
Abstract
Background
Identification of single nucleotide polymorphisms (SNPs) for specific genes involved in reproduction might improve reliability of genomic estimates for these low-heritability traits. Semen from 550 Holstein bulls of high (≥ 1.7; n = 288) or low (≤ −2; n = 262) daughter pregnancy rate (DPR) was genotyped for 434 candidate SNPs using the Sequenom MassARRAY® system. Three types of SNPs were evaluated: SNPs previously reported to be associated with reproductive traits or physically close to genetic markers for reproduction, SNPs in genes that are well known to be involved in reproductive processes, and SNPs in genes that are differentially expressed between physiological conditions in a variety of tissues associated in reproductive function. Eleven reproduction and production traits were analyzed.
Results
A total of 40 SNPs were associated (P < 0.05) with DPR. Among these were genes involved in the endocrine system, cell signaling, immune function and inhibition of apoptosis. A total of 10 genes were regulated by estradiol. In addition, 22 SNPs were associated with heifer conception rate, 33 with cow conception rate, 36 with productive life, 34 with net merit, 23 with milk yield, 19 with fat yield, 13 with fat percent, 19 with protein yield, 22 with protein percent, and 13 with somatic cell score. The allele substitution effect for SNPs associated with heifer conception rate, cow conception rate, productive life and net merit were in the same direction as for DPR. Allele substitution effects for several SNPs associated with production traits were in the opposite direction as DPR. Nonetheless, there were 29 SNPs associated with DPR that were not negatively associated with production traits.
Conclusion
SNPs in a total of 40 genes associated with DPR were identified as well as SNPs for other traits. It might be feasible to include these SNPs into genomic tests of reproduction and other traits. The genes associated with DPR are likely to be important for understanding the physiology of reproduction. Given the large number of SNPs associated with DPR that were not negatively associated with production traits, it should be possible to select for DPR without compromising production.
Similar content being viewed by others
Background
There is a negative genetic correlation between milk yield and fertility in dairy cattle [1–3]. Partly as a result, the large improvement in milk yield over the last 40 years was accompanied by a decline in fertility [4–6]. Genetic selection for fertility is hampered by low heritability. For example, the heritability for daughter pregnancy rate (DPR), the fertility trait most widely measured in the United States, has been estimated at 0.04% [2]. Genetic estimates of fertility can be improved by genome-wide single nucleotide polymorphism (SNP) arrays. Utilization of the BovineSNP50 chip from Illumina (San Diego, CA, USA) improved reliability for DPR [7, 8] but the low heritability and polygenic nature of the trait has meant that improvements in reliabilities achieved by incorporation of genomic information was less than for other traits. Thus, while the incorporation of information from the SNP50 chip increased reliability of DPR by 17% in Holsteins, this improvement was one of the lowest of the 12 traits examined [8].
One possible way to improve the accuracy of genomic estimates of fertility is to incorporate SNPs for specific genes involved in reproduction into SNP panels. The bovine genome contains over 20,000 genes, and over 14,000 of those do not contain a single SNP on the BovineSNP50 chip [9]. Incorporation of candidate gene SNPs into genomic tests for reproduction would allow selection of causative SNPs or SNPs physically more close to causative SNPs. Such an approach has been successful for improving ability to detect genomic associations with disease [10].
Many genes have been associated with reproduction in the dairy cow. Among these are SNPs related to in vitro fertilization or development, such as STAT5A[11], FGF2[12, 13] and PGR[14]), DPR (CAST[15]), sire conception rate including STAT5A[16], FGF2[16], and ITGB5[17], calving interval (GHR[18]), superovulation response (FSHR[19]), twinning rate (IGF1[20]) and incidence of still birth (NLRP9[21] and LEP[22]). In beef cattle, SNPs related to reproductive function include those in HSPA1A, associated with calving rate [23], and PAPPA2, associated with calving interval [24].
The previously mentioned SNPs only represent a small portion of the genes involved in reproductive processes. Recent studies have revealed genes whose expression in tissues or cells of importance to reproduction vary with reproductive status; these genes are candidates for containing SNPs that impact fertility. For example, genes were identified that were differentially regulated in the brain of cows displaying strong estrus compared to those displaying weak estrus [25], in the endometrium of heifers which produced viable embryos compared to those which produced non-viable embryos [26], and in biopsies from embryos that resulted in live calves as compared to embryos that died following embryo transfer [27]. Genetic variants in the genes differentially expressed in the aforementioned studies and others may be responsible for differences in fertility among animals.
The goal of the current study was to identify SNPs in candidate genes affecting reproductive processes. The approach was to evaluate effectiveness of SNPs in candidate genes for explaining genetic variation in DPR. Three types of SNPs were evaluated: SNPs previously reported to be associated with reproductive traits of dairy or beef cattle or physically close to genetic markers for reproduction, SNPs in genes that are well known to be involved in reproductive processes, and SNPs in genes reported to be differentially expressed between physiological conditions in a variety of tissues associated in reproductive function. As an additional goal, SNPs were also evaluated for their relationship to other traits. Given the negative genetic correlation between milk yield and reproduction [1–3], it was hypothesized that some SNPs associated with DPR would have an antagonistic relationship with production traits.
Methods
Selection of bulls
Straws and ampules of semen were obtained from 550 Holstein bulls born between 1962 and 2010. Bulls were chosen based on their predicted transmitting ability (PTA) and reliability for DPR. In particular, bulls were chosen to have either a high PTA for DPR (≥ 1.7) or low PTA for DPR (≤ −2) with reliability as high as possible. The PTA for the low DPR group (n = 262) ranged from −5.9 to −2 (average = −3.5), and the PTA for the high DPR group (n = 288) ranged from 1.7 to 5.3 (average = 2.87). Reliabilities ranged from 0.46 to 0.99 (3 bulls < 50%, 17 between 50 and 60%, 150 between 60 and 70%, 213 between 70 and 80%, 47 between 80 and 90%, and 120 greater than 90%). The distribution of reliabilities was similar between the low (average = 79%) and high (average = 77%) DPR groups. Predicted transmitting abilities for a variety of traits of the bulls are presented in Additional file 1: Table S1. Semen was obtained from the Cooperative Dairy DNA Repository [CDDR (Beltsville, MD, USA; 445 bulls)], Alta Genetics (Watertown, WI, USA; 38 bulls), Genex Cooperative Inc. (Shawano, WI, USA; 31 bulls), Taurus-service Inc. (Mehoopany, PA, USA; 26 bulls), Foundation Sires Inc. (Listowel, ON, CAN; 5 bulls), Accelerated Genetics (Baraboo, WI, USA, 2 bulls), Interglobe Genetics (Pontiac, IL, USA, 2 bulls), and Nebraska Bull Service (McCook, NE, USA, 1 bull). Five bulls were born in the 1960s, 15 in the 1970s, 54 in the 1980s, 154 in the 1990s, and 322 in the 2000s.
SNP discovery
The choice of 434 SNPs to be used for genotyping was made using a three-step process: candidate gene selection, SNP identification, and SNP selection. A list of candidate genes affecting reproduction was compiled using two methods. The first was to include genes commonly known to affect reproductive processes such as steroidogenesis (STAR, HSD17B3, etc.), follicular development (LHB, FSHB, etc.), oocyte maturation (BMP15, GDF9, etc.), and early embryonic development (CSF2, IGF1, etc.), as well as nutritional genes including orexins (NPY, HCRT, etc.) and anorexins (CCK, LEP, etc.). Furthermore, genes that were in physical proximity to SNPs related genetically to interval to insemination (IGFBP7, IRF9, etc. [28]) and 56 d non-return rate (BAIAP2, SCRN1, etc. [29]) were included. In addition, genes reported to be differentially expressed between physiological conditions in a variety of tissues associated with reproductive function were incorporated. This list included genes differentially regulated in the following conditions: the brain of cows displaying strong vs. weak estrus (CALCR, POMC, etc. [25]), embryos after cryopreservation (BAX, DSC2, etc. [29]), superovulated embryos compared to embryos from unstimulated dams (GOLGA4, KIT, etc. [30]), embryos which survived to term compared to embryos that died in vivo after embryo transfer (ATP5A1, OCLN, etc. [27, 31]), embryos treated with CSF2 (CACNA1G, MADD, etc. [32]) or IGF1 (COQ9, CREG1, etc. [33]) compared to control embryos, embryos cultured in vitro in the well-of-the-well system compared to embryos cultured in groups (CSNK1E, ZP4, etc. [34]), oocytes compared to 8-cell embryos (CLIC4, PDGFR, etc. [35]) and blastocysts (GJA1, TAF9, etc. [36]), oocytes at different stages of oocyte maturation (CPS1F, ZP2, etc. [37]), endometrium related to embryo survival (DGKA, BSP3, etc. [26]), endometrium in lactating cows compared to non-lactating cows (APBB1, ST13, etc. [38]) or pregnant cows compared to non-pregnant cows (ASL, GPLD1, etc. [38]), cumulus cells regulated by the LH surge (DHCR24, HAS2, etc. [39]), at different stages of oocyte maturation (AP3B1, CLU, etc. [40]), or from embryos produced in vivo embryos compared to embryos produced in vitro [LPL, MAGED1, etc. [41]), dominant follicles compared to subordinate follicles (CYP19A1, FST, etc. [42–45]), liver during the transition period (ACLY, PCCB, etc. [46]), mammary tissue during lactation (ABCA1, INSR, etc. [47]), and oviduct at diestrus compared to estrus (C3, OVGP1, etc. [48]).
Using the procedures described above, a total of 1532 candidate genes were identified. The SNPs in each of these genes were identified by querying the SNP database maintained by the National Center for Biotechnology Information (dbSNP; http://www.ncbi.nlm.nih.gov/snp). Then, SNPs were screened to only include those in the coding region of the gene which resulted in a nonsense, frameshift, or missense mutation. Of the 1532 genes screened, 553 genes containing a total of 1644 SNPs fit those criteria. In addition to these markers, SNPs previously linked to fertility were considered for inclusion. That list of candidate SNPs included CAST[15], FGF2[16], FSHR[19], GHR[18], HSPA1A[23], ITGB5[17], LEP[22], NLRP9[21], PAPPA2[24], PGR[14], SERPINA14[49], and STAT5A[11, 16].
In order to determine the final list of SNPs to be used in the assay, each SNP was graded based on primer designability and predicted change in protein function. Each SNP causing an amino acid change was evaluated for the likelihood that the SNP would change the structure of the encoded protein using an exchangeability matrix [50]. The average exchangeability value was calculated for each substitution of pairs of amino acids, and SNPs were ranked in order of exchangeability. For final selection of 434 SNPs, a maximum of one SNP per gene was selected. Nonsense mutations were selected first, then frameshifts, followed by SNPs with the lowest score in the exchangeability matrix (those most likely to cause a change in protein function). The selection criteria were also applied to SNPs already linked genetically to reproduction. Of the final selected SNPs, 5 were the exact SNPs used in the literature: STAT5A[11], FGF2[16], PGR[14], HSPA1A[23], and PAPPA2[24], and 7 SNPs were replaced with the best option using the criteria mentioned above (ITGB5, GHR, FSHR, NLRP9, LEP, CAST, and SERPINA14). The final list of genes used in the assay is shown in Additional file 1: Table S2 and the SNPs that were chosen from those genes are shown in Additional file 1: Table S3. The SNP panel included 10 nonsense, 22 frameshift, 397 missense, 1 synonymous, 3 intron region, and 1 promoter region SNPs.
SNP genotyping
Total DNA was extracted from each straw of semen using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Amount of double-stranded DNA was assessed using the Quant-itTM Picogreen® dsDNA kit (Invitrogen, Grand Island, NY, USA), and DNA was resuspended to a concentration of 50 ng/μL. Genotyping was performed by GeneSeek Inc. (Lincoln, NE, USA) using the Sequenom MassARRAY® system (iPLEX GOLD; Sequenom, San Diego, CA, USA) according to the manufacturer’s instructions. The technique is based on the analysis of DNA products using matrix-assisted laser desorption ionization time-of-flight mass spectrometry [51]. The region of DNA containing the SNP was amplified by PCR, a primer extension reaction was performed to generate allele-specific DNA products, and the size and amount of each allele-specific product was determined using chip-based mass spectrometry.
Quality control
Samples with call rates < 70% were removed from all analyses. The average call rate prior to removing those samples was 88.2%. After removing the failed samples, the average call rate was 91.2%. Reliability was assessed by duplicating 18 SNPs for every DNA sample, and by assaying 63 DNA samples twice. Of the duplicated SNPs, 16 were selected based on interest (CAST, CSF2, CYP19A1, FGF2, GHR, HSPA1A, IFNT, ITGB5, LEP, LHCGR, NALP9, PAPPA2, PGR, POU5F1, STAT5A, and UTMP) and the other two were selected based on poor primer designability (ETF1 and POMC). The primers for the duplicated SNPs were designed based on the sequence of the opposite DNA strand of where the original primer was designed. The duplicated DNA samples were randomly selected. There was 99.2% identity between SNPs duplicated within an assay and 98.6% identity between duplicated samples. After quality control was assessed, duplicated samples were merged. If any genotype at a given SNP did not match between samples, both genotypes were deleted and treated as a no call. Duplicated SNPs were merged in the same manner. The call rate after merging samples and SNPs was 91.5%.
Statistical analysis
Minor allele frequency (MAF) was determined using the FREQ procedure of SAS (V9.3; SAS Institute Inc., Cary, NC). Distributions of genotypes were tested for deviation from Hardy-Weinberg equilibrium (HWE) using a chi-square test. In addition, chi-square was used to determine whether MAF differed between high and low DPR bulls.
The association of genetic variants with each trait was evaluated using the MIXED procedure of SAS. The full model included:
where Y i is the deregressed PTA of the trait of interest for the ith bull (i = 1, 2, …, 550) , byrj is the fixed effect of the jth birth year (j = 1, 2, …, 5; where birth year is grouped by decade: 1960 to 2010) of the ith bull, β is the linear regression coefficient for the kth SNP, SNP k is the number of copies (k = 0, 1, or 2) of the major allele, POLY l is the random polygenic effect (including all available pedigree information) of the ith bull, and ϵ i is the random residual effect. The POLY l ~ Aσa2 and ϵ i ~ Iσe2, where A is the numerator relationship matrix, I is an identity matrix, σa2 is the additive genetic variance of the trait of interest, and σe2 is the residual error variance. All of the available pedigree information for each bull was used when modeling the covariance among the polygenic effects [52].
SNP effects were estimated using two analyses. In the first, genotype was considered a continuous variable to determine the allele substitution effect (the linear effect of the number of copies of the major allele; least-squares means represent values for 0,1 and 2 copies of the major allele). In the second, genotype was considered a categorical variable, and an orthogonal contrast was used to estimate dominance effects [(AA + aa)/2 vs. Aa]. SNPs in which the linear or dominance effect was P < 0.05 were noted. To control for multiple testing, false discovery rate was controlled for by calculating the Q value using the Q-value package in R [53]. The acceptable false discovery rate for the Q value analysis was chosen as 0.05.
Pathway analysis
The list of genes significantly related to DPR was subjected to pathway analysis using Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, http://www.ingenuity.com). The reference set was the Ingenuity Knowledge Base (genes only) and both direct and indirect relationships that were experimentally observed were included. Three analyses were conducted. The first was to identify canonical pathways in which 2 or more genes were overrepresented. The program was also used to build customized networks of genes based on direct and indirect relationships. Finally, upstream regulators in which genes related to DPR were overrepresented were identified. A P value of 0.05 or less was considered significant for all analyses.
Results
Genetic characteristics of bulls used for genotyping
The range of PTAs for bulls are shown in Additional file 1: Table S1, while the effect of DPR class (high or low) on PTAs are shown in Table 1. Daughter pregnancy rate class had a significant effect on all other traits examined. In particular, bulls in the high DPR class had higher PTAs for heifer conception rate (HCR), cow conception rate (CCR), productive life (PL), net merit (NM), fat percent (FPC), and protein percent (PPC) and lower PTAs for milk yield (MY), fat yield (FY), protein yield (PY), and somatic cell score (SCS) than bulls in the low DPR class (Table 1). Correlations among PTAs are shown in Additional file 2: Table S4. Daughter pregnancy rate was significantly and positively correlated with HCR (0.61), CCR (0.91), PL (0.81), NM (0.49), FPC (0.16), and PPC (0.31) and was significantly and negatively correlated with MY (−0.45), FY (−0.35), PY (−0.34), SCS (−0.55), and birth year (BY; -0.15). These results are consistent with correlations reported earlier [54] for traits included in the lifetime net merit selection index. Since the bulls were selected from the two extremes of DPR, correlations within DPR class (DPRC) were also examined (Additional file 2: Table S5). Within the high DPRC, DPR was positively correlated with HCR and CCR and negatively correlated with NM, MY, FY, PY, and BY. Within the low DPRC, DPR was positively correlated with CCR, PL, and NM and was negatively correlated with SCS and BY.
Minor allele frequencies
Of the 434 SNPs, only 107 had MAF ≥ 5% and only 98 of those that had MAF ≥ 5% and had a call rate > 70%. Nine SNPs had MAF ≥ 5% but failed the genotyping process (call rate < 70%; AHCYL2, APBB1IP, FXC1, HSF1, PHGDH, POMC, SLC1A5, ST13, and TTF1) and were removed from all further analyses. The probability that the MAF was ≥ 5% was dependent upon the type of SNP. Four of the 5 genes in which the SNP was in the non-coding regions or was synonymous had a MAF ≥ 5% (80%) whereas only 20% (2/10) of the nonsense, 25% of the missense (99/397), and 9% (2/22) of the frameshift mutations had ≥ 5% MAF (χ2 for non-coding/synonymous vs others, 8.34, P < 0.01).
Hardy-Weinberg equilibrium
Characteristics of the 98 SNPs in which MAF ≥ 5% and call rate was > 70% are shown in Additional file 2: Table S6. A total of 26 SNPs were not in equilibrium (AVP, BOLA-DMB, C17H22orf25, CCDC88B, CCT8, CD2, CFDP2, COQ9, DEPDC7, DTX2, FUT1, HSD17B6, IBSP, IFNT2, MARVELD1, NEU3, RALGPS1, SEC14L1, SREBF1, STAT5A, SYTL2, TAF9, TSPYL1, UHRF1, WBP1, and ZP2). All but one of these SNPs caused a missense mutation. The exception was for UHRF1, which was a frameshift mutation where the mutation causing the frameshift had a frequency of 91.7%. The genes most out of equilibrium were CCT8, MARVELD1 and SYTL2, in which the number of minor allele homozygotes was lower than expected, CD2, DTX2, NEU3, and RALGPS1, in which the number of heterozygotes was lower than expected, and TAF9 and TSPYL1, in which the number of heterozygotes was greater than expected.
SNP effects on daughter pregnancy rate
Each of the 98 SNPs with MAF ≥ 5% and a call rate > 70% were analyzed for effects on DPR and other genetic traits. Two types of analyses were performed: a regression analysis to determine the allele substitution effect of each SNP (0, 1 or 2 copies of the major allele) and use of an orthogonal contrast to determine the dominance effect (heterozygote vs. the average of the two homozygotes). Both P values and Q values corrected for multiple testing were determined. Since the Q value correction for multiple testing is highly conservative in cases where few tests are significant, both the P value and the Q value were used to identify SNPs associated with genetic traits.
Results for DPR are shown in Table 2. Allele substitution effects were different from 0 for 40 genes [ACAT2, AP3B1, APBB1, BSP3, C17H22orf25 (interim symbol TANGO2), C1QB, C7H19orf60, CACNA1D, CAST, CCDC86, CD14, CD40, CFDP2, COQ9, CPSF1, CSNK1E, CSPP1, DEPDC7, DSC2, DYRK3, FUT1, GPLD1, HSD17B12, HSD17B7, LDB3, MARVELD1, MON1B, MRGPRF, MS4A8B, NEU3, NFKBIL1, NLRP9, OCLN, PARM1, PCCB, PMM2, RABEP2, TBC1D24, TDRKH, and ZP2]. These effects were significant based on both P and Q values. In addition, there were 4 genes exhibiting dominance based on P values, including two in which the allele substitution effect was significant (CD14 and FUT1) and two in which the allele substitution was not significant (ARL6IP1 and TSHB). After correcting for multiple testing, none of the dominance effects achieved significance.
SNP effects on other fertility traits
For HCR, there were allele substitution effects for 19 SNPs (AP3B1, APBB1, C1QB, CACNA1D, CD14, CPSF1, CSNK1E, DEPDC7, DSC2, FSHR, FYB, GPLD1, HSD17B7, LDB3, MS4A8B, NFKBIL1, PARM1, TDRKH, and ZP2) and dominance effects for 5 SNPs (ARPL6IP1,CACNA1D, CD14, DZIP3, and GOLGA4; Table 3). None of the dominant effects remained significant after correcting for multiple testing. The only SNPs significant after correcting for multiple testing were allele substitution effects for DEPDC7, LDB3, MS4A8B, PARM1, and TDRKH.
For CCR, there were allele substitution effects for 29 SNPs (ACAT2, AP3B1, APBB1, BCAS1, C1QB, CAST, CCDC86, CCT8, CFDP2, COQ9, CPSF1, CSNK1E, CSPP1, FUT1, GPLD1, HSD17B7, LDB3, MARVELD1, MON1B, NEU3, NFKBIL1, OCLN, PARM1, PMM2, RABEP2, TBC1D24, TDRKH, WBP1, and ZP2) and dominance effects for 4 SNPs (ARL6IP1, SEC14L1, SERPINE2, and SLC18A2; Table 4). All but one of the allele substitution effects were significant after correction for multiple testing, the exception being for ARL6IP1, but none of the dominance effects were significant based on Q values.
SNP effects on productive life and net merit
For PL, there were allele substitution effects for 33 SNPs (ACAT2, AP3B1, ASL, CCDC86, CD40, CFDP2, COQ9, CSPP1, DEPDC7, DSC2, FSHR, FUT1, GPLD1, HSD17B12, HSD17B6, HSD17B7, HSPA1A, LDB3, LHCGR, MARVELD1, MON1B, MS4A8B, NEU3, OCLN, PARM1, PCCB, PMM2, RABEP2, SYTL2, TBC1D24, TDRKH, WBP1, and ZP2) and dominance effects for 5 SNPs (ARL6IP1, AVP, CSPP1, DEPDC7, and IBSP; Table 5). After correcting for multiple testing, none of the dominant effects were significant.
For NM, there were allele substitution effects for 30 SNPs (ACAT2, AP3B1, ASL, C17H22orf25, CCT8, CD2, CD40, COQ9, CSNK1E, DEPDC7, EPAS1, FST, FUT1, HSD17B12, HSD17B6, HSPA1A, IBSP, LDB3, LHCGR, MON1B, MRPL48, MS4A8B, NEU3, OCLN, PARM1, PCCB, PMM2, RABEP2, TBC1D24, and TDRKH) and dominance effects for 6 SNPs (SNPs in ARL6IP1, CD14, DEPDC7, FGF2, IBSP, and SLC18A2; Table 6). Except for HSPA1A, the allele substitution effects were significant after correcting for multiple testing, but dominance effects were not significant.
SNP effects on production traits
There were fewer effects on production traits compared to fertility traits, which is consistent with the conclusion of Cole et al. [55] that yield traits generally are consistent with an infinitesimal model, in which the trait is controlled by many alleles of small effect. For MY, there were allele substitution effects for 18 SNPs and dominance effects for 6 SNPs (Table 7). Only linear effects of CD14, CPSF1, FAM5C, and PARM1 were significant after correcting for multiple testing. For FY, there were allele substitution effects for 13 SNPs and dominance effects for 7 SNPs (Table 8). Only the linear effects of CPSF1 and PARM1 were significant after correcting for multiple testing. For FPC, there were allele substitution effects for 10 SNPs and dominance effects for 4 SNPs (Table 9). After correcting for multiple testing, only linear effects of CPSF1, DEPDC7, FAM5C, MS4A8B, and SREBF1 were significant.
For PY, there were allele substitution effects for 17 SNPs and dominance effects for 4 SNPs (Table 10). None of the effects were significant after correcting for multiple testing. For PPC, there were linear effects of 21 SNPs and 1 SNP with a dominance effect (Table 11). After correcting for multiple testing, only the linear effects of BSP3, CPSF1, FAM5C, FCER1G, FUT1, HSPA1A, MS4A8B, PARM1, and TDRKH were significant.
Results for SCS are shown in Table 12. There were allele substitution effects of 8 SNPs and dominance effects for 6 SNPs. After correcting for multiple testing, the linear effects of CFDP2, CPSF1, DSC2, FST, PMM2, SEC14L1, TXN2 and the dominance effect of NFKBIL1 were significant.
Relationship between allele substitution effects for SNPs related to DPR with effects on other traits
It was determined whether SNPs affecting DPR had association with other traits and, if so, whether the allele substitution effect was in the same or opposite direction as for DPR. Results are shown in Figure 1. As expected, many SNPs associated with DPR were also associated with HCR and CCR and in the same direction as for DPR. Of 40 SNPs in which there was a linear effect on DPR (Q < 0.05), 13 also were associated with HCR and 25 were associated with CCR. In all cases, the allele substitution effect was in the same direction for DPR and either HCR or CCR. Similar results were observed for PL and NM. Of the 40 SNPs associated with DPR, 26 were also associated with PL and 20 with NM and the allele substitution effect was in the same direction for DPR and either PL and NM.

Allele substitution effects of SNPs on fertility and production traits. Only effects with P values and/or Q values < 0.05 are included. Numbers represent the magnitude of the allele substitution effect. Dark blue rectangles are linear effects in the same direction as DPR (Q < 0.05), light blue rectangles are linear effects in the same direction as DPR (P < 0.05 but Q value ≥ 0.05), red rectangles are linear effects in the opposite direction as DPR (P and Q value < 0.05), and pink rectangles are linear effects in the opposite direction as DPR (P < 0.05 but Q ≥ 0.05). Empty cells indicate that there was no significant effect of the SNP on the trait based on P or Q values. Abbreviations are as follows: CCR, cow conception rate; DPR, daughter pregnancy rate; FPC, fat percent; FY, fat yield; HCR, heifer conception rate; MY, milk yield; NM, net merit; PL, productive life; PPC, protein percent; PY, protein yield; SCS, somatic cell score.
Fewer SNPs associated with DPR were also associated with production traits. Furthermore, when occurring, the direction of the effect was often in the opposite direction as for DPR, especially for yield traits. There were 10 SNPs associated with MY and all but one (CPSF1) were in the opposite direction as for DPR (i.e., genotypes favoring DPR were unfavorable for milk yield). There were 7 SNPs associated with FY and 5 of these were in the opposite direction as for DPR. There were 7 SNPs associated with PY and 5 of these were in the opposite direction as for DPR. For other production traits, however, there were fewer negative relationships between allele substitution effects on DPR. For FPC, there were 5 SNPs but only 1 was in the opposite direction as DPR. For PPC, there were 13 SNPs but only 1 was in the opposite direction as DPR. For SCS, there were 5 SNPs with three in the same direction as DPR and two in the opposite direction.
Of the 40 SNPs related to DPR, there were 29 that were not negatively associated with yield traits (milk, fat and protein). Thus, it should be possible to select for specific SNPs affecting DPR without compromising yield traits.
Relationship between SNPs associated with DPR and SNPs reported previously to be related to fertility
Of the 434 SNPs analyzed, 17 were chosen because they had previously been reported to be associated with reproduction or to be close to SNPs related to interval to insemination or 56-d non-return rate (Additional file 1: Table S2). Of these, only 8 had a MAF ≥ 5% (CAST, FGF2, FSHR, HSPA1A, IRF9, NLRP9, PGR and STAT5A) and only 2 (CAST and NLRP9) were significantly associated with DPR (Table 2).
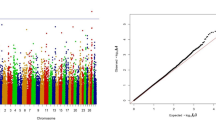
The physical location of each SNP associated with DPR in the current study was compared to markers from the BovineSNP50 chip previously associated with DPR [7]. Figure 2 shows the relative location of the SNPs and the SNP50 marker effects. The SNP effects from the current custom array have a much greater effect on DPR than those found on the BovineSNP50 chip. The largest genetic standard deviation on the BovineSNP50 chip for DPR was 0.07 genetic standard deviations [7]; however, in the current study, the marker effect ranged from 0.44 to 1.78 (Additional file 3: Table S7).

Manhattan plots comparing SNP effects on daughter pregnancy rate from the current study (panel A; UF SNP) to marker effects from the BovineSNP50 chip in a previous study (panel B; USDA SNP) [[7]]. Each chromosome is represented in a different color along the x-axis. The y-axis is the marker effect on daughter pregnancy rate (genetic standard deviations). The markers are color coordinated according to their chromosome location.
A literature search was conducted to determine if any SNPs previously related to fertility were within 100,000 bases of any of the SNPs related to DPR in the current study. The literature provided evidence for 3 other SNPs located close to SNPs from the current study. A SNP in DGAT1, which is about 65,000 bp from the SNP in CPSF1, was associated with 28 and 56 day nonreturn rate to first service, age at puberty, number of inseminations per conception, and conception rate [56–58]. A SNP in TNF, which is about 25,000 bp from the SNP in NFKBIL1, was associated with early first ovulation in postpartum cows [59]. Also, a SNP in HSD14B14, which is about 60,000 bp from the SNP in FUT1, was associated with DPR [7]. Since these SNPs are close in distance, there could be linkage disequilibrium between them. Therefore, it is possible that either gene in each of the previous locations could contain the causative SNP.
Effect of tissue type used for SNP discovery on probability of identifying SNPs associated with DPR
An analysis was performed to determine whether the tissue type used to identify genes for SNP discovery affected the probability that a gene was related to DPR (Additional file 3: Table S8). Using chi-square analysis, fewer SNPs identified in genes identified as expressed in the brain or pituitary were significantly associated with DPR (18%) than for embryo genes (51%), endometrium or oviduct genes (50%) or ovary genes (43%) (χ2 for brain/pituitary vs others, 3.74, P = 0.05).
Pathway analysis of genes with SNPs associated with DPR
There were a total of 5 canonical pathways in which 2 or more genes were overrepresented (P < 0.05). These were Estrogen Biosynthesis (HSD17B7 and HSD17B12), Estrogen-Dependent Breast Cancer Signaling (HSD17B7 and HSD17B12), Hepatic Fibrosis/Hepatic Stellate Activation (CD14 and CD40), Tight Junction Signaling (CDSF1 and OCLN), and Dopamine-DARPP32 Feedback in cAMP Signaling (CACNAID and CSNK1E). The IPA software also built 4 networks of genes related to DPR. The most revealing was one that included 16 genes (ACAT2, AP3B1, COQ9, CPSF1, CSPP1, DEPDC7, DSC2, GPLD1, HSD17B12, MARVELD1, MON1B, NFKBIL1, PMM2, RABEP2, TBC1D24, and TDRKH) which interacted directly or indirectly with UBC (Additional file 3: Figure S1).
The list of genes related to DPR was also examined for upstream regulators in which regulated genes were significantly (P < 0.05) overrepresented. A total of 5 transcription factors were identified (Additional file 3: Figure S2) including HNF4A, which regulates 8 genes associated with DPR, TCF3, which regulates 3 DPR genes, and CTBP2, FOSB, and SP100, which each regulate one gene. Additional regulators of genes associated with DPR were two hormones (estradiol and prostaglandin E1) and one growth factor (TGFB1). Estradiol regulates 10 DPR genes, TGFB1 regulates 6 genes, and prostaglandin E1 regulates 2 genes (Figure 3).

Growth factors and steroids which regulate daughter pregnancy rate genes. Only significant pathways are shown (P < 0.05). Red symbols are genes in which SNPs were associated with daughter pregnancy rate and arrows represent regulation.
Discussion
The results of this study verified that the candidate gene approach could be a successful method of determining markers for DPR. It was anticipated that, since the SNPs used for genotyping were specifically chosen for their function in reproductive processes, a larger proportion of them would be associated with reproductive traits than for production traits. Such a result was obtained. Of the 98 genes that met the criteria for analysis (MAF ≥ 5% and call rate ≥ 70%) and where effects were P < 0.05, there were 42 genes associated with DPR (Table 2) but only 23 associated with MY (Table 7). Moreover, all of the significant SNP effects for DPR in this study were between 5 and 25 times greater than the largest marker effect from the BovineSNP50 chip [7] (Figure 2 and Additional file 3: Table S7). This result is probably due to the differences in SNP selection between the two methods. The majority of SNPs on the BovineSNP50 chip are between genes (63%) and over 14,000 genes are not represented by a SNP on the Bovine SNP50 chip [9]. In the current study, almost all of the SNPs examined were located within the coding region of the gene and the remainder were close physically to the coding region. Moreover, SNPs were chosen to maximize the probability that there would be a change in the characteristic of the protein encoded for the gene. Thus, it is likely that many of the SNPs that have large effects on DPR do so because they are causative SNPs resulting in changes in protein function. The remainder may represent linkages to causative SNPs. The SNPs identified in this study may be closer to the causative SNPs than the SNPs on the BovineSNP50 chip. Allele substitution effects were estimated individually with a linear mixed model, rather than simultaneously as described in Cole et al. [7], which also could explain some of the differences.
Polymorphisms in the current study were chosen for having the greatest probability of changing protein function. In order to maximize the possibility of finding causative SNPs, we prioritized the selection of SNPs within a gene to favor those causing the greatest change in protein function. This decision may have been one reason why there was a high rate (75%) of SNPs with MAF < 5% because the SNP would be subjected to purifying selection. Only 20% of the nonsense, 25% of the missense and 9% of the frameshift mutations had MAF ≥ 5% whereas this frequency was 80% of the 5 SNPs that were in a non-coding region or did not result in an amino acid substitution. Many of the SNPs were not in Hardy-Weinberg equilibrium and this, too, may reflect the effect of the SNPs on protein function.
Of the 9 SNPs most out of equilibrium, only 3 (CCT8, MARVELD1 and SYTL2) had less than expected frequencies of minor allele homozygotes. The interpretation is that few of the mutations in which MAF was ≥ 5% were lethal. Interestingly, for six genes, the heterozygote was more or less frequent than expected. Some of the decrease in heterozygosity could be due to inbreeding, which is high in Holstein cattle [60]. Other changes in heterozygosity could be due to either an advantage or disadvantage of the heterozygote. Heterozygote advantage could be due to the ability of receptors to recognize more forms of the peptides they bind (e.g. MHC class I [61]), heterozygotes having the optimal level of gene expression [62], or in theory, the optimal allele being different for different cell types. A reason for heterozygote disadvantage is not clear.
The antagonistic genetic relationship between fertility traits and milk production [1–3] was verified here. There was a negative correlation between DPR and MY across DPR classes (Additional file 2: Table S4) and within cows in the high DPR class (Additional file 2: Table S5). Nonetheless, there were many SNP related to DPR (and often, other reproductive and health traits) that were not antagonistic for MY. Accordingly, it should be possible to select for DPR without reducing MY. Of the 40 SNPs linearly related to DPR, only 11 were negatively associated with MY, FY, or PY (Figure 1).
SNPs that affected DPR were also positively related with other fertility traits (HCR, CCR, and NM). Other studies have also shown a positive genetic correlation among fertility traits [3, 28, 63]. It is not surprising that these traits are affected by the SNPs associated with DPR. One determinant of DPR is CCR. In addition, PL depends in part on the probability of culling for reproduction. The equation to calculate NM includes DPR and PL. The fact that SNPs associated with DPR are also associated with HCR, CCR, PL and NM means that selection of genes that improve DPR are likely to improve other reproductive traits and traits that depend upon reproduction.
SNPs linked to traits in the current study that were previously linked to other traits are summarized in Table 13. Of the 17 genes with SNPs previously linked to fertility or close to SNPs related to fertility traits, 9 SNPs had MAF < 5% (BAIAP2, GHR, LEP, IGF1, IGFBP7, ITGB5, PAPPA2, SCRN1, and SERPINA14) and were not analyzed. Of the other 8, 2 were significantly associated with DPR (CAST and NLRP9) and one tended to be (FGF2). The exact SNP in CAST analyzed here was previously associated with DPR, PL, and NM [15]. A different SNP in NLRP9 than the one studied here was associated with incidence of still birth [21]. Another gene, FGF2, tended to have an association with DPR (P = 0.08), with the AA genotype being superior to the GG genotype. Previously, the AA genotype of FGF2 was associated with higher estimated relative conception rate in bulls [16] although, surprisingly, associated with lower in vitro embryo development [12]. Another SNP, in PGR, was previously associated with in vitro fertilization rate and development [14] and in vivo fertilization [64] and pregnancy rates [65], and while not significant (P = 0.16), the GG genotype was superior to the CC genotype for DPR in agreement with the superior genotype seen earlier [14, 64, 65]. A SNP in FSHR was previously associated with superovulation response [19, 66] and, while not significantly associated with DPR in the current study, was associated with HCR and PL. There was no significant effect of genotype for four other SNPs in genes previously associated with reproductive traits, including HSPA1A, associated with calving rate in beef cattle [23], IRF9, which was physically close to a SNP for interval to insemination [28], and STAT5A, associated with in vitro embryo development [11] and sire conception rate [16]. Note that HSPA1A was significantly associated with PL and NM (Table 13) and both of these traits depend upon reproductive function.
The genes in the current study with SNPs that were associated with DPR participate in a wide range of physiological functions associated with reproductive processes. Many function in the endocrine system, either in synthesis of hormones or in cell signaling. The estrogen biosynthesis pathway was one of the pathways in which genes associated with DPR were significantly overrepresented. The gene ACAT2 is involved in cholesterol metabolism [67], and expression of ACAT2 in cumulus cells is increased for infertile women as compared to fertile women [68]. The gene HSD17B12 encodes for an enzyme that converts estrone to estradiol [69]. It is also involved in the synthesis of arachidonic acid and is essential for embryo survival in mice [70]. Another gene related to DPR, HSD17B7, also converts estrone to estradiol [71] and is essential for de novo cholesterol synthesis in the fetus [72–74]. In addition to genes involved in steroid synthesis, TSHB, a gene which codes for the β strand of the pituitary hormone, TSH, was associated with DPR. Thyroid function, which is under the control of TSH [75], can impact reproductive function in cattle [76, 77]. Some genes related to DPR may also affect release of neurotransmitters controlling hypothalamic-pituitary function. One, AP3B1, is involved in formation of synaptic vesicles [78], and APBB1 controls GnRH-1 neurogenesis [79]. Another, TBC1D24, stimulates primary axonal arborization [80, 81]. Polymorphisms in TBC1D24 have been associated with shortened axons and epileptic seizures [80, 81].
Among the DPR genes involved in cell signaling are the G protein-coupled receptors MRGPRF and MS4A8B[82], GPLD1, which cleaves cell surface proteins anchored by phosphatidylinositol glycans [83], the sialidase NEU3, which is important for insulin signaling [84], CACNA1D, a component of calcium channels [85], and DSC2, an important component of membrane rafts and cell-cell junctions [86] and which is involved in blastocoel formation [87]. Similarly, OCLN is a major component of tight junctions and is involved in barrier stability [88]. Another gene involved in cell-cell binding related to DPR is PMM2, which isomerizes mannose 6-phosphate into mannose 1-phosphate [89], which eventually is converted to GDP-fucose and used to make fucosylated glycans [90]. Fucosylated glycans serve several functions, including leukocyte-endothelial adhesion, host-microbe interactions, embryo compaction, and signal transduction [90].
One gene associated with DPR, CSNK1E, is involved in paracrine regulation of cell function as a positive regulator of the canonical WNT/β-catenin pathway [91, 92]. The WNT pathway plays important roles in cell differentiation [93, 94], preimplantation development [95], formation of the epiblast [96] and implantation [97]. Moreover, CSNK1E regulates circadian rhythm by controlling nuclear entry of PER1, a regulator of CLOCK[98]. Expression of PER1 was associated with depth of anestrus at the start of the breeding season in beef cattle [99].
Three genes related to DPR are involved with the function of spermatozoa in the female tract. The gene BSP3 aids in maintaining sperm motility during storage in the oviduct [100]. Protein concentrations are associated with bull fertility [101] and the mRNA is down-regulated in the endometrium of heifers which carried a pregnancy to term compared to those in which the embryo died after transfer [26]. Another gene, CAST, plays an important role in sperm capacitation and the acrosome reaction [102–104] and may play a role in oocyte calcium-mediated processes that occur during oocyte activation [105]. The same SNP in CAST found to be associated with DPR in this study was earlier associated with DPR, PL, NM and SCS [15]. The embryonic gene ZP2 encodes for a protein that makes up part of the zona pellucida and is the location that sperm bind on the zona pellucida [106, 107]. One of the genes related to DPR, NLRP9, is likely to play an important function in the oocyte. The gene is expressed in the oocyte, and steady-state amounts of NLRP9 mRNA decline after fertilization and become undetectable after the maternal to zygote transition [108–110].
There is much evidence to implicate immune function in the establishment of pregnancy [111]. Seven of the genes with SNPs associated with DPR are involved in immune function. The gene C1QB is involved in complement activation [112], CD14 is a co-receptor for recognition of bacteria [113], CD40 regulates cell surface receptor signaling [114], and NFKBIL1 regulates dendritic cell function [115]. Additionally, MON1B and RABEP2 help regulate phagocytosis and endocytosis [116, 117] and mutations in FUT1 have been associated with disease resistance [118–120]. Polymorphisms in FUT1 have also been associated with total number of piglets born [121, 122] and number of piglets alive at weaning [119]. It is possible that allelic variants in these genes that are positively associated with DPR improve immune function and decrease incidence of diseases such as endometritis, metritis, and mastitis that disrupt reproduction [123–125].
Three genes related to DPR are anti-apoptotic: ARL6IP1, DYRK3 and PARM1I[126–128]. Induction of apoptosis in the oocyte and associated cumulus cells is associated with reduced fertilization rate [129–131]. Two molecules that improve embryo competence for establishment of pregnancy after transfer into recipients, CSF2 and IGF1 [132, 133], are anti-apoptotic in embryos [32, 134].
A variety of other roles are also represented by the genes with SNPs associated with DPR. Two genes are involved in energy pathways (COQ9 and PCCB). The COQ9 protein is necessary for the synthesis of CoQ10 [135], which is needed for generating ATP [136]. PCCB is an enzyme that converts proponyl CoA to methylmalonyl CoA during gluconeogenesis [137]. The CSPP1 gene plays a role in spindle formation and cytokinesis [138], MARVELD1 inhibits cell cycle progression and migration [139], and LDB3 helps organize actin and α-actinin binding in sarcomeres [140]. Finally, CPSF1 is involved in 3′ end-processing of pre-messenger RNAs into messenger RNAs [140].
Several gene networks were significant among the genes related to DPR but most contained only two genes. The exceptions were estrogen biosynthesis, discussed earlier, and a network of genes associated with ubiquitin C (UBC). It is not surprising that the proteins encoded for by so many genes bind to UBC because ubiquitin is involved in a large number of intracellular functions [141–144]. Five transcription factors (HNF4A, TCF3, CTBP2, FOSB, and SP100), two hormones (estradiol and prostaglandin E1), and one growth factor (TGFB1) were determined by the IPA software to be significantly overrepresented as regulators of DPR genes. Each of these upstream regulators could be studied further for the potential to improve fertility by regulating activation of pathways controlled by these molecules.
Conclusions
In conclusion, SNPs in a total of 40 genes associated with DPR were identified as well as SNPs for other traits. It might be feasible to include these SNPs into genomic tests of reproduction and other traits. The genes associated with DPR are likely to be important for understanding the physiology of reproduction and manipulating reproduction function in cattle. Given the large number of SNPs associated with DPR that were not negatively associated with production traits, it should be possible to select for DPR without compromising production.
Abbreviations
- CCR:
-
Cow conception rate
- DPR:
-
Daughter pregnancy rate
- ERCR:
-
Estimated relative conception rate
- FPC:
-
Fat percent, FY, Fat yield
- HCR:
-
Heifer conception rate
- MAF:
-
Minor allele frequency
- MY:
-
Milk yield
- NM:
-
Net merit
- PL:
-
Productive life
- PPC:
-
Protein percent
- PTA:
-
Predicted transmitting ability, PY, Protein yield
- SCS:
-
Somatic cell score
- SNP:
-
Single nucleotide polymorphism.
References
Van Arendonk JAM, Hovenier R, De Boer W: Phenotypic and genetic association between fertility and production in dairy cows. Livestock Production Sci. 1989, 21: 1-12.
VanRaden PM, Sanders AH, Tooker ME, Miller RH, Norman HD, Kuhn MT, Wiggans GR: Development of a national genetic evaluation for cow fertility. J Dairy Sci. 2004, 87: 2285-2292.
Pritchard T, Coffey M, Mrode R, Wall E: Genetic parameters for production, health, fertility and longevity traits in dairy cows. Animal. 2013, 2013 (7): 34-46.
Washburn SP, Silvia WJ, Brown CH, McDaniel BT, McAllister AJ: Trends in reproductive performance in Southeastern Holstein and Jersey DHI herds. J Dairy Sci. 2002, 85: 244-251.
Hare E, Norman HD, Wright JR: Trends in calving ages and calving intervals for dairy cattle breeds in the United States. J Dairy Sci. 2006, 89: 365-70.
Norman HD, Wright JR, Hubbard SM, Miller RH, Hutchison JL: Reproductive status of Holstein and Jersey cows in the United States. J Dairy Sci. 2009, 92: 3517-3528.
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, Van Tassell CP, Yang J, Wang S, LK M: Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011, 12: 408-
Wiggans GR, Vanraden PM, Cooper TA: The genomic evaluation system in the United States: past, present, future. J Dairy Sci. 2011, 94: 3202-3211.
Michelizzi VN, Wu X, Dodson MV, Michal JJ, Zambrano-Varon J, McLean DJ, Jiang Z: A global view of 54,001 single nucleotide polymorphisms (SNPs) on the Illumina BovineSNP50 BeadChip and their transferability to water buffalo. Int J Biol Sci. 2011, 7: 18-27.
Amos W, Driscoll E, Hoffman JI: Candidate genes versus genome-wide associations: which are better for detecting genetic susceptibility to infectious disease?. Proc Biol Sci. 2011, 278: 1183-1188.
Khatib H, Monson RL, Schutzkus V, Kohl DM, Rosa GJ, Rutledge JJ: Mutations in the STAT5A gene are associated with embryonic survival and milk composition in cattle. J Dairy Sci. 2008, 91: 784-793.
Khatib H, Maltecca C, Monson RL, Schutzkus V, Wang X, Rutledge JJ: The fibroblast growth factor 2 gene is associated with embryonic mortality in cattle. J Anim Sci. 2008, 86: 2063-2067.
Wang X, Schutzkus V, Huang W, Rosa GJ, Khatib H: Analysis of segregation distortion and association of the bovine FGF2 with fertilization rate and early embryonic survival. Anim Genet. 2009, 40: 722-728.
Driver AM, Huang W, Gajic S, Monson RL, Rosa GJ, Khatib H: Short communication: Effects of the progesterone receptor variants on fertility traits in cattle. J Dairy Sci. 2009, 92: 4082-4085.
Garcia MD, Michal JJ, Gaskins CT, Reeves JJ, Ott TL, Liu Y, Jiang Z: Significant association of the calpastatin gene with fertility and longevity in dairy cattle. Anim Genet. 2006, 37: 304-305.
Khatib H, Monson RL, Huang W, Khatib R, Schutzkus V, Khateeb H, Parrish JJ: Short communication: Validation of in vitro fertility genes in a Holstein bull population. J Dairy Sci. 2010, 93: 2244-2249.
Feugang JM, Kaya A, Page GP, Chen L, Mehta T, Hirani K, Nazareth L, Topper E, Gibbs R, Memili E: Two-stage genome-wide association study identifies integrin beta 5 as having potential role in bull fertility. BMC Genomics. 2009, 10: 176-
Waters SM, McCabe MS, Howard DJ, Giblin L, Magee DA, MacHugh DE, Berry DP: Associations between newly discovered polymorphisms in the Bos taurus growth hormone receptor gene and performance traits in Holstein-Friesian dairy cattle. Anim Genet. 2011, 42: 39-49.
Yang WC, Li SJ, Tang KQ, Hua GH, Zhang CY, Yu JN, Han L, Yang LG: Polymorphisms in the 5′ upstream region of the FSH receptor gene, and their association with superovulation traits in Chinese Holstein cows. Anim Reprod Sci. 2010, 119: 172-1777.
Kim ES, Shi X, Cobanoglu O, Weigel K, Berger PJ, Kirkpatrick BW: Refined mapping of twinning-rate quantitative trait loci on bovine chromosome 5 and analysis of insulin-like growth factor-1 as a positional candidate gene. J Anim Sci. 2009, 87: 835-843.
Ponsuksili S, Brunner RM, Goldammer T, Kühn C, Walz C, Chomdej S, Tesfaye D, Schellander K, Wimmers K, Schwerin M: Bovine NALP5, NALP8, and NALP9 genes: assignment to a QTL region and the expression in adult tissues, oocytes, and preimplantation embryos. Biol Reprod. 2006, 2006 (74): 577-584.
Brickell JS, Pollott GE, Clempson AM, Otter N, Wathes DC: Polymorphisms in the bovine leptin gene associated with perinatal mortality in Holstein-Friesian heifers. J Dairy Sci. 2010, 93: 340-347.
Rosenkrans C, Banks A, Reiter S, Looper M: Calving traits of crossbred Brahman cows are associated with heat shock protein 70 genetic polymorphisms. Anim Reprod Sci. 2010, 119: 178-182.
Luna-Nevarez P, Rincon G, Medrano JF, Riley DG, Chase CC, Coleman SW, Vanleeuwen DM, DeAtley KL, Islas-Trejo A, Silver GA, Thomas MG: Single nucleotide polymorphisms in the growth hormone-insulin-like growth factor axis in straightbred and crossbred Angus, Brahman, and Romosinuano heifers: population genetic analyses and association of genotypes with reproductive phenotypes. J Anim Sci. 2011, 89: 926-934.
Kommadath A, Woelders H, Beerda B, Mulder HA, de Wit AA, Veerkamp RF, te Pas MF, Smits MA: Gene expression patterns in four brain areas associate with quantitative measure of estrous behavior in dairy cows. BMC Genomics. 2011, 12: 200-
Beltman ME, Forde N, Furney P, Carter F, Roche JF, Lonergan P, Crowe MA: Characterisation of endometrial gene expression and metabolic parameters in beef heifers yielding viable or non-viable embryos on Day 7 after insemination. Reprod Fertil Dev. 2010, 22: 987-999.
El-Sayed A, Hoelker M, Rings F, Salilew D, Jennen D, Tholen E, Sirard MA, Schellander K, Tesfaye D: Large-scale transcriptional analysis of bovine embryo biopsies in relation to pregnancy success after transfer to recipients. Physiol Genomics. 2006, 28: 84-96.
Pimentel EC, Bauersachs S, Tietze M, Simianer H, Tetens J, Thaller G, Reinhardt F, Wolf E, König S: Exploration of relationships between production and fertility traits in dairy cattle via association studies of SNPs within candidate genes derived by expression profiling. Anim Genet. 2011, 42: 251-262.
Kuzmany A, Havlicek V, Wrenzycki C, Wilkening S, Brem G, Besenfelder U: Expression of mRNA, before and after freezing, in bovine blastocysts cultured under different conditions. Theriogenology. 2011, 75: 482-494.
Gad A, Besenfelder U, Rings F, Ghanem N, Salilew-Wondim D, Hossain MM, Tesfaye D, Lonergan P, Becker A, Cinar U, Schellander K, Havlicek V, Hölker M: Effect of reproductive tract environment following controlled ovarian hyperstimulation treatment on embryo development and global transcriptome profile of blastocysts: implications for animal breeding and human assisted reproduction. Hum Reprod. 2011, 26: 1693-1707.
Salilew-Wondim D, Hölker M, Rings F, Ghanem N, Ulas-Cinar M, Peippo J, Tholen E, Looft C, Schellander K, Tesfaye D: Bovine pretransfer endometrium and embryo transcriptome fingerprints as predictors of pregnancy success after embryo transfer. Physiol Genomics. 2010, 42: 201-218.
Loureiro B, Oliveira LJ, Favoreto MG, Hansen PJ: Colony-stimulating factor 2 inhibits induction of apoptosis in the bovine preimplantation embryo. Am J Reprod Immunol. 2011, 65: 578-588.
Bonilla AQ, Oliveira LJ, Ozawa M, Newsom EM, Lucy MC, Hansen PJ: Developmental changes in thermoprotective actions of insulin-like growth factor-1 on the preimplantation bovine embryo. Mol Cell Endocrinol. 2011, 332: 170-179.
Hoelker M, Rings F, Lund Q, Ghanem N, Phatsara C, Griese J, Schellander K, Tesfaye D: Effect of the microenvironment and embryo density on developmental characteristics and gene expression profile of bovine preimplantative embryos cultured in vitro. Reproduction. 2009, 137: 415-425.
Misirlioglu M, Page GP, Sagirkaya H, Kaya A, Parrish JJ, First NL, Memili E: Dynamics of global transcriptome in bovine matured oocytes and preimplantation embryos. Proc Natl Acad Sci USA. 2006, 103: 18905-18910.
Mamo S, Sargent CA, Affara NA, Tesfaye D, El-Halawany N, Wimmers K, Gilles M, Schellander K, Ponsuksili S: Transcript profiles of some developmentally important genes detected in bovine oocytes and in vitro-produced blastocysts using RNA amplification and cDNA microarrays. Reprod Domest Anim. 2006, 41: 527-534.
Fair T, Carter F, Park S, Evans AC, Lonergan P: Global gene expression analysis during bovine oocyte in vitro maturation. Theriogenology. 2007, 68 (Suppl 1): S91-S97.
Cerri RL, Thompson IM, Kim IH, Ealy AD, Hansen PJ, Staples CR, Li JL, Santos JE, Thatcher WW: Effects of lactation and pregnancy on gene expression of endometrium of Holstein cows at day 17 of the estrous cycle or pregnancy. J Dairy Sci. 2012, 95: 5657-5675.
Assidi M, Dieleman SJ, Sirard MA: Cumulus cell gene expression following the LH surge in bovine preovulatory follicles: potential early markers of oocyte competence. Reproduction. 2010, 140: 835-852.
Salhab M, Tosca L, Cabau C, Papillier P, Perreau C, Dupont J, Mermillod P, Uzbekova S: Kinetics of gene expression and signaling in bovine cumulus cells throughout IVM in different mediums in relation to oocyte developmental competence, cumulus apoptosis and progesterone secretion. Theriogenology. 2011, 75: 90-104.
Tesfaye D, Worku D, Rings F, Phatsara C, Tholen E, Schellander K, Hoelker M: Identification and expression profiling of microRNAs during bovine oocyte maturation using heterologous approach. Mol Reprod Dev. 2009, 76: 665-677.
Fayad T, Lévesque V, Sirois J, Silversides DW, Lussier JG: Gene expression profiling of differentially expressed genes in granulosa cells of bovine dominant follicles using suppression subtractive hybridization. Biol Reprod. 2004, 70: 523-533.
Pfeffer PL, Sisco B, Donnison M, Somers J, Smith C: Isolation of genes associated with developmental competency of bovine oocytes. Theriogenology. 2007, 68 (Suppl 1): S84-S90.
Zielak AE, Forde N, Park SD, Doohan F, Coussens PM, Smith GW, Ireland JJ, Lonergan P, Evans AC: Identification of novel genes associated with dominant follicle development in cattle. Reprod Fertil Dev. 2007, 19: 967-975.
Luo W, Gumen A, Haughian JM, Wiltbank MC: The role of luteinizing hormone in regulating gene expression during selection of a dominant follicle in cattle. Biol Reprod. 2011, 84: 369-378.
Graber M, Kohler S, Kaufmann T, Doherr MG, Bruckmaier RM, van Dorland HA: A field study on characteristics and diversity of gene expression in the liver of dairy cows during the transition period. J Dairy Sci. 2010, 93: 5200-5215.
Mani O, Körner M, Sorensen MT, Sejrsen K, Wotzkow C, Ontsouka CE, Friis RR, Bruckmaier RM, Albrecht C: Expression, localization, and functional model of cholesterol transporters in lactating and nonlactating mammary tissues of murine, bovine, and human origin. Am J Physiol Regul Integr Comp Physiol. 2010, 299: R642-R654.
Bauersachs S, Rehfeld S, Ulbrich SE, Mallok S, Prelle K, Wenigerkind H, Einspanier R, Blum H, Wolf E: Monitoring gene expression changes in bovine oviduct epithelial cells during the oestrous cycle. J Mol Endocrinol. 2004, 32: 449-466.
Khatib H, Schutzkus V, Chang YM, Rosa GJ: Pattern of expression of the uterine milk protein gene and its association with productive life in dairy cattle. J Dairy Sci. 2007, 90: 2427-2433.
Yampolsky LY, Stoltzfus A: The exchangeability of amino acids in proteins. Genetics. 2005, 170: 1459-1472.
Abel K, Reneland R, Kammerer S, Mah S, Hoyal C, Cantor CR, Nelson MR, Braun A: Genome-wide SNP association: identification of susceptibility alleles for osteoarthritis. Autoimmun Rev. 2006, 5: 258-263.
Verschoor CP, Pant SD, Schenkel FS, Sharma BS, Karrow NA: SNPs in the bovine IL-10 receptor are associated with somatic cell score in Canadian dairy bulls. Mamm Genome. 2009, 20: 447-454.
Storey JD: A direct approach to false discovery rates. J Roy Stat Soc Ser B. 2002, 64: 479-498.
Cole JB, VanRaden PM, Multi-State Project S-1040: Net merit as a measure of lifetime profit: 2010 revision. AIPL Res Rep NM$4(12–09). http://aipl.arsusda.gov/reference/nmcalc-2010.htm,
Cole JB, VanRaden PM, O’Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, Taylor JF, Wiggans GR: Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009, 92: 2931-2946.
Schennink A, Stoop W, Visker M, Heck J, Bovenhuis H, van der Poel J, van Valenberg H, van Arendonk J: DGAT1 underlies large genetic variation in milk-fat composition of dairy cows. Anim Genet. 2007, 38: 467-473.
Oikonomou G, Angelopoulou K, Arsenos G, Zygoyiannis D, Banos G: The effects of polymorphisms in the DGAT1, leptin and growth hormone receptor gene loci on body energy, blood metabolic and reproductive traits of Holstein cows. Anim Genet. 2009, 40: 10-17.
Collis E, Fortes M, Zhang Y, Tier B, Schutt K, Barendse W, Hawken R: Genetic variants affecting meat and milk production traits appear to have effects on reproduction traits in cattle. Anim Genet. 2012, 43: 442-446.
Shirasuna K, Kawashima C, Murayama C, Aoki Y, Masuda Y, Kida K, Matsui M, Shimizu T, Miyamoto A: Relationships between the first ovulation postpartum and polymorphism in genes relating to function of immunity, metabolism and reproduction in high-producing dairy cows. J Reprod Dev. 2011, 57: 135-142.
VanRaden PM, Olson KM, Wiggans GR, Cole JB, Tooker ME: Genomic inbreeding and relationships among Holsteins, Jerseys, and Brown Swiss. J Dairy Sci. 2011, 94: 5673-5682.
Spurgin LG, Richardson DS: How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc Biol Sci. 2010, 277: 979-988.
Lippman ZB, Zamir D: Heterosis: revisiting the magic. Trends Genet. 2007, 23: 60-66.
Kuhn MT, Hutchison JL, Wiggans GR: Characterization of Holstein heifer fertility in the United States. J Dairy Sci. 2006, 89: 4907-4920.
Yang W, Tang K, Li S, Yang L: Association analysis between variants in bovine progesterone receptor gene and superovulation traits in Chinese Holstein cows. Reprod Domest Anim. 2011, 46: 1029-1034.
Tang K, Yang W, Pai B, Li S, Chen L, Yang L: Effects of PGR and ESRα genotypes on the pregnancy rates after embryo transfer in Luxi cattle. Mol Biol Rep. 2013, 40: 579-584.
Cory AT, Price CA, Lefebvre R, Palin MF: Identification of single nucleotide polymorphisms in the bovine follicle-stimulating hormone receptor and effects of genotypes on superovulatory response traits. Anim Genet. 2013, 44: 197-201.
Chang TY, Li BL, Chang CC, Urano Y: Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab. 2009, 297: E1-E9.
McReynolds S, Dzieciatkowska M, McCallie BR, Mitchell SD, Stevens J, Hansen K, Schoolcraft WB, Katz-Jaffe MG: Impact of maternal aging on the molecular signature of human cumulus cells. Fertil Steril. 2012, 98: 1574-1580.
Luu-The V, Tremblay P, Labrie F: Characterization of type 12 17β-hydroxysteroid dehydrogenase, an isoform of type 3 17β-hydroxysteroid dehydrogenase responsible for estradiol formation in women. Mol Endocrinol. 2006, 20: 437-443.
Rantakari P, Lagerbohm H, Kaimainen M, Suomela JP, Strauss L, Sainio K, Pakarinen P, Poutanen M: Hydroxysteroid (17β) dehydrogenase 12 is essential for mouse organogenesis and embryonic survival. Endocrinology. 2010, 151: 1893-1901.
Nokelainen P, Peltoketo H, Vihko R, Vihko P: Expression cloning of a novel estrogenic mouse 17β-hydroxysteroid dehydrogenase/17-ketosteroid reductase (m17HSD7), previously described as a prolactin receptor-associated protein (PRAP) in rat. Mol Endocrinol. 1998, 12: 1048-1059.
Breitling R, Krazeisen A, Möller G, Adamski J: 17β-hydroxysteroid dehydrogenase type 7–an ancient 3-ketosteroid reductase of cholesterogenesis. Mol Cell Endocrinol. 2001, 171: 199-204.
Shehu A, Mao J, Gibori GB, Halperin J, Le J, Devi YS, Merrill B, Kiyokawa H, Gibori G: Prolactin receptor-associated protein/17β-hydroxysteroid dehydrogenase type 7 gene (Hsd17b7) plays a crucial role in embryonic development and fetal survival. Mol Endocrinol. 2008, 22: 2268-2277.
Jokela H, Rantakari P, Lamminen T, Strauss L, Ola R, Mutka AL, Gylling H, Miettinen T, Pakarinen P, Sainio K, Poutanen M: Hydroxysteroid (17β) dehydrogenase 7 activity is essential for fetal de novo cholesterol synthesis and for neuroectodermal survival and cardiovascular differentiation in early mouse embryos. Endocrinology. 2010, 151: 1884-1892.
Stathatos N: Thyroid physiology. Med Clin North Am. 2012, 96: 165-173.
Bernal A, DeMoraes GV, Thrift TA, Willard CC, Randel RD: Effects of induced hypothyroidism on ovarian response to superovulation in Brahman (Bos indicus) cows. J Anim Sci. 1999, 77: 2749-2756.
Spicer LJ, Alonso J, Chamberlain CS: Effects of thyroid hormones on bovine granulosa and thecal cell function in vitro: dependence on insulin and gonadotropins. J Dairy Sci. 2001, 84: 1069-1076.
Nakatsu F, Ohno H: Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct. 2003, 28: 419-429.
Forni PE, Fornaro M, Guénette S, Wray S: A role for FE65 in controlling GnRH-1 neurogenesis. J Neurosci. 2011, 31: 480-491.
Corbett MA, Bahlo M, Jolly L, Afawi Z, Gardner AE, Oliver KL, Tan S, Coffey A, Mulley JC, Dibbens LM, Simri W, Shalata A, Kivity S, Jackson GD, Berkovic SF, Gecz J: A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am J Hum Genet. 2010, 87: 371-375.
Falace A, Filipello F, La Padula V, Vanni N, Madia F, De Pietri TD, de Falco FA, Striano P: Dagna Bricarelli F, Minetti C, Benfenati F, Fassio A, Zara F: TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet. 2010, 87: 365-370.
Ross PC, Figler RA, Corjay MH, Barber CM, Adam N, Harcus DR, Lynch KR: RTA, a candidate G protein-coupled receptor: cloning, sequencing, and tissue distribution. Proc Natl Acad Sci USA. 1990, 87: 3052-3056.
Low MG, Prasad AR: A phospholipase D specific for the phosphatidylinositol anchor of cell-surface proteins is abundant in plasma. Proc Natl Acad Sci USA. 1988, 85: 980-984.
Miyagi T, Wada T, Yamaguchi K, Hata K, Shiozaki K: Plasma membrane-associated sialidase as a crucial regulator of transmembrane signalling. J Biochem. 2008, 144: 279-285.
Baldelli P, Hernández-Guijo JM, Carabelli V, Novara M, Cesetti T, Andrés-Mateos E, Montiel C, Carbone E: Direct and remote modulation of L-channels in chromaffin cells: distinct actions on α1C and α1D subunits?. Mol Neurobiol. 2004, 29: 73-96.
Resnik N, Sepcic K, Plemenitas A, Windoffer R, Leube R, Veranic P: Desmosome assembly and cell-cell adhesion are membrane raft-dependent processes. J Biol Chem. 2011, 286: 1499-1507.
Fleming TP, Garrod DR, Elsmore AJ: Desmosome biogenesis in the mouse preimplantation embryo. Development. 1991, 112: 527-539.
Cummins PM: Occludin: one protein, many forms. Mol Cell Biol. 2012, 32: 242-250.
Guha SK, Rose ZB: The synthesis of mannose 1-phosphate in brain. Arch Biochem Biophys. 1985, 243: 168-173.
Becker DJ, Lowe JB: Fucose: biosynthesis and biological function in mammals. Glycobiology. 2003, 13: 41R-53R.
Sakanaka C, Sun TQ, Williams LT: New steps in the Wnt/β-catenin signal transduction pathway. Recent Prog Horm Res. 2000, 55: 225-236.
McKay RM, Peters JM, Graff JM: The casein kinase I family in Wnt signaling. Dev Biol. 2001, 235: 388-396.
He S, Pant D, Schiffmacher A, Meece A, Keefer CL: Lymphoid enhancer factor 1-mediated Wnt signaling promotes the initiation of trophoblast lineage differentiation in mouse embryonic stem cells. Stem Cells. 2008, 26: 842-849.
Granier C, Gurchenkov V, Perea-Gomez A, Camus A, Ott S, Papanayotou C, Iranzo J, Moreau A, Reid J, Koentges G, Sabéran-Djoneidi D, Collignon J: Nodal cis-regulatory elements reveal epiblast and primitive endoderm heterogeneity in the peri-implantation mouse embryo. Dev Biol. 2011, 349: 350-362.
Denicol AC, Dobbs KB, McLean KM, Carambula SF, Loureiro B, Hansen PJ: Canonical WNT signaling regulates development of bovine embryos to the blastocyst stage. Sci Rep. 2013, 3: 1266-
Kemler R, Hierholzer A, Kanzler B, Kuppig S, Hansen K, Taketo MM, de Vries WN, Knowles BB, Solter D: Stabilization of β-catenin in the mouse zygote leads to premature epithelial-mesenchymal transition in the epiblast. Development. 2004, 131: 5817-5824.
Xie H, Tranguch S, Jia X, Zhang H, Das SK, Dey SK, Kuo CJ, Wang H: Inactivation of nuclear Wnt-β-catenin signaling limits blastocyst competency for implantation. Development. 2008, 135: 717-727.
Vielhaber E, Eide E, Rivers A, Gao ZH, Virshup DM: Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon. Mol Cell Biol. 2000, 20: 4888-4899.
Fields SD, Perry BL, Perry GA, Cushman RA: Association between Period 1 mRNA levels in leukocytes and initiation of estrous cycles in peripubertal beef heifers. Biol Reprod. 2008, 78: 654-
Gwathmey TM, Ignotz GG, Mueller JL, Manjunath P, Suarez SS: Bovine seminal plasma proteins PDC-109, BSP-A3, and BSP-30-kDa share functional roles in storing sperm in the oviduct. Biol Reprod. 2006, 2006 (75): 501-507.
Killian GJ, Chapman DA, Rogowski LA: Fertility-associated proteins in Holstein bull seminal plasma. Biol Reprod. 1993, 49: 1202-1207.
Rojas FJ, Brush M, Moretti-Rojas I: Calpain-calpastatin: a novel, complete calcium-dependent protease system in human spermatozoa. Mol Hum Reprod. 1999, 5: 520-526.
Yudin AI, Goldberg E, Robertson KR, Overstreet JW: Calpain and calpastatin are located between the plasma membrane and outer acrosomal membrane of cynomolgus macaque spermatozoa. J Androl. 2000, 21: 721-729.
Bastián Y, Roa-Espitia AL, Mújica A, Hernández-González EO: Calpain modulates capacitation and acrosome reaction through cleavage of the spectrin cytoskeleton. Reproduction. 2010, 140: 673-684.
Ben-Aharon I, Haim K, Shalgi R, Ben-Yosef D: Expression and possible involvement of calpain isoforms in mammalian egg activation. Reproduction. 2005, 130: 165-175.
Bleil JD, Greve JM, Wassarman PM: Identification of a secondary sperm receptor in the mouse egg zona pellucida: role in maintenance of binding of acrosome-reacted sperm to eggs. Dev Biol. 1998, 128: 376-385.
Baibakov B, Boggs NA, Yauger B, Baibakov G, Dean J: Human sperm bind to the N-terminal domain of ZP2 in humanized zonae pellucidae in transgenic mice. J Cell Biol. 2012, 197: 897-905.
Dalbiès-Tran R, Papillier P, Pennetier S, Uzbekova S, Monget P: Bovine mater-like NALP9 is an oocyte marker gene. Mol Reprod Dev. 2005, 71: 414-421.
Thélie A, Papillier P, Pennetier S, Perreau C, Traverso JM, Uzbekova S, Mermillod P, Joly C, Humblot P, Dalbiès-Tran R: Differential regulation of abundance and deadenylation of maternal transcripts during bovine oocyte maturation in vitro and in vivo. BMC Dev Biol. 2007, 7: 125-
Romar R, De Santis T, Papillier P, Perreau C, Thélie A, Dell’Aquila ME, Mermillod P, Dalbiès-Tran R: Expression of maternal transcripts during bovine oocyte in vitro maturation is affected by donor age. Reprod Domest Anim. 2011, 46: e23-e30.
Hansen PJ: The immunology of early pregnancy in farm animals. Reprod Domest Anim. 2011, 46 (Suppl 3): 18-30.
Galvan MD, Greenlee-Wacker MC, Bohlson SS: C1q and phagocytosis: the perfect complement to a good meal. J Leukoc Biol. 2012, 92: 489-497.
Landmann R, Müller B, Zimmerli W: CD14, new aspects of ligand and signal diversity. Microbes Infect. 2000, 2: 295-304.
Ferrari S, Plebani A: Cross-talk between CD40 and CD40L: lessons from primary immune deficiencies. Curr Opin Allergy Clin Immunol. 2002, 2: 489-494.
Chiba T, Matsuzaka Y, Warita T, Sugoh T, Miyashita K, Tajima A, Nakamura M, Inoko H, Sato T, Kimura M: NFKBIL1 confers resistance to experimental autoimmune arthritis through the regulation of dendritic cell functions. Scand J Immunol. 2011, 73: 478-485.
Rios EJ, Piliponsky AM, Ra C, Kalesnikoff J, Galli SJ: Rabaptin-5 regulates receptor expression and functional activation in mast cells. Blood. 2008, 112: 4148-4157.
Kinchen JM, Ravichandran KS: Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010, 464: 778-782.
Meijerink E, Fries R, Vögeli P, Masabanda J, Wigger G, Stricker C, Neuenschwander S, Bertschinger HU, Stranzinger G: Two α(1,2) fucosyltransferase genes on porcine chromosome 6q11 are closely linked to the blood group inhibitor (S) and Escherichia coli F18 receptor (ECF18R) loci. Mamm Genome. 1997, 8: 736-741.
Bao WB, Ye L, Zhu J, Pan ZY, Zhu GQ, Huang XG, Wu SL: Evaluation of M307 of FUT1 gene as a genetic marker for disease resistance breeding of Sutai pigs. Mol Biol Rep. 2012, 39: 4223-4228.
Wang SJ, Liu WJ, Yang LG, Sargent CA, Liu HB, Wang C, Liu XD, Zhao SH, Affara NA, Liang AX, Zhang SJ: Effects of FUT1 gene mutation on resistance to infectious disease. Mol Biol Rep. 2012, 39: 2805-2810.
Horák P, Urban T, Dvorák J: The FUT1 and ESR genes–their variability and associations with reproduction in Prestice Black-Pied sows. J Anim Breed Genet. 2005, 122: 210-213.
Zhu H-Y, Zhao S, Zhao W, Su Y-H: Polymorphism of FUT1 gene and its relationship with litter size in northeast Hebao pigs. Animal Husbandry and Feed Science. 2009, 1: 3-
Hansen PJ, Soto P, Natzke RP: Mastitis and fertility in cattle - possible involvement of inflammation or immune activation in embryonic mortality. Am J Reprod Immunol. 2004, 51: 294-301.
Sheldon IM, Price SB, Cronin J, Gilbert RO, Gadsby JE: Mechanisms of infertility associated with clinical and subclinical endometritis in high producing dairy cattle. Reprod Domest Anim. 2009, 44 (Suppl 3): 1-9.
Santos JEP, Bisinotto RS, Ribeiro ES, Lima FS, Greco LF, Staples CR, Thatcher WW: Applying nutrition and physiology to improve reproduction in dairy cattle. Reproduction in Domestic Ruminants VII. Edited by: Smith MF, Lucy MC, Pate JL, Spencer TE. 2011, Nottingham, United Kingdom: Nottingham University Press, 387-403.
Lui HM, Chen J, Wang L, Naumovski L: ARMER, apoptotic regulator in the membrane of the endoplasmic reticulum, a novel inhibitor of apoptosis. Mol Cancer Res. 2003, 1: 508-18.
Guo X, Williams JG, Schug TT, Li X: DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010, 285: 13223-13232.
Isodono K, Takahashi T, Imoto H, Nakanishi N, Ogata T, Asada S, Adachi A, Ueyama T, Oh H, Matsubara H: PARM-1 is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS One. 2010, 5: e9746-
Høst E, Mikkelsen AL, Lindenberg S, Smidt-Jensen S: Apoptosis in human cumulus cells in relation to maturation stage and cleavage of the corresponding oocyte. Acta Obstet Gynecol Scand. 2000, 79: 936-940.
Lee KS, Joo BS, Na YJ, Yoon MS, Choi OH, Kim WW: Cumulus cells apoptosis as an indicator to predict the quality of oocytes and the outcome of IVF-ET. J Assist Reprod Genet. 2001, 18: 490-498.
Feng WG, Sui HS, Han ZB, Chang ZL, Zhou P, Liu DJ, Bao S, Tan JH: Effects of follicular atresia and size on the developmental competence of bovine oocytes: a study using the well-in-drop culture system. Theriogenology. 2007, 67: 1339-1350.
Block J, Hansen PJ: Interaction between season and culture with insulin-like growth factor-1 on survival of in vitro produced embryos following transfer to lactating dairy cows. Theriogenology. 2007, 67: 1518-1529.
Loureiro B, Bonilla L, Block J, Fear JM, Bonilla AQ, Hansen PJ: Colony-stimulating factor 2 (CSF-2) improves development and posttransfer survival of bovine embryos produced in vitro. Endocrinology. 2009, 150: 5046-5054.
Jousan FD, Hansen PJ: Insulin-like growth factor-I promotes resistance of bovine preimplantation embryos to heat shock through actions independent of its anti-apoptotic actions requiring PI3K signaling. Mol Reprod Dev. 2007, 74: 189-196.
Duncan AJ, Bitner-Glindzicz M, Meunier B, Costello H, Hargreaves IP, López LC, Hirano M, Quinzii CM, Sadowski MI, Hardy J, Singleton A, Clayton PT, Rahman S: A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009, 84: 558-566.
Crane FL: Biochemical functions of coenzyme Q10. J Am Coll Nutr. 2001, 20: 591-598.
Aschenbach JR, Kristensen NB, Donkin SS, Hammon HM, Penner GB: Gluconeogenesis in dairy cows: the secret of making sweet milk from sour dough. IUBMB Life. 2010, 62: 869-677.
Asiedu M, Wu D, Matsumura F, Wei Q: Centrosome/spindle pole-associated protein regulates cytokinesis via promoting the recruitment of MyoGEF to the central spindle. Mol Biol Cell. 2009, 20: 1428-1440.
Zeng F, Tian Y, Shi S, Wu Q, Liu S, Zheng H, Yue L, Li Y: Identification of mouse MARVELD1 as a microtubule associated protein that inhibits cell cycle progression and migration. Mol Cells. 2011, 31: 267-274.
Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, Bortoletto G, Scannapieco P, Salamon M, Mouly V, Valle G, Lanfranchi G: ZASP: a new Z-band alternatively spliced PDZ-motif protein. J Cell Biol. 1999, 146: 465-475.
Martinson HG: An active role for splicing in 3′-end formation. Wiley Interdiscip Rev RNA 1011. 2011, 2: 459-470.
Tuoc TC, Stoykova A: Roles of the ubiquitin-proteosome system in neurogenesis. Cell Cycle. 2010, 9: 3174-3180.
Shang F, Taylor A: Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 2011, 51: 5-16.
Voutsadakis IA: The ubiquitin-proteasome system and signal transduction pathways regulating epithelial mesenchymal transition of cancer. J Biomed Sci. 2012, 19: 67-
Acknowledgements
Research was supported by the Florida Dairy Checkoff Program and by Agriculture and Food Research Initiative Competitive Grant no. 2013-68004-20365 from the USDA National Institute of Food and Agriculture. The authors thank the USDA CDDR and USDA National Animal Germplasm Program for donating semen used in the current study, Taurus Service Inc. for reducing the price of semen, and Maureen Long (University of Florida) for assistance with the IPA analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: SDC, PJH, JBC. Performed the experiments: SDC. Analyzed the data: SDC, DJN, JBC, PJH. Wrote initial drafts of the paper: SDC, PJH. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2013_1120_MOESM1_ESM.pdf
Additional file 1: Table S1: Predicted transmitting ability for selected traits of bulls used for genotyping. Table S2. Source of genes included in SNP array. Table S3. Full list of SNPs used in array. (PDF 42 KB)
12863_2013_1120_MOESM2_ESM.pdf
Additional file 2: Table S4: Correlations among predicted transmitting ability for traits on bulls used for genotyping. Table S5. Correlations, within DPR class, among predicted transmitting ability for traits on bulls used for genotyping. Table S6. Information for all SNPs with minor allele frequencies (MAF) > 5% and call rates > 70%. (PDF 71 KB)
12863_2013_1120_MOESM3_ESM.pdf
Additional file 3: Table S7: Genetic standard deviations of SNPs associated with daughter pregnancy rate. Table S8. Effect of tissue type in which genes were identified on the percent of genes that were significantly associated with daughter pregnancy rate (DPR). Figure S1. The ubiquitin pathway contains an overrepresentation of daughter pregnancy rate genes. Figure S2. Transcription factors which regulate daughter pregnancy rate genes. (PDF 164 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cochran, S.D., Cole, J.B., Null, D.J. et al. Discovery of single nucleotide polymorphisms in candidate genes associated with fertility and production traits in Holstein cattle. BMC Genet 14, 49 (2013). https://doi.org/10.1186/1471-2156-14-49
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-14-49