 10.1093/nar/gkn180.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Chevenet F, Brun C, Bañuls A, Jacq B, Christen R: TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006, 7: 439-10.1186/1471-2105-7-439.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-63. 10.1093/bib/5.2.150.
Phylogeny.fr: Robust Phylogenetic Analysis For The Non-Specialist. [http://www.phylogeny.fr/]
Acknowledgements
We acknowledge the use of the PACA-Bioinfo Bioinformatics platform. Work in the IGS laboratory is supported by recurrent funding from the CNRS, and grants (to J-M. C.) from the National Genopole Network, the PACA Region, and IBiSA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HCS designed the project, performed the experiments and wrote the manuscript. SA, ML and JMC analyzed the data and wrote the manuscript.
Electronic supplementary material
12864_2009_2236_MOESM1_ESM.txt
Additional file 1: Supplementary File 1: Whole genome alignment with superimposed annotation of the large chromosomes of B. microti and B. suis 1330. Letter x in the consensus line denotes an indel or a point mutation. In protein coding regions, the reading frame is indicated. (TXT 14 MB)
12864_2009_2236_MOESM2_ESM.txt
Additional file 2: Supplementary File 2: Whole genome alignment with superimposed annotation of the small chromosomes of B. microti and B. suis 1330. Letter x in the consensus line denotes an indel or a point mutation. In protein coding regions, the reading frame is indicated. (TXT 8 MB)
12864_2009_2236_MOESM3_ESM.pdf
Additional file 3: Supplementary Table 1: List of indels between the genomes of B. microti and B. suis 1330. The coordinates in B. microti and B. suis are given as well as the localization and the putative effect of the indels on gene products. (PDF 206 KB)
12864_2009_2236_MOESM4_ESM.pdf
Additional file 4: Supplementary Table 2: Distinctive VNTR typing of B. microti as confirmed by the genome sequence. For each tandem repeat we listed the primer pair, the theoretical length of the amplimer in B. suis 1330, B. melitensis 16M, B. abortus 9–941 and B. microti CCM 4915, and the location of the theoretical PCR product in the genome of B. microti. (PDF 61 KB)
12864_2009_2236_MOESM5_ESM.pdf
Additional file 5: Supplementary Figure 2: Alignment of the 23S ribosomal RNA gene sequences in O. anthropi and other Brucella studied in this work. In addition, we included the sequences of B. ceti and Brucella inopinata B01. Abbreviations: oan, O. anthropi; bcs, B. canis; bmt, B. suis ATCC 23445; bms, B. suis 1330; bmb, B. abortus 9–941; bmf, B. melitensis biovar abortus 2308; bmc, B. abortus S19; bce, B. ceti; bme, B. melitensis 16M; bov, B. ovis; bmi, B. microti and B01, Brucella inopinata B01. (PDF 2 MB)
12864_2009_2236_MOESM6_ESM.pdf
Additional file 6: Supplementary Figure 1: Phylogenetic representation of the alignment of the regions corresponding to the first 10,000 nucleotides of B. microti genome sequence, showing that Brucella inopinata sp. nov. strain B01 diverged earlier than the other Brucella studied in this work. The sequence of B. ceti is also included. B. ceti and Brucella inopinata B01 sequences were obtained from the PATRIC web site [18]. (PDF 76 KB)
12864_2009_2236_MOESM7_ESM.pdf
Additional file 7: Supplementary Table 3: Extended list of orthologous genes exhibiting annotation differences between B. microti and B. suis 1330 (one, or both being annotated as pseudogene, or presenting notable difference). For each gene, we give its identification in B. microti and B. suis 1330 as well as its status in other Brucella. Abbreviations include: * for internal stop, a number indicates multiple stops, fs for frameshift, + for an intact sequence, Mult. Diffs for multiple difference, Mult. Fs for multiple frameshifts, NF for not found. (PDF 322 KB)
12864_2009_2236_MOESM8_ESM.pdf
Additional file 8: Supplementary Figure 4: Phylogenetic tree for the 4 genes conserved in B. microti and O. anthropi and impaired in the other Brucella . The trees were built using the Phylogeny.fr Web Server [65] using defaults settings. A) BMI_I149, malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) and its paralog BMI_I1020 intact in other Brucella; B) BMI_I1566, aspartyl/asparaginyl beta-hydroxylase; C) BMI_I1599, extracellular solute-binding protein belonging to an ABC-type transport system involved probably in dipeptide transport and D) BMI_II978, MarR family transcriptional regulator. (PDF 216 KB)
12864_2009_2236_MOESM9_ESM.pdf
Additional file 9: Supplementary Figure 3: Genomic representation of the region around the cluster of flagella assembly genes that is contiguous in O. anthropi and interrupted in Brucella. Intact genes are represented as black arrows, pseudogenes as red arrows. (PDF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Audic, S., Lescot, M., Claverie, JM. et al. Brucella microti: the genome sequence of an emerging pathogen. BMC Genomics 10, 352 (2009). https://doi.org/10.1186/1471-2164-10-352
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-352