
Abstract
Background
Recent phylogenetic studies have revealed that the mitochondrial genome of the angiosperm Silene noctiflora (Caryophyllaceae) has experienced a massive mutation-driven acceleration in substitution rate, placing it among the fastest evolving eukaryotic genomes ever identified. To date, it appears that other species within Silene have maintained more typical substitution rates, suggesting that the acceleration in S. noctiflora is a recent and isolated evolutionary event. This assessment, however, is based on a very limited sampling of taxa within this diverse genus.
Results
We analyzed the substitution rates in 4 mitochondrial genes (atp1, atp9, cox3 and nad9) across a broad sample of 74 species within Silene and related genera in the tribe Sileneae. We found that S. noctiflora shares its history of elevated mitochondrial substitution rate with the closely related species S. turkestanica. Another section of the genus (Conoimorpha) has experienced an acceleration of comparable magnitude. The phylogenetic data remain ambiguous as to whether the accelerations in these two clades represent independent evolutionary events or a single ancestral change. Rate variation among genes was equally dramatic. Most of the genus exhibited elevated rates for atp9 such that the average tree-wide substitution rate for this gene approached the values for the fastest evolving branches in the other three genes. In addition, some species exhibited major accelerations in atp1 and/or cox3 with no correlated change in other genes. Rates of non-synonymous substitution did not increase proportionally with synonymous rates but instead remained low and relatively invariant.
Conclusion
The patterns of phylogenetic divergence within Sileneae suggest enormous variability in plant mitochondrial mutation rates and reveal a complex interaction of gene and species effects. The variation in rates across genomic and phylogenetic scales raises questions about the mechanisms responsible for the evolution of mutation rates in plant mitochondrial genomes.
Similar content being viewed by others


Background
Substitution rates in plant mitochondrial genomes are generally low relative to their nuclear and chloroplast counterparts, as well as relative to the mitochondrial genomes of other organisms [1–3]. In fact, absolute rates of sequence evolution in seed plant mitochondrial DNA (mtDNA) are among the slowest ever estimated (Figure 1; [4]). A series of recent studies, however, has revealed notable exceptions to this generalization [4–7]. There are angiosperm species that not only deviate from the slow substitution rates typical of plant mtDNA but also exhibit some of the highest eukaryotic substitution rates ever documented (Figure 1). With such a substantial fraction of known rate variation captured in a relatively small twig within the tree of life, plant mitochondrial genomes represent an intriguing system for investigating the evolutionary forces that shape substitution rates [8–14].

Diversity in substitution rates. Synonymous substitution rates per site per billion years (SSB) for different organisms and genomes plotted on a log scale. Black bars represent seed plant mitochondrial genomes. Average rates for animal taxa from Lynch et al. [20]; angiosperm chloroplast and nuclear estimates from Wolfe et al. [74]; mitochondrial rates for individual plant species taken from Cho et al. [5] and Mower et al. [4].
Studies of rate accelerations in plant mitochondrial genomes have consistently shown that these effects are most pronounced at so-called synonymous sites, which do not affect the corresponding amino acid sequence (e.g. [5]). One of the pillars of the neutral theory of molecular evolution is that the rate of neutral substitutions (i.e. those with no fitness effect) is expected to equal the mutation rate [15]. Synonymous substitutions are not completely neutral, however. They are subject to a variety of selection pressures including translational efficiency, mRNA stability and the conservation of regulatory motifs (reviewed in [16]), and direct measurements of mutation rates can be more than an order of magnitude higher than those estimated from synonymous substitution rates [17]. Nevertheless, synonymous sites still offer one of our best approximations of the underlying mutation rate. Therefore, considering the absence of well-supported alternative hypotheses, the extreme synonymous substitution rates observed in certain plant mitochondrial genomes are most likely a result of mutational acceleration.
Silene noctiflora (Caryophyllaceae) is a recent addition to a growing list of angiosperms exhibiting major accelerations in mitochondrial synonymous substitution rate [4, 7]. In other well-documented examples (e.g. Plantago and Pelargonium), rate accelerations appear relatively old (ca. 30-80 million years) having preceded the divergence of large clades or even an entire genus [6]. In contrast, the extreme mitochondrial substitution rates of S. noctiflora appear unique relative to other Silene species, suggesting a very recent acceleration. Estimates of mitochondrial substitution rate, however, are available for only a few Silene species, representing a tiny fraction of this large and diverse genus. The sparse sampling severely limits the phylogenetic resolution to detect historical changes in substitution rate.
The scarcity of mitochondrial sequence data within Silene reflects a broader under-representation of plant mtDNA in studies of molecular evolution. Whereas chloroplast DNA (cpDNA) and animal mtDNA are utilized extensively in phylogenetic studies, the low baseline substitution rates and growing evidence for rate heterogeneity in plant mtDNA often limit its utility in this context--particularly at local phylogenetic scales [18]. Understanding the causes and consequences of mutation rate variation is a fundamental problem in evolutionary biology [12, 19–21], but the lack of plant mtDNA sequence data is a hindrance to investigating this question. To characterize the pattern of mitochondrial substitution rate variation throughout Silene and related genera, we sequenced four mitochondrial loci in a sample of 74 species that were selected to capture the phylogenetic diversity of this genus and its closest relatives (Table 1). To our knowledge, this effort represents the most extensive species-level sampling to date of mitochondrial sequence divergence in a plant genus.
To compare absolute substitution rates in a gene across lineages requires an estimate of the genealogy with dated nodes (i.e. divergence times). In cases of extreme rate variation, generating such a tree directly from the gene in question is problematic. With rate variation, slowly-evolving taxa can be difficult to resolve, and long branch attraction can favor incorrect topologies [22]. Even with an accurate topology, rate variation can bias the estimate of divergence times with molecular clock based methods. For this reason, previous studies of substitution rate variation in plant mitochondrial genomes have constrained their analyses based on phylogenies and divergence times inferred from nuclear and chloroplasts sequences.
Because both mitochondrial and chloroplast genomes are predominantly maternally inherited in Silene, they are expected to share a common genealogy [23, 24] (although breakdowns in uniparental inheritance may potentially disrupt this relationship [25–27]). Therefore, we chose the chloroplast gene matK to estimate phylogenetic relationships and divergence times. This gene has proven to be highly informative in phylogenetic reconstruction, partly because of its high rates of substitution [28, 29]. It has also been used in two recent analyses of divergence times within Silene and the Caryophyllaceae [4, 30].
We identified substantial rate accelerations in multiple lineages within the Silene phylogeny as well as major rate differences among mitochondrial genes. Here, we discuss the complex patterns of mitochondrial rate variation in the genus Silene and the implications they have for the evolution of mitochondrial mutation rates and the patterns of selection on mtDNA at the sequence level.
Results
Chloroplast DNA phylogeny
Likelihood, parsimony and Bayesian phylogenetic methods were in general agreement for the matK dataset. The 70% parsimony bootstrap consensus tree (Figure 2) did not conflict with any of the nodes from either the ML or Bayesian analysis. The results were also generally consistent with previous cpDNA studies of the tribe Sileneae [31, 32]. The analysis recovered the two previously identified subgenera (e.g. [32])--Silene and Behenantha (Otth.) Endl. (=subgenus Behen (Dumort.) Rohrb.)--along with the relationships among the major clades in subgenus Silene. There was, however, incomplete resolution in some parts of the tree--particularly among the major lineages within subgenus Behenantha, which appear as a large radiation. Four Silene species were not grouped with either of the two major subgenera. As found in the analysis of other chloroplast loci, S. sordida was placed in a clade with Lychnis [32]. Silene odontopetala was also assigned to this clade with strong support. The relationships between subgenus Behenantha, subgenus Silene, the Lychnis/S. odontopetala/S. sordida clade, and a fourth lineage consisting solely of S. cordifolia could not be confidently resolved. Finally, there was unexpected support for a sister relationship between S. delicatula and the rest of our Silene/Lychnis sample.

Chronogram showing divergence times estimated in BEAST based on full-length matK coding sequence. Time scale is in millions of years. Error bars at each node show 95% HPD for node age. Values to the right of each node show Bayesian posterior probability and parsimony bootstrap support (in that order) for the corresponding clade. Tree topology was constrained based on 70% parsimony bootstrap consensus.
Divergence times
We used three different dating methods, which produced roughly similar estimates of divergence times, but there was a consistent pattern distinguishing them [see Additional files 1 and 2]. Specifically, the Langley-Fitch method produced the youngest estimates of divergence times within Sileneae, while a penalized likelihood method produced the oldest. For example, the estimated age of the root node for the entire Silene/Lychnis clade differed by 50% between the two methods (21.0 vs. 14.0 Myr). The BEAST model (Figure 2) generally produced intermediate estimates of divergence time relative to the other two methods. Only the BEAST values were used for subsequent rate analyses, so the uncertainty in divergence times should be considered when interpreting absolute substitution rate estimates.
Mitochondrial rate variation
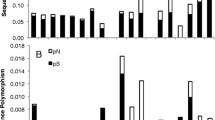
Branch lengths in terms of both synonymous (d S ) and non-synonymous (d N ) substitutions per site for each mitochondrial gene are shown in Figure 3. All four genes show little divergence at non-synonymous sites across the entire tree (Table 2). In addition, they all share a pattern of extreme synonymous divergence in six Silene species that can be divided into two clear clades: (1) the previously characterized S. noctiflora along with its close relative S. turkestanica and (2) S. ammophila, S. conica, S. conoidea, and S. macrodonta, which all belong to section Conoimorpha. Beyond those similarities, the four mitochondrial genes differ markedly in synonymous branch lengths (Figure 3). Very little divergence is observed in nad9 outside of the aforementioned six species. Synonymous divergence is similarly low throughout much of the cox3 and atp1 trees, but there are a number of species that exhibit substantial divergence, particularly within subgenus Silene. This group includes S. nutans which, despite showing no sign of abnormal divergence in cox3 and nad9, is highly divergent for atp1. Finally, synonymous divergence in atp9 is extreme and highly variable throughout most of the genus Silene, although many of the outgroup genera exhibit typically low levels of divergence. The total synonymous tree length is approximately 9-fold larger for atp9 than the slowly-evolving nad9. This gap widens to 41-fold if the six taxa that have accelerated rates across all genes are excluded from the analysis (Table 2).

d N and d S trees for mitochondrial genes. Branch lengths are in terms of non-synonymous (d N ) or synonymous (d S ) substitutions per site as estimated by PAML under a constrained topology. The scale is the same for all trees.
As expected given the enormous variation in mitochondrial divergence across species, absolute synonymous substitution rates (R S ) differ dramatically throughout the tribe Sileneae (Figure 4, [see Additional files 3 and 4]). The outgroups to Silene tend to have R S values of less than 0.5 substitutions per site per billion years (SSB), and certain branches have an estimated R S of 0 because they lack a single synonymous substitution (Figure 4). Many lineages within Silene have maintained these low rates. At the other extreme, the rapidly-evolving Silene lineages have R S values that are more than two orders of magnitude greater than the low rates of Beta vulgaris and other outgroups. The fastest rate estimates observed in the entire dataset were found in the atp9 tree. The internal branch subtending the minimally inclusive clade that contains S. succulenta and S. imbricata had an estimated R S value of 392 SSB. The fastest terminal branch in the atp9 tree was that of S. schafta with a rate of 292 SSB, although it should be noted that the error associated with atp9 rate estimates for individual branches was generally large [see Additional file 4]. R S and R N values were both positively correlated across atp1, cox3 and nad9, but this correlation broke down in comparisons with atp9 and matK (Table 3). In addition, R S and R N values were significantly correlated with each other within genes for atp1, cox3 and nad9 but not for the other two loci.

Phylogenetic variation in R S . Branches labelled with absolute synonymous substitution rates and approximate standard errors based on concatenation of nad9, cox3 and atp1. Branch colors indicate fast (red) and slow (blue) rates.
Evolutionary congruence between mitochondrial and chloroplast genomes
We utilized a constrained topology derived from cpDNA to analyze evolutionary rates in mtDNA, reflecting the assumption that the two organelle genomes share a single genealogy. Although the mitochondrial genes often yielded limited phylogenetic signal because of the dual problems of low variation and long branch attraction, there was some evidence to support phylogenetic congruence between these genomes [see Additional file 5]. For example, S. hookeri and S. menziesii were consistently paired by both chloroplast and mitochondrial genes, suggesting that the allopolyploid S. hookeri inherited both of its cytoplasmic genomes from the S. menziesii parental lineage [33]. In addition, the rapidly evolving atp9 gene produced a tree that generally agreed with the chloroplast matK topology at younger nodes, which are presumably less susceptible to saturation at synonymous sites.
There were a large number of incongruencies between chloroplast and mitochondrial trees, but they were generally lacking in support. Perhaps the most suspicious example of conflict between mitochondrial and chloroplast topologies was the placement of S. samojedora, S. seoulensis, S. zawadzkii and the major accelerated species from subgenus Behenantha in a clade otherwise populated by subgenus Silene in the cox3 tree [see Additional file 5]. Although this clade was supported by 74% of bootstrap replicates, inspection of the alignments showed that the grouping was based entirely on a single 6 bp region with 5 substitutions, raising doubts about the independence of those characters. Overall, we found no overwhelming evidence of conflicts between mitochondrial and chloroplast topologies, but the lack of mitochondrial divergence in many lineages gave us little statistical power. Therefore, it is possible that topological inaccuracies in our constraint tree could have led to misidentification of small mitochondrial rate accelerations, but given the phylogenetic scale of our analysis, it is unlikely that any of the major rate changes was an artifact of topological conflicts.
To date, studies of angiosperms with major increases in plant mitochondrial substitution rates (e.g. Plantago, Pelargonium and Silene) have concluded that the observed accelerations are largely independent of evolutionary rates in the chloroplast or nuclear genomes (although there is growing evidence for accelerated sequence and structural evolution in the chloroplast genomes of species with high mitochondrial substitution rates [34–36]). In our dataset, we found little substitution rate variation among species for the chloroplast matK gene and no significant correlation between mitochondrial and chloroplast rates (Table 3).
Discussion
Mutation rate variation among species
Silene noctiflora has been shown to have dramatically accelerated rates of mitochondrial evolution relative to its congeners [4, 7]. We examined the phylogenetic distribution of this rate acceleration within the tribe Sileneae and identified six Silene species grouped into two clades that exhibited major increases in synonymous substitution rates across all four loci examined (Figure 3). As an illustration of the magnitude of these accelerations, we note the average synonymous pairwise divergence between these two closely-related clades within Silene subgenus Behenantha exceeds the divergence typically observed between flowering plants and liverworts--the deepest split in the land plant phylogeny [37]. Based on the currently available data in seed plants, the synonymous substitution rates exhibited by these rapidly-evolving lineages (Figure 4) are exceeded only by the fastest lineages of Plantago (Figure 1) [5]. In addition, the observed rates are on par with average estimates for mammalian mtDNA, although they still fall well below the fastest mammalian rates [38]. As discussed above (see Background), the observed differences in synonymous substitution rates most likely reflect differences in the underlying mutation rate.
The phylogenetic data remain ambiguous with respect to whether the two clades with rate accelerations represent independent evolutionary events. The matK tree does not strongly support or reject a monophyletic relationship between S. noctiflora/S. turkestanica and section Conoimorpha (Figure 2; [see Additional files 6 and 7]). More thorough phylogenetic analyses of these taxa have recently been conducted, utilizing both chloroplast and nuclear loci [39]. These studies have found that, while cpDNA sequences suggest phylogenetic independence between the two clades, at least some nuclear loci support monophyly. Therefore it is possible but inconclusive that both high rate clades are sister taxa that inherited an accelerated mitochondrial substitution rate from a common ancestor. If so, the two clades must have split shortly after that acceleration, because internal branches shared by the two lineages in the mitochondrial gene trees are quite short relative to the divergence between the lineages [see Additional file 5]. Resolving these phylogenetic relationships could prove difficult because previous studies have shown that the evolutionary history of subgenus Behenantha may be complicated by reticulation [32, 40], such that relationships differ across genes and genomes
Comparisons of mitochondrial sequences from multiple populations of S. noctiflora have revealed very low levels of polymorphism, suggesting that the historically high mutation rates in this lineage may have undergone a reversion to more typical levels ([41] and unpublished data). This conclusion was, at least partially, supported by our phylogenetic data. The terminal branches for S. noctiflora and S. turkestanica exhibited a marked reduction in R S values relative to the ancestral rate for that clade (Figure 4). In contrast, the patterns of divergence within section Conoimorpha gave little indication of rate reversions.
The genus Silene is characterized by great diversity in breeding system and life history, and there has been substantial interest in how these traits may be related to molecular evolution in mitochondrial genomes [14, 26, 41–44]. There is no clear correlation between breeding system/life history and rate acceleration. The species exhibiting rate acceleration across all four mitochondrial genes are all hermaphroditic/gynomonoecious annuals with the exception of S. turkestanica, which is perennial. However, there are at least ten additional annual lineages represented in our sampling, and breeding system (hermaphroditic/gynomonoecious or gynodioecious) has yet to be determined for most species.
Mutation rate variation among genes
Substitution rates commonly differ among regions within a genome because of variation in selection and/or mutational pressure, and a previous study had already identified substantial rate heterogeneity among Silene mitochondrial genes [45]. Nevertheless, the differences in synonymous substitution rates among mitochondrial genes in the current study are surprisingly large. If the six species that show universal acceleration across all four mitochondrial genes are excluded, atp9 appears to be evolving more than 40 times faster than nad9 at synonymous sites, while cox3 and atp1 fall in between these extremes.
The extreme elevation in atp9 substitution rates calls into question whether a biological mechanism other than an increase in the mutation rate might be responsible. The obvious alternatives to explain high levels of divergence include horizontal gene transfer (HGT) from distantly related species [46], maintenance of ancient, trans-specific polymorphism by balancing selection [26, 41, 44, 47], re-localization of the gene to the higher mutation rate environment of the nuclear genome [48], or relaxed selection in a non-functional pseudogene [49].
None of these explanations, however, are fully consistent with the data. To explain the observed levels of divergence based on HGT without an increase in evolutionary rates would require multiple phylogenetically distant donor species (i.e. outside the angiosperms). Phylogenetic analysis of atp9, however, clearly places these sequences within the Caryophyllaceae ([Additional file 5] and unpublished data; note that this argument also applies to the lineage-specific divergence in S. noctiflora/S. turkestanica and section Conoimorpha). Likewise, in the absence of rate acceleration, an explanation based on balancing selection alone would require that polymorphism be maintained for hundreds of millions of years. Such a model seems extremely unlikely and even still could not explain the retention of partial phylogenetic congruence between atp9 and matK. Of course, the fact that balancing selection alone cannot explain the pattern of divergence in atp9 does not rule out the possibility that balancing selection has been acting on atp9 and other mitochondrial genes in Silene.
It is unlikely that atp9 has been functionally transferred to the nucleus in at least four Silene species--S. latifolia, S. noctiflora, S. vulgaris and S. paradoxa. Whole mitochondrial genome sequences confirm that atp9 is mitochondrially encoded in both S. latifolia and S. noctiflora (Sloan et al., unpublished data). In addition, the gene has been shown to be maternally inherited in S. vulgaris [26]. Comparing cDNA and genomic sequence also confirms that atp9 contains a site that undergoes C-to-U RNA editing in S. paradoxa--a process that is characteristic of organellar but not nuclear genes in plants (Sloan et al., unpublished data). Although we cannot definitively rule out the possibility of nuclear transfer, these data strongly suggest that nuclear transfer is not the driving force behind the pattern of elevated substitution rate observed in atp9.
It is also clear that atp9 is functional based on its low ω values and the absence of internal stop codons. Therefore, we conclude that the most likely explanation for the high levels of divergence is an increased mutation rate that is specific to atp9 (or a subset of the mitochondrial genome that includes atp9).
The molecular evolution of atp9 could be influenced by the presence of multiple gene copies in at least some species (see Methods). The existence of multiple copies could reflect heteroplasmy resulting from paternal leakage [25], non-functional paralogs in the mitochondria or other genomes [50], or the existence of multiple functional mitochondrial copies [51]. It is conceivable that atp9 is located in a region of active recombination within the Silene mitochondrial genome or is experiencing frequent retroprocessing back into the genome from mRNA. Both of these processes may be mutagenic as well as lead to gene duplication and, therefore, would be consistent with our observations [6, 52, 53]. Alternatively, high mutations rates in atp9 may have simply increased divergence between heteroplasmic and/or paralogous copies, thereby enhancing our ability to detect multiple copies of atp9 even though they exist for other genes as well. Sequencing complete mitochondrial genomes, analyzing relative copy number of atp9 variants, and sampling multiple individuals per species would help distinguish between these possibilities. In a sample of individuals from 40 different populations of S. vulgaris, we found 4 individuals with multiple atp9 copies, and certain variants were only found in multi-copy individuals (unpublished data). This result suggests there is polymorphism for the presence of a paralogous copy within S. vulgaris, although heteroplasmy involving a rare haplotype is also plausible.
The acceleration in atp9 appears to be common to most of Silene/Lychnis. In contrast, most of the other Sileneae genera exhibit more conventional substitution rates for atp9, although their rates are still elevated on average. This pattern is consistent with an atp9-specific increase in substitution rate very early in the divergence of Silene, which may have been magnified by further accelerations in local areas of the genus.
A previous study of mitochondrial substitution rate variation across the seed plant phylogeny identified a handful of individual species exhibiting elevated divergence in one gene but not others [4]. Our observations of rate variation in atp9 within Silene indicate that such gene-specific effects can be maintained across large clades of species over millions of years. We also found that these effects can occur quite locally. Most notably, S. nutans exhibited an R S value of 80 SSB for atp1 (a rate that exceeds all other species for that gene), but it showed no sign of acceleration in nad9 or cox3 (Figure 3). A number of other species showed more modest rate increases in atp1 and/or cox3 without correlated accelerations in other genes. These patterns may reflect local mutational effects within the genome. Alternatively, given the mounting evidence for recombination in plant mtDNA [41, 46, 54] and the existence of rate variation both within and among species [7], rate discrepancies between genes may be the result of recombination between genomes with different mutational histories. Finally, the possibility of nuclear transfer for a gene such as atp1 in S. nutans should also be considered [4].
Evolution at synonymous and non-synonymous sites
Despite the massive variation in synonymous substitution rates among genes and species, we found that rates of non-synonymous substitution generally remained low (although there was a positive correlation between R N and R S across branches: Figure 3, Table 3). Across genes, R S values vary by 9 to 41-fold (depending on whether the six species with apparent genome-wide accelerations are included), while R N values vary by only 2 to 3-fold (Table 2). As a result, there is an apparent negative relationship between R S and the ratio of non-synonymous to synonymous changes (ω). While R S is commonly interpreted as a measure of the mutation rate, ω is used as an estimate of the intensity/efficacy of purifying selection (i.e. "the selective sieve" [55]). Under these interpretations, our data would suggest that genes experiencing high mutations rates also face greater purifying selection. In contrast, the opposite pattern has been observed in comparisons of nuclear genes in mammals [56].
The relationship between R S and ω among mitochondrial genes in Silene should be confirmed in a larger sample, because we have examined only 4 loci in the present study, and atp9 may generally be subject to strong purifying selection [57]. Whether R S and ω can be reliably interpreted as measures of mutation rate and purifying selection depends on the distribution of fitness effects for mutations at synonymous and non-synonymous sites, which are not well understood in plant mitochondrial genomes. These distributions will dictate how the synonymous and non-synonymous substitution rates scale with the mutation rate.
In a comparison of sequence divergence in 15 protein-coding mitochondrial genes between angiosperms and the liverwort Marchantia, Laroche et al. [57] found much greater variation among genes in d N than in d S --the opposite of what we observed. This discrepancy highlights the importance of phylogenetic scale in these studies. Across deep nodes in the land plant phylogeny, local differences in gene-specific mutation rates are apparently averaged out, and variation in the magnitude of purifying/positive selection among genes becomes the primary determinant of evolutionary rates. In contrast, at the local phylogenetic scale of our study, the signature of gene-specific differences in mutation rate is apparently maintained.
Because of the high variance and abundance of 0 values associated with short branches in our analysis, it is difficult to test for the same relationship between R S and ω across lineages that we observed across genes. We did see, however, that removing the rapidly evolving branches from each tree raised the ω ratio for all four genes, suggesting that the same pattern may hold. Mower et al. [4] conducted a broad phylogenetic survey of seed plants in which short branch lengths would be expected to be less of a problem. Their data showed a strong negative relationship between R S and ω across lineages (see also [53]). Therefore, it appears that major increases in mitochondrial synonymous substitution rate--either gene or taxon-specific--are accompanied by a less than proportional increase in non-synonymous substitution rate such that the effects of an apparent increased mutational pressure on amino acid sequences are greatly dampened.
Uncertainty in divergence time estimates
We used molecular clock based methods to estimate divergence times in our matK gene tree. The estimated ages were generally older than estimates from two previous studies [4, 30]. The discrepancy between these studies is likely attributable to two major differences. First, there is simple difference in calibration age between our study and the analysis of Mower et al. (2007), which utilized an age of 38 Myr for the Beta/Silene divergence derived from a broader molecular clock analysis of the angiosperms [58]. This date appears to be in conflict with our fossil calibration point, as all three of our analyses estimate the age of the Beta/Silene split to be at least 52 Myr old. This distinction, however, cannot explain the contrasting results between our study and that of Frajman et al. [30], because we used essentially the same calibration point. Instead, we note that there was a significant difference in sampling schemes between these two studies. Our focus on the genus Silene produced a very imbalanced topology with much denser branching in certain parts of the tree than others. In contrast, Frajman et al. [30] utilized a much more balanced phylogenetic sampling. Because it is easier to detect multiple substitutions at the same site in regions with lots of branching, there is a tendency to estimate longer branch lengths in species-rich parts of a phylogeny (the "node density effect" [59]). This effect may contribute to our older age estimates within the tribe Sileneae. Because of this potential bias, the uncertainty over calibration points and the many assumptions associated with molecular clock based dating, it is important to stress that the divergence times used in this analysis should be considered only as approximations.
Divergence time estimates are necessary to calculate absolute substitution rates, so dating uncertainty should be considered in comparing absolute rates across studies. For example, re-calibrating node ages within Silene to correspond with the 12 Myr divergence time for the genus estimated by Frajman et al. [30] would increase our substitution rate estimates by approximately 50%. In relative terms, however, our three dating analyses were quite consistent within Sileneae. Therefore, our estimates of proportional variation in substitution rate across species and genes are less sensitive to dating method.
Conclusion
Based on our analysis of mitochondrial divergence within the tribe Sileneae, we conclude that mutational acceleration is not restricted to a single species nor is it completely confined to a small number of high rate lineages. The patterns of divergence in atp9 illustrated that elevated rates have been maintained throughout much of the genus Silene for at least one mitochondrial gene, highlighting a complex gene × species interaction in the distribution of rate variation. The diversity in phylogenetic and genomics scale suggests that there is no simple rule or single mechanism underlying mutation rate variation in plant mitochondrial genomes. Elucidating the mechanistic forces that shape mutation rate variation should represent a high priority in the field of plant mitochondrial genomics. Silene was targeted for this in-depth sampling of species-level mitochondrial divergence because of a priori knowledge of the rate acceleration in S. noctiflora. Determining whether the patterns of rate variation among species and among genes in Silene are broadly representative of angiosperm genera or represent something unique about the molecular evolution of Silene will require similar levels of sampling in taxa that currently show no evidence of rate increase.
Methods
Study species
Silene (Caryophyllaceae) comprises approximately 700 predominantly herbaceous species that vary substantially in life history and breeding system [60]. The genus has become a model system for diverse areas of research with a particular focus on the molecular evolution of organelle genomes, including studies of population genetics [61, 62], organelle transmission [25, 27], evolutionary rates [4, 7, 35, 45], and cytoplasmic male sterility [26, 44]. Silene belongs to the tribe Sileneae, which has been the subject of extensive and ongoing phylogenetic analysis [30–32, 63–65]. Taxa were selected so as to represent major groups that will appear in a forthcoming revised taxonomy of the genus (Oxelman et al. in prep). For this study, we used a combination of field collected samples and preserved herbarium specimens along with previously published sequence data. Sample collection and voucher information are summarized in Table 1.
DNA extraction, PCR and Sequencing
We extracted total genomic DNA from each sample. For silica-dried samples and herbarium specimens, we followed the protocol described by Oxelman et al. [31] and performed subsequent purification using the Qiagen QIAquick Purification Kit protocol, Ultra Silica Bead kit (ABgene), or GFX PCR DNA and Gel Band Purification Kit (Amersham Biosciences). For fresh tissue samples, extractions were performed using the Qiagen Plant DNeasy Kit.
We PCR amplified the full-length coding sequence of the chloroplast gene maturase K (matK) and portions of four mitochondrial protein coding genes: ATP synthase subunit 1 (atp1), ATP synthase subunit 9 (atp9), cytochrome c oxidase subunit 3 (cox3) and NADH dehydrogenase subunit 9 (nad9). [See Additional file 8 for PCR primer sequences.]
PCR products were cleaned with Exonuclease I and shrimp alkaline phosphatase (USB Corporation), cycle sequenced with BigDye v3.1 (Applied Biosystems), and analyzed on an ABI 3130 × l capillary sequencer. Automated basecalls were edited manually using published Beta vulgaris sequences as a reference for reading frame, and sequences were assembled into contigs using Sequencher v4.5 (Gene Codes). All sequences obtained for S. sorensenis were identical to those from S. involucrata, so S. sorensenis was excluded to simplify subsequent analysis. DNA sequences have been submitted to GenBank [see Additional file 9 for accession numbers]. Sequence alignments were generated using the Clustal function imbedded in MEGA v4.0 [66] and edited manually [see Additional file 10].
matKphylogenetic analysis and dating
We estimated the phylogeny of our sample based on the matK dataset, using both maximum likelihood (ML) and maximum parsimony (MP) criteria in PAUP* v4.0b10 [67]. In addition to the species listed in Table 1, our analysis also included matK sequences from GenBank for the following outgroups: Beta vulgaris (Amaranthaceae), Illecebrum verticillatum (Caryophyllaceae) and Scleranthus perennis (Caryophyllaceae). Our ML search employed a GTR+Γ substitution model with fixed parameter values identified based on an analysis of our full matK dataset (including outgroups) using the AIC method in ModelTest v3.7 [68]. The ML topology was identified with a heuristic search using the TBR branch swapping algorithm, the MULTREES option in effect, and random addition of sequences with 10 replicates. A MP bootstrap analysis based on 1000 replicate datasets was performed using the same heuristic search settings except with MULTREES off. We performed ML and MP analyses in the same fashion for each mitochondrial gene.
We used three different techniques to estimate divergence times from our matK gene tree: (1) a Bayesian relaxed clock model implemented in BEAST v1.4.8 [69], (2) a penalized likelihood (PL) method, and (3) the Langley-Fitch (LF) method. The latter two methods were both performed in r8s v1.71 [70]. The LF model is a maximum likelihood strict molecular clock method that enforces a constant substitution rate over the entire tree. The other two methods allow for rate variation among branches. The PL approach assumes that rates are correlated across adjacent branches and penalizes models that require rapid rate changes within the tree. In contrast, the BEAST analysis constrained the rate variation among branches to a lognormal distribution but placed no restriction on correlations between adjacent branches. All dating analyses incorporated an extra outgroup, Nepenthes glabrata (Nepenthaceae), which was added solely to determine the position of the root along the Beta vulgaris branch. It was pruned from the resulting trees and discarded from all subsequent analyses. We used a calibration time of 34 million years (Myr) for the split between Scleranthus and Sileneae, which corresponds to the recent analysis of Frajman et al. [30] and the fossil evidence described therein.
The BEAST analysis was conducted with a GTR+Γ model of substitution with 4 rate categories, empirical base frequencies and a birth-death process tree prior. We defined a monophyletic ingroup to include all species except Nepenthes, Beta and Illecebrum. The calibration date was effectively fixed by specifying a normal distribution with mean of 34 and standard deviation of 0.00001 as the prior for the time to most recent common ancestor (TMRCA) of the pre-defined ingroup. We ran 3 MCMC chains of length 50 million each with trees saved every 25,000 iterations. The first 1000 trees (50%) from each chain were discarded as burn in, and chains were combined after verifying convergence among runs. We generated a maximum credibility tree with mean node heights as well as a tree that was constrained to the 70% bootstrap consensus topology from our parsimony analysis. BEAST reported the estimated node ages along with the associated 95% high probability densities (HPDs), as well as the posterior probability for each node.
For the PL and LF analyses in r8s, we used the 70% parsimony bootstrap consensus topology with branch lengths optimized under ML in PAUP*. We fixed the root age of the tree to an arbitrary value of 1 and calibrated the resulting output tree with the 34 Myr fossil age for the Scleranthus/Sileneae split. Both analyses utilized the TN search algorithm with 10 restarts, 10 time guesses, and the checkgradient option on. The cross-validation procedure was used to determine an optimal smoothing parameter of 0.0022 for the PL analysis. Error in divergence times for both methods was estimated based on the distribution of 100 bootstrap replicate datasets, following the recommendations in the r8s documentation.
Estimating d N and d S
We estimated the branch lengths of matK and all 4 mitochondrial genes individually in terms of synonymous (d S ) and non-synonymous (d N ) substitutions per site, using a codon-based model of substitution within the codeml application in PAML v4.0 [71]. We also analyzed a concatenation of all 4 mitochondrial genes and a concatenation of atp1, cox3 and nad9 only. Tree topologies were constrained based on the matK 70% parsimony bootstrap consensus. Codon frequencies were determined by an F1x4 model. The parameters values for ω and transition/transversion ratio were estimated from the data with initial values of 0.4 and 2 respectively. Separate ω values were estimated for each branch. As in the dating analysis, Nepenthes glabrata was used as an outgroup in the matK dataset to identify the position of the root along the Beta vulgaris branch. Arabidopsis thaliana served a similar purpose for the mitochondrial genes. These outgroups were pruned from the resulting trees and not considered further. Because the process of C-to-U RNA editing can bias the estimation of d N and d S values in plant mitochondrial genomes [72], we excluded all codons known to undergo RNA editing in Beta vulgaris [73].
In the matK dataset, 6 species (Heliosperma pusillum, S. conica, S. paradoxa, S. samojedora, S. seoulensis and S. conoidea) produced sequences with an apparent frameshift indel in one of two homopolymer regions, raising the possibility that we sequenced pseudogenes in these species. In addition, S. otites did not have a start codon at the conserved position in the matK alignment. These sequences showed little indication of elevated rates or other abnormal substitution patterns, and their phylogenetic placement was consistent with a priori information. Therefore, they were retained in the dataset, and codons that contained frameshift indels were removed to keep all sequences in frame.
For the mitochondrial dataset, we obtained sequences for atp1, cox3, and nad9 from all sampled species, but only a subset of atp9 sequences were successfully generated. For 5 of the 74 species in our sample, we failed to successfully amplify and sequence atp9, and an additional 9 species appeared to have multiple atp9 copies. In the latter group, sequencing electropherograms indicated the presence of two different nucleotides in between 1.2 and 19.5% of sites. These samples were excluded from the analysis.
Estimates of absolute substitution rates (R N and R S )
Following the basic methodology described by Cho et al. [5], absolute substitution rates (substitutions per site per year) can be obtained by dividing branch lengths (defined in terms of substitution per site) by the age of the branch. We calculated branch ages using the divergence times estimated by BEAST. We divided the d N and d S values reported by PAML for each branch by the respective branch age to obtain absolute substitution rates in terms of non-synonymous and synonymous sites (R N and R S , respectively). Standard errors for R N and R S were calculated as described by Parkinson et al. [6] where the standard errors for node ages were approximated as one quarter of the 95% HPD. Pearson correlations coefficient for R N and R S values across and within genes were calculated with PROC CORR in SAS Software v9.1.
References
Wolfe KH, Li WH, Sharp PM: Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci. 1987, 84 (24): 9054-9058. 10.1073/pnas.84.24.9054.
Palmer JD, Herbon LA: Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 1988, 28 (1-2): 87-97. 10.1007/BF02143500.
Drouin G, Daoud H, Xia J: Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol Phylogenet Evol. 2008, 49 (3): 827-831. 10.1016/j.ympev.2008.09.009.
Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD: Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 2007, 7 (1): 135-10.1186/1471-2148-7-135.
Cho Y, Mower JP, Qiu YL, Palmer JD: Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci. 2004, 101 (51): 17741-17746. 10.1073/pnas.0408302101.
Parkinson CL, Mower JP, Qiu YL, Shirk AJ, Song K, Young ND, DePamphilis CW, Palmer JD: Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol Biol. 2005, 5: 73-10.1186/1471-2148-5-73.
Sloan DB, Barr CM, Olson MS, Keller SR, Taylor DR: Evolutionary rate variation at multiple levels of biological organization in plant mitochondrial DNA. Mol Biol Evol. 2008, 25 (2): 243-246. 10.1093/molbev/msm266.
Laird CD, McConaughy BL, McCarthy BJ: Rate of fixation of nucleotide substitutions in evolution. Nature. 1969, 224 (5215): 149-154. 10.1038/224149a0.
Britten RJ: Rates of DNA sequence evolution differ between taxonomic groups. Science. 1986, 231 (4744): 1393-1398. 10.1126/science.3082006.
Martin AP, Palumbi SR: Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci. 1993, 90 (9): 4087-4091. 10.1073/pnas.90.9.4087.
Gaut BS, Morton BR, McCaig BC, Clegg MT: Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci. 1996, 93 (19): 10274-10279. 10.1073/pnas.93.19.10274.
Sniegowski PD, Gerrish PJ, Johnson T, Shaver A: The evolution of mutation rates: separating causes from consequences. Bioessays. 2000, 22 (12): 1057-1066. 10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W.
Foury F, Hu J, Vanderstraeten S: Mitochondrial DNA mutators. Cell Mol Life Sci. 2004, 61 (22): 2799-2811. 10.1007/s00018-004-4220-y.
Smith SA, Donoghue MJ: Rates of molecular evolution are linked to life history in flowering plants. Science. 2008, 322 (5898): 86-89. 10.1126/science.1163197.
Kimura M: The Neutral Theory of Molecular Evolution. 1983, Cambridge: Cambridge University Press
Chamary JV, Parmley JL, Hurst LD: Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006, 7 (2): 98-108. 10.1038/nrg1770.
Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK: High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science. 2000, 289 (5488): 2342-2344. 10.1126/science.289.5488.2342.
Muse SV: Examining rates and patterns of nucleotide substitution in plants. Plant Mol Biol. 2000, 42 (1): 25-43. 10.1023/A:1006319803002.
Drake JW, Charlesworth B, Charlesworth D, Crow JF: Rates of spontaneous mutation. Genetics. 1998, 148 (4): 1667-1686.
Lynch M, Koskella B, Schaack S: Mutation pressure and the evolution of organelle genomic architecture. Science. 2006, 311 (5768): 1727-1730. 10.1126/science.1118884.
Baer CF, Miyamoto MM, Denver DR: Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007, 8: 619-631. 10.1038/nrg2158.
Bergsten J: A review of long-branch attraction. Cladistics. 2005, 21 (2): 163-193. 10.1111/j.1096-0031.2005.00059.x.
Desplanque B, Viard F, Bernard J, Forcioli D, Saumitou-Laprade P, Cuguen J, Van Dijk H: The linkage disequilibrium between chloroplast DNA and mitochondrial DNA haplotypes in Beta vulgaris ssp. maritima (L.): the usefulness of both genomes for population genetic studies. Mol Ecol. 2000, 9 (2): 141-154. 10.1046/j.1365-294x.2000.00843.x.
Olson MS, McCauley DE: Linkage disequilibrium and phylogenetic congruence between chloroplast and mitochondrial haplotypes in Silene vulgaris. Proc R Soc Lond B. 2000, 267 (1454): 1801-1808. 10.1098/rspb.2000.1213.
McCauley DE, Bailey MF, Sherman NA, Darnell MZ: Evidence for paternal transmission and heteroplasmy in the mitochondrial genome of Silene vulgaris, a gynodioecious plant. Heredity. 2005, 95 (1): 50-58. 10.1038/sj.hdy.6800676.
Houliston GJ, Olson MS: Nonneutral evolution of organelle genes in Silene vulgaris. Genetics. 2006, 174 (4): 1983-1994. 10.1534/genetics.106.060202.
McCauley DE, Sundby AK, Bailey MF, Welch ME: Inheritance of chloroplast DNA is not strictly maternal in Silene vulgaris (Caryophyllaceae): evidence from experimental crosses and natural populations. Am J Bot. 2007, 94 (8): 1333-10.3732/ajb.94.8.1333.
Hilu KW, Liang H: The matK gene: sequence variation and application in plant systematics. Am J Bot. 1997, 84 (6): 830-830. 10.2307/2445819.
Barthet MM, Hilu KW: Evaluating evolutionary constraint on the rapidly evolving gene matK using protein composition. J Mol Evol. 2008, 66 (2): 85-97. 10.1007/s00239-007-9060-6.
Frajman B, Eggens F, Oxelman B: Hybrid origins and homoploid reticulate evolution within Heliosperma (Sileneae, Caryophyllaceae) - a multigene phylogenetic approach with relative dating. Systematic Biology. 2009, 58 (3): 328-345. 10.1093/sysbio/syp030.
Oxelman B, Liden M, Berglund D: Chloroplast rps16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Plant Syst Evol. 1997, 206 (1-4): 393-410. 10.1007/BF00987959.
Erixon P, Oxelman B: Reticulate or tree-like chloroplast DNA evolution in Sileneae (Caryophyllaceae)?. Mol Phylogenet Evol. 1997, 48 (1): 313-325. 10.1016/j.ympev.2008.04.015.
Popp M, Oxelman B: Origin and evolution of North American polyploid Silene (Caryophyllaceae). Am J Bot. 2007, 94 (3): 330-10.3732/ajb.94.3.330.
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK: The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006, 23 (11): 2175-10.1093/molbev/msl089.
Erixon P, Oxelman B: Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE. 2008, 3 (1): e1386-10.1371/journal.pone.0001386.
Guisinger MM, Kuehl JV, Boore JL, Jansen RK: Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc Natl Acad Sci. 2008, 105: 18424-18429. 10.1073/pnas.0806759105.
Qiu YL, Li L, Wang B, Chen Z, Knoop V, Groth-Malonek M, Dombrovska O, Lee J, Kent L, Rest J: The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci. 2006, 103 (42): 15511-15516. 10.1073/pnas.0603335103.
Nabholz B, Glemin S, Galtier N: Strong variations of mitochondrial mutation rate across mammals--the longevity hypothesis. Mol Biol Evol. 2008, 25 (1): 120-130. 10.1093/molbev/msm248.
Rautenberg A: Phylogenetic relationships of Silene sect. Melandrium and allied taxa (Caryophyllaceae), as deduced from multiple gene trees. Ph.D. thesis. 2009, Uppsala University; Uppsala
Rautenberg A, Filatov D, Svennblad B, Heidari N, Oxelman B: Conflicting phylogenetic signals in the SlX1/Y1 gene in Silene. BMC Evol Biol. 2008, 8 (1): 299-10.1186/1471-2148-8-299.
Touzet P, Delph LF: The effect of breeding system on polymorphism in mitochondrial genes of Silene. Genetics. 2009, 181 (2): 631-644. 10.1534/genetics.108.092411.
Lenaz G: Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta. 1998, 1366 (1-2): 53-67. 10.1016/S0005-2728(98)00120-0.
Ingvarsson PK, Taylor DR: Genealogical evidence for epidemics of selfish genes. Proc Natl Acad Sci. 2002, 99 (17): 11265-11269. 10.1073/pnas.172318099.
Stadler T, Delph LF: Ancient mitochondrial haplotypes and evidence for intragenic recombination in a gynodioecious plant. Proc Natl Acad Sci USA. 2002, 99 (18): 11730-11735. 10.1073/pnas.182267799.
Barr CM, Keller SR, Ingvarsson PK, Sloan DB, Taylor DR: Variation in mutation rate and polymorphism among mitochondrial genes in Silene vulgaris. Mol Biol Evol. 2007, 24 (8): 1783-1791. 10.1093/molbev/msm106.
Richardson AO, Palmer JD: Horizontal gene transfer in plants. J Exp Bot. 2007, 58 (1): 1-9. 10.1093/jxb/erl148.
Ioerger TR, Clark AG, Kao T: Polymorphism at the self-incompatibility locus in Solanaceae predates speciation. Proc Natl Acad Sci. 1990, 87 (24): 9732-9735. 10.1073/pnas.87.24.9732.
Adams KL, Qiu YL, Stoutemyer M, Palmer JD: Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci. 2002, 99 (15): 9905-9912. 10.1073/pnas.042694899.
Li WH, Gojobori T, Nei M: Pseudogenes as a paradigm of neutral evolution. Nature. 1981, 292: 237-239. 10.1038/292237a0.
Lin X, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M: Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999, 402: 761-768. 10.1038/45471.
Marienfeld J, Unseld M, Brandt P, Brennicke A: Genomic recombination of the mitochondrial atp6 gene in Arabidopsis thaliana at the protein processing site creates two different presequences. DNA Research. 1996, 3 (5): 287-290. 10.1093/dnares/3.5.287.
Lercher MJ, Hurst LD: Human SNP variability and mutation rate are higher in regions of high recombination. Trends in Genetics. 2002, 18 (7): 337-340. 10.1016/S0168-9525(02)02669-0.
Bakker FT, Breman F, Merckx V: DNA sequence evolution in fast evolving mitochondrial DNA nad1 exons in Geraniaceae and Plantaginaceae. Taxon. 2006, 55 (4): 887-896.
Pearl SA, Welch ME, McCauley DE: Mitochondrial heteroplasmy and paternal leakage in natural populations of Silene vulgaris, a gynodioecious plant. Mol Biol Evol. 2009, 26 (3): 537-545. 10.1093/molbev/msn273.
Lynch M, Blanchard JL: Deleterious mutation accumulation in organelle genomes. Genetica. 1998, 103: 29-39. 10.1023/A:1017022522486.
Wyckoff GJ, Malcom CM, Vallender EJ, Lahn BT: A highly unexpected strong correlation between fixation probability of nonsynonymous mutations and mutation rate. Trends in Genetics. 2005, 21 (7): 381-385. 10.1016/j.tig.2005.05.005.
Laroche J, Li P, Maggia L, Bousquet J: Molecular evolution of angiosperm mitochondrial introns and exons. Proc Natl Acad Sci. 1997, 94 (11): 5722-5727. 10.1073/pnas.94.11.5722.
Wikström N, Savolainen V, Chase MW: Evolution of the angiosperms: calibrating the family tree. Proc R Soc Lond B. 2001, 268 (1482): 2211-10.1098/rspb.2001.1782.
Venditti C, Meade A, Pagel M: Detecting the node-density artifact in phylogeny reconstruction. Syst Biol. 2006, 55 (4): 637-643. 10.1080/10635150600865567.
Brach AR, Song H: eFloras: New directions for online floras exemplified by the Flora of China Project. Taxon. 2006, 55 (1): 188-
McCauley DE: Contrasting the distribution of chloroplast DNA and allozyme polymorphism among local populations of Silene alba: implications for studies of gene flow in plants. Proc Natl Acad Sci. 1994, 91 (17): 8127-8131. 10.1073/pnas.91.17.8127.
Taylor DR, Keller SR: Historical range expansion determines the phylogenetic diversity introduced during contemporary species invasion. Evolution. 2007, 61 (2): 334-345. 10.1111/j.1558-5646.2007.00037.x.
Oxelman B, Liden M: Generic boundaries in the tribe Sileneae (Caryophyllaceae) as inferred from nuclear rDNA sequences. Taxon. 1995, 44 (4): 525-542. 10.2307/1223498.
Popp M, Oxelman B: Evolution of a RNA polymerase gene family in Silene (Caryophyllaceae) - Incomplete concerted evolution and topological congruence among paralogues. Syst Biol. 2004, 53 (6): 914-932. 10.1080/10635150490888840.
Frajman B, Oxelman B: Reticulate phylogenetics and phytogeographical structure of Heliosperma (Sileneae, Caryophyllaceae) inferred from chloroplast and nuclear DNA sequences. Mol Phylogenet Evol. 2007, 43 (1): 140-155. 10.1016/j.ympev.2006.11.003.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods). Version 4. 1998, Sunderland, MA: Sinauer Associates
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818. 10.1093/bioinformatics/14.9.817.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Sanderson MJ: r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003, 19 (2): 301-302. 10.1093/bioinformatics/19.2.301.
Yang Z: PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007, 24 (8): 1586-1591. 10.1093/molbev/msm088.
Lu MZ, Szmidt AE, Wang XR: RNA editing in gymnosperms and its impact on the evolution of the mitochondrial coxI gene. Plant Mol Biol. 1998, 37 (2): 225-234. 10.1023/A:1005972513322.
Mower JP, Palmer JD: Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genomics. 2006, 276 (3): 285-293. 10.1007/s00438-006-0139-3.
Wolfe KH, Sharp PM, Li WH: Rates of synonymous substitution in plant nuclear genes. J Mol Evol. 1989, 29 (3): 208-211. 10.1007/BF02100204.
Holmgren PK, Holmgren NH, Barnett LC: Index Herbariorum: Part 1. The herbaria of of the world. 1990, New York: New York Botanical Garden
Acknowledgements
We thank Stephanie Goodrich for providing Agrostemma seeds and Nahid Heidari and Vivian Aldén for assistance in the lab. We also appreciate comments on an earlier version of this manuscript from Steve Keller, Magnus Lidén, Matt Olson, and the members of the Taylor lab. This study was supported by NSF DEB-0808452 (to DBS and DRT), NSF DEB-0349558 (to DRT) and grants from the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (to BO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
DBS conceived of the study, participated in its design, conducted the bulk of the sequencing and data analysis, and drafted the manuscript. BO participated in design of the study, collected and identified specimens, and helped with data analysis and drafting of the manuscript. AR collected and identified specimens and helped with sequencing, data analysis, and drafting of the manuscript. DRT participated in design of the study and helped with data analysis and drafting the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2009_1174_MOESM1_ESM.xls
Additional file 1: Estimated divergence times (in millions of years) from three different dating methods. [See Additional file 2 for definitions of node names]. (XLS 22 KB)
12862_2009_1174_MOESM3_ESM.xls
Additional file 3: Detailed data on d N , d S , R N , R S and associated error for each gene and concatenated dataset (all species). Each row corresponds to a phylogenetic branch defined by its basal node and derived node/tip. R N and R S values are in terms of SSB. Approximated standard errors (SEs) are provided for R N and R S . Note that SE approximations are undefined and reported as 0 for any absolute rate estimate of 0. This table does not include atp9 because it was not sequenced in all species. [See Additional file 2 for definitions of node names]. (XLS 72 KB)
12862_2009_1174_MOESM4_ESM.xls
Additional file 4: Detailed data on d N , d S , R N , R S and associated error for each gene and concatenated dataset ( atp9 subset). Same setup as Additional file 2. Only the 61 species for which atp9 was sequenced are included, so that data for atp9 and the concatenation of all four mitochondrial genes could be presented. [See Additional file 2 for definitions of node names]. (XLS 40 KB)
12862_2009_1174_MOESM5_ESM.pdf
Additional file 5: Maximum likelihood trees for each of the 4 mitochondrial genes (generated without topological constraint). Parsimony bootstrap values are noted to the left of the corresponding node. Only values > 0.5 are shown. Branch lengths are in terms of substitutions per site. (PDF 2 MB)
12862_2009_1174_MOESM6_ESM.pdf
Additional file 6: Maximum likelihood tree for matKdataset. Branch lengths are in terms of substitutions per site. (PDF 445 KB)
12862_2009_1174_MOESM7_ESM.pdf
Additional file 7: BEAST analysis of matKdataset with unconstrained topology. Time scale is in millions of years. Posterior support is shown to the right of each node. (PDF 43 KB)
12862_2009_1174_MOESM8_ESM.xls
Additional file 8: Sequences and references for PCR primers. Nucleotide sequences (5' to 3') of primers used for PCR amplification and DNA sequencing. (XLS 12 KB)
12862_2009_1174_MOESM9_ESM.xls
Additional file 9: GenBank accession numbers for all sequences. Accession numbers in bold are not from the voucher listed in Table 1. (XLS 29 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sloan, D.B., Oxelman, B., Rautenberg, A. et al. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae. BMC Evol Biol 9, 260 (2009). https://doi.org/10.1186/1471-2148-9-260
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-260