
Abstract
Background
The Neogastropoda is a highly diversified group of predatory marine snails (Gastropoda: Caenogastropoda). Traditionally, its monophyly has been widely accepted based on several morphological synapomorphies mostly related with the digestive system. However, recent molecular phylogenetic studies challenged the monophyly of Neogastropoda due to the inclusion of representatives of other caenogastropod lineages (e.g. Littorinimorpha) within the group. Neogastropoda has been classified into up to six superfamilies including Buccinoidea, Muricoidea, Olivoidea, Pseudolivoidea, Conoidea, and Cancellarioidea. Phylogenetic relationships among neogastropod superfamilies remain unresolved.
Results
The complete mitochondrial (mt) genomes of seven Neogastropoda (Bolinus brandaris, Cancellaria cancellata, Conus borgesi, Cymbium olla, Fusiturris similis, Nassarius reticulatus, and Terebra dimidiata) and of the tonnoidean Cymatium parthenopeum (Littorinimorpha), a putative sister group to Neogastropoda, were sequenced. In addition, the partial sequence of the mitochondrial genome of the calyptraeoidean Calyptraea chinensis (Littorinimorpha) was also determined. All sequenced neogastropod mt genomes shared a highly conserved gene order with only two instances of tRNA gene translocation. Phylogenetic relationships of Neogastropoda were inferred based on the 13 mt protein coding genes (both at the amino acid and nucleotide level) of all available caenogastropod mitochondrial genomes. Maximum likelihood (ML) and Bayesian inference (BI) phylogenetic analyses failed to recover the monophyly of Neogastropoda due to the inclusion of the tonnoidean Cymatium parthenopeum within the group. At the superfamily level, all phylogenetic analyses questioned the taxonomic validity of Muricoidea, whereas the monophyly of Conoidea was supported by most phylogenetic analyses, albeit weakly. All analyzed families were recovered as monophyletic except Turridae due to the inclusion of Terebridae. Further phylogenetic analyses based on either a four mt gene data set including two additional Littorinimorpha or combining mt and nuclear sequence data also rejected the monophyly of Neogastropoda but rendered rather unresolved topologies. The phylogenetic performance of each mt gene was evaluated under ML. The total number of resolved internal branches of the reference (whole-mt genome) topology was not recovered in any of the individual gene phylogenetic analysis. The cox2 gene recovered the highest number of congruent internal branches with the reference topology, whereas the combined tRNA genes, cox1, and atp8 showed the lowest phylogenetic performance.
Conclusion
Phylogenetic analyses based on complete mt genome data resolved a higher number of internal branches of the caenogastropod tree than individual mt genes. All performed phylogenetic analyses agreed in rejecting the monophyly of the Neogastropoda due to the inclusion of Littorinimorpha lineages within the group. This result challenges morphological evidence, and prompts for further re-evaluation of neogastropod morphological synapomorphies. The important increase in number of analyzed positions with respect to previous studies was not enough to achieve conclusive results regarding phylogenetic relationships within Neogastropoda. In this regard, sequencing of complete mtDNAs from all closely related caenogastropod lineages is needed. Nevertheless, the rapid radiation at the origin of Neogastropoda may not allow full resolution of this phylogeny based only on mt data, and in parallel more nuclear sequence data will also need to be incorporated into the phylogenetic analyses.
Similar content being viewed by others


Background
Neogastropoda [1, 2], also known as Stenoglossa [3], comprises a highly diverse group of predatory marine shelled gastropods with more than 16,000 living species [4] including e.g., cone snails (Conidae), balers (Volutidae), purple dye murex snails (Muricidae), augers (Terebridae), and whelks (Buccinidae) [5]. Neogastropoda are dominant in many benthic environments, and attain their maximum diversity in the tropical seas [6, 7]. The most prominent feature of Neogastropoda is their active predatory behavior (most species are carnivorous), which was achieved after important morphological changes including e.g., the elongation of the siphonal canal, a shift in the mouth opening to a terminal position on the head, and the formation of a well-developed proboscis [5, 8–10].
Ever since Thiele [1], Neogastropoda have been considered a natural group, clearly differentiated from other Caenogastropoda. The monophyly of the group is widely accepted among morphologists [5], and it is based on several synapomorphies mostly related with the anatomy of the digestive system [5, 8, 10–13]. Current classifications of Neogastropoda generally recognize up to six superfamilies: Buccinoidea, Muricoidea, Olivoidea, Pseudolivoidea, Conoidea, and Cancellarioidea [1, 2, 8, 12, 14] (Additional file 1). Phylogenetic relationships among neogastropod superfamilies based on morphological characters are rather unstable, and for instance, Cancellarioidea [8] or Buccinoidea [5] have been alternatively proposed as the sister group of the remaining Neogastropoda.
Thus far, molecular phylogenetic analyses based on relatively short fragments of both nuclear and mitochondrial (mt) DNA recovered rather unresolved topologies, which contradicted morphological evidence, and failed to support the monophyly of Neogastropoda [15–20]. For instance, the most complete molecular phylogenetic study thus far performed on this subject [21] was based on a multigene data set that included both nuclear (18S rRNA, 28S rRNA, EF1-α, and Histone H3; 2,707 bp) and mt (cox1 and 12S rRNA; 1,288 bp) sequences of 29 Caenogastropoda. The reconstructed tree failed to recover the monophyly of Neogastropoda, and two Littorinimorpha lineages (Tonnoidea and Calyptraeoidea) were recovered as the closest sister groups of two Neogastropoda lineages (Volutidae and Cancellariidae, respectively), although with low statistical support. In contrast, more recently, a phylogenetic analysis combining both morphological, and molecular data (using the same genes as in [21]) recovered Neogastropoda as monophyletic [5] stimulating the debate.
The available fossil record of Neogastropoda is quite thorough, and supports a widely accepted evolutionary scenario of an Early Cretaceous origin of the group followed by two rapid diversification rounds in the late Cretaceous and the Paleocene, respectively [22–24]. The successive bursts of cladogenesis in the evolutionary history of the group could be hampering successful recovery of phylogenetic relationships within the group. Given the relatively short internodes connecting main superfamilies of Neogastropoda and related caenogastropod lineages, it seems worthwhile gathering and analyzing larger sequence data sets in order to obtain more phylogenetic informative sites for trying to resolve the question at hand.
Phylogenetic analyses based on complete mt sequence data have proved to enhance resolution, and statistical confidence of inferred phylogenetic trees in vertebrates when compared with analyses based only on partial mt genes [25–27]. Within gastropods, mitogenomic data have demonstrated to be useful in recovering phylogenetic relationships within opisthobranchs (sea slugs), and in demonstrating the non-monophyly of pulmonates (snails) [28]. Moreover, gastropod mitochondrial genomes show a wide variety of gene arrangements, which, if shared-derived, could be used to infer phylogenetic relationships [5, 28]. Hence, sequencing and analyzing mt genome data seems to be a promising tool for further addressing the controversy on neogastropod monophyly and phylogenetic relationships. Nevertheless, it is important to note that phylogenetic analyses based on mt genomes show limitations (particularly when addressing deep phylogenetic relationships but not only), and can render odd results that are incongruent with other evidences [29–31]. Unorthodox phylogenetic relationships based on mt genome data can be explained as artefacts due to e.g. incorrect rooting [32] or low signal-to-noise ratio [33], but are difficult to correct. Hence, it is widely accepted that phylogenetic results derived from mt genome data need to be confirmed with evidence based on nuclear genes, which have a slower evolutionary rate, and may show in some cases better phylogenetic performance (e.g. [34]). Thus far, however, only few nuclear markers such as e.g. 18S rRNA, 28S rRNA, EF1-α, and Histone H3 have been developed for phylogenetic studies in gastropods with limited success [15, 16, 20, 21].
To date, only five entire mt genomes of Neogastropoda have been determined: Conus textile NC_009797, [35]; Lophiotoma cerithiformis NC_008098, [36]; Ilyanassa obsoleta NC_007781, [37], Thais clavigera NC_010090[38] and Rapana venosa NC_011193[39]. These mt genomes represent only two (Conoidea and Buccinoidea) out of the six currently recognized superfamilies of Neogastropoda. Moreover, no mt genomes are available for any of the putative caenogastropod sister group lineages of Neogastropoda.
In this study, we sequenced the entire mt genome of another seven neogastropod species that represent four out of the six currently recognized superfamilies (Muricoidea, Buccinoidea, Cancellarioidea, and Conoidea), as well as a representative of Tonnoidea (Littorinimorpha), one of the proposed sister groups to the Neogastropoda [11, 40–42]. In addition, we sequenced a fragment of approximately 8,500 base pairs of the mt genome of a representative of Calyptraeoidea (Littorinimorpha), another potentially closely related taxon of Neogastropoda [21]. The new sequence data were used (both at the amino acid and nucleotide level) to reconstruct the phylogeny of Neogastropoda, and test its monophyly. The phylogenetic performance of individual mt genes was also investigated, and compared with that of whole mt genomes. In addition, evolution of mitochondrial gene arrangements within Gastropoda was revisited in the light of the new mt genomic data.
Results
Mitochondrial genome organization
The complete mt genomes of seven neogastropods (Bolinus brandaris, Cancellaria cancellata, Conus borgesi, Cymbium olla, Fusiturris similis, Nassarius reticulatus, and Terebra dimidiata) and of Cymatium parthenopeum (Littorinimorpha:Tonnoidea) were sequenced. The total lengths of the new sequences ranged between 15,270 and 16,648 bp (Table 1). All newly sequenced complete mt genomes encoded for 13 protein-coding, 22 transfer RNA (tRNA), and two ribosomal RNA (rRNA) genes. All neogastropod mt genomes shared the same gene order with only two exceptions (Fig. 1): (1) the trnS (ucn) gene from Fusiturris similis was positioned between nad6 and cob genes, whereas in the other neogastropod mt genomes is found between cob and trnT; (2) the position of the trnV in Terebra dimidiata was located between trnS (ucn) and trnT, whereas in the other neogastropod mt genomes is found between both rRNA genes. The mt genome of the tonnoidean Cymatium parthenopeum presented the neogastropod mt genome consensus gene order (Fig. 1). The partial sequence of the mt genome of Calyptraea chinensis (Littorinimorpha: Calyptraeoidea) was 8,530 bp in length, and comprised 25 out of the 37 mt genes. The deduced gene order of this partial mt genome was consistent with the neogastropod mt genome consensus gene order (from trnK to trnL (uur)) (Fig. 1). Moreover, the gene order of the partial sequence of Littorina saxatilis (Littorinomorpha: Littorinoidea) mt genome, which is available in GenBank (AJ_132137, [43], is also consistent with the neogastropod mt genome consensus gene order (from cox1 to cob) (Fig. 1).

Hypothesized gene rearrangements of gastropod mt genomes. Genes encoded by the minor strand are underlined. Genes are colored to facilitate following main inversion and transposition events between Cephalopoda, Vetigastropoda, and Caenogastropoda. The many gene rearrangements that potentially occurred at the origin of Patellogastropoda and Heterobranchia, or between both groups and Caenogastropoda, are not shown. Ce.: Cephalopoda; Ve.: Vetigastropoda; Ca.: Caenogastropoda; He.: Heterobranchia; Pa.: Patellogastropoda; Oc.: Octopoda; Pl.: Pleurotomarioidea; Ne.: Neogastropoda; Li.: Littorinimorpha; Pu.: Pulmonata; Op.: Opisthobranchia; Ac.: Acmaeoidea.
All genes of the newly sequenced complete mt genomes used ATG as start codon except nad4, which used other start codons (ATA in Bolinus brandaris, Nassarius reticulatus, and Cymbium olla, ATT in Cancellaria cancellata and Conus borgesi, and TTG in Fusiturris similis, Cymatium parthenopeum, and Terebra dimidiata). In these mt genomes, only eigth out of 296 genes ended with incomplete stop codons (notably nad4 gene in Fusiturris similis, Nassarius reticulatus, and Terebra dimidiata). Two noncoding regions were found in all analyzed mitochondrial genomes between trnF and cox3 (14 – 848 bp in length), and between atp6 and trnM (27 – 44 bp in length). Another two noncoding regions were found in all analyzed mt genomes except Cancellaria cancellata between cox1 and cox2 (15 – 132 bp in length), and between cox3 and trnK (12 – 83 bp in length). In addition, a large noncoding region located between nad1 and trnP (869 bp in length) was found in Cancellaria cancellata. The potential origin of replication was located between trnF and cox3 (15 – 848 bp in length) by comparison with other gastropod mt genomes. Instances of overlapping between adjacent genes occurred in all newly sequenced mt genomes (Table 1). The cox2 and trnD genes overlap in every genome except that of Calyptraea chinensis whereas trnW and trnQ overlap in every genome except that of Cymbium olla. Overall base compositions of the newly sequenced mt genomes are shown in Table 1.
Phylogenetic relationships of Neogastropoda based on complete mt genome sequences
Phylogenetic relationships among gastropod main lineages were inferred based on complete mt genome data both at the amino acid (13-protein data set) and nucleotide (allnuc data set) level using Lottia digitalis (Patellogastropoda), and Haliotis rubra (Vetigastropoda) as outgroups, respectively. Both data sets were analyzed under maximum likelihood (ML) and Bayesian inference (BI).
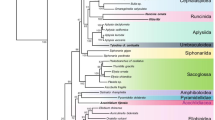
Both, the ML (-LnL = 46918.42) and the BI (-LnL = 47950.14) trees that were reconstructed based on the 13-protein data set failed to recover the monophyly of Neogastropoda (Fig. 2) due to the inclusion of Cymatium parthenopeum (Littorinimorpha: Tonnoidea) within the group with strong support (74% bootstrap support (BP), and 100% Bayesian posterior probability (BPP), respectively). The monophyly of the superfamily Conoidea was only recovered in the BI analysis (with maximal support; not shown). Both, ML and BI analyses failed to recover the monophyly of the superfamily Muricoidea (Fig. 2). The status of other neogastropod superfamilies could not be tested in the present study since only one family represented them. Internal branches connecting neogastropod families were rather short, and phylogenetic relationships among superfamilies were rather unresolved. Only the relative most basal position of Cancellarioidea with respect to other superfamilies was strongly supported by both, ML and BI analyses (Fig. 2). At a lower taxonomic level, the monophyly of all analyzed neogastropod families was strongly supported by the 13-protein data set with the exception of Turridae, which was strongly rejected (90% BP and 100% BPP in the ML and BI analyses, respectively) due to the inclusion of Terebridae (Fig. 2).

Phylogenetic relationships within Gastropoda. ML phylogram inferred from a single concatenated data set of deduced amino acid sequences of all 13 mitochondrial protein-coding genes (13-protein data set). Species whose complete mt genome was sequenced in this study are presented in bold. Lottia digitalis (Patellogastropoda) was used as outgroup. Numbers in the nodes correspond to ML bootstrap proportions (above branches) and BI posterior probabilities (below branches). Only values above 70% are represented. The inset shows a ML topology based on fragments of mt and nuclear data (adapted from Fig. two of Colgan et al., 2007).
Phylogenetic relationships within Caenogastropoda were also analyzed based on complete mt genome nucleotide sequence data (allnuc data set). Neogastropoda were not recovered as a monophyletic group due to the inclusion of the tonnoidean Cymatium parthenopeum, which was placed as sister group of Cancellariidae in both, ML (-lnL = 125810.87) and BI (-lnL = 124904.38) analyses (Fig. 3). However, this result was only strongly supported by BI when third codon positions of all protein coding genes were removed from the analysis (not shown). Both, ML and BI analyses based on the allnuc data set recovered Connoidea as a monophyletic group but without strong statistical support. In contrast, both ML and BI analyses rejected the monophyly of Muricoidea because Volutidae failed to group together with Muricidae (Fig. 3). This result only received strong bootstrap support when third codon positions of all protein coding genes were removed from the analysis (not shown). Phylogenetic relationships among superfamilies were largely unresolved in the reconstructed tree based on the allnuc data set (Fig. 3).

Phylogenetic relationships within Gastropoda. ML phylogram based on the nucleotide sequences of the rRNA, tRNA and protein-coding genes of all available complete mt genomes of neogastropods, and one Vetigastropoda (Haliotis rubra), chosen as outgroup (allnuc data set). Species whose complete mt genome was sequenced in this study are presented in bold. Numbers in the nodes correspond to ML bootstrap proportions (above branches) and BI posterior probabilities (below branches). Only values above 70% are depicted. The inset shows a ML topology based on fragments of mt and nuclear data (adapted from Fig. Two of Colgan et al., 2007). Letters A-K label nodes used in the analysis of phylogenetic performance.
Phylogenetic relationships of Neogastropoda based on partial mt genome sequences
To further test the monophyly of Neogastropoda, we analyzed a 4-protein data set that included two extra Littorinimorpha species (Calyptraea chinensis and Littorina saxatilis), using Lottia digitalis as outgroup (see Material and Methods). Both, the ML (-Ln L = 12497.47) and BI (-Ln L = 12694.60) reconstructed trees could not recover the monophyly neither of Neogastropoda nor of Littorinimorpha, albeit these results lacked statistical support (Fig. 4). As in the phylogenetic analyses based on the 13-protein and allnuc data sets, the monophyly of each analyzed family was recovered with strong support with the exception of that of Turridae (due to the inclusion of Terebridae). Phylogenetic relationships among families were largely unresolved, and monophyly at the superfamily level could not be recovered.

Phylogenetic relationships within Gastropoda. ML phylogram inferred from a single concatenated data set of deduced amino acid sequences of 4 mitochondrial protein-coding genes (4-protein data set). Lottia digitalis (Patellogastropoda) was used as outgroup. Numbers in the nodes correspond to ML bootstrap proportions (above branches) and BI posterior probabilities (below branches). Only values above 70% are represented. The inset shows a ML topology based on fragments of mt and nuclear data (adapted from Fig. Two of Colgan et al., 2007).
Phylogenetic relationships of Neogastropoda based on combined mt and nuclear sequences
Phylogenetic relationships within Caenogastropoda were further evaluated based on a data set (all combined) that concatenated the complete nucleotide sequences of all mt genes with partial ones of four nuclear genes (Fig. 5A). The reconstructed ML (-lnL = 102152.98) and BI (-lnL = 100152.06) trees based on the all combined data set (using different models of evolution for each partition; see Table 2) were congruent with those based on the 13-protein and all nuc data sets (Figs. 2 and 3). They only differed in the relative phylogenetic position of Conidae, (Fig. 5A versus Figs. 2 and 3). The nucleotide sequences of the genes of the four mt protein data set were also combined with partial sequences of four nuclear genes [21] into a single data set (partial combined). The reconstructed ML (-lnL = 52671.57) and BI (-lnL = 51166.67) trees were rather unresolved. Among analyzed Littorinimorpha lineages, Littorinidae and Calyptraeidae were recovered in a relative basal position, and only Ranellidae (Tonnoidea) was placed within Neogastropoda, as sister group of Cancellariidae (Fig. 5B). Conoidea was recovered as a monophyletic group, although without statistical support.

Phylogenetic relationships within Gastropoda. (A) ML phylogram based on the all combined data set. (B) ML phylogram based on the partial combined data set. Numbers in the nodes correspond to ML bootstrap proportions (above branches) and BI posterior probabilities (below branches). Only values above 70% are represented. The inset shows a ML topology based on fragments of mt and nuclear data (adapted from Fig. Two of Colgan et al., 2007).
Testing alternative phylogenetic hypotheses
Given that phylogenetic analyses based on the most resolving (13-protein) data set failed to recover consistently the monophyly of several traditional morphological groups including Neogastropoda, Conoidea, Muricoidea, and Turridae, we further tested their validity evaluating different alternative topologies (Additional file 2). We also tested several alternative phylogenetic hypotheses on neogastropod relationships previously reported in the literature (Additional file 2; [8, 9, 21]). Alternative topologies were built imposing constraints on the ML topology (Fig. 2). For instance, the monophyly of Neogastropoda was imposed by forcing the tonnoidean to be positioned in a basal position with respect to the remaining Neogastropoda, and by keeping identical all remaining phylogenetic relationships as recovered in the ML tree.
The SH, AU and KH tests rejected the following alternative topologies: (1) the ML hypothesis of Colgan (topology adapted from Fig. Four of Colgan et al., 2007); (2) the morphology-based phylogeny of Kantor [8], and (3) monophyly of Neogastropoda, and all analyzed superfamilies/families within the group. In contrast, several alternative topologies were found to be not significantly different from the ML tree: (1) the monophyly of Conoidea; (2) the morphology-based phylogeny adapted from Ponder and Lindberg [9]; (3) the monophyly of Turridae, and (4) the monophyly of Muricoidea (Additional file 2). Finally, it is noteworthy that the monophyly of Neogastropoda was rejected under the AU and KH tests but not by the SH test (Additional file 2).
Phylogenetic performance of mitochondrial genes
ML phylogenies were reconstructed at the nucleotide level based on each of the mt protein-coding (including all codon positions), each the two rRNA genes, and a combined data set including all mt tRNA genes. The number of congruent internal branches (i.e. nodes) between the individual phylogenies, and the whole-mt genome topology (Fig. 3) was used as a measure of the phylogenetic performance of mt genes (Table 3). None of the individual gene analysis recovered the 12 nodes (A-L) of the reference topology (Table 3 and Fig. 3). Individual genes typically recovered between 2–5, and 0–6 congruent nodes to the reference topology with bootstrap supports above/equal or below 50%, respectively. The three nodes (F, H, and L) were consistently recovered by most genes whereas others (A-D, G, I, J) were recovered only by 1–2 genes. The cox2, atp6, and nad4 genes recovered the highest number of congruent nodes, whereas the combined tRNA genes cox1, and atp8 showed the lowest phylogenetic performance.
Discussion
Monophyly of Neogastropoda
Recent molecular phylogenetic analyses (e.g. [28]) are challenging our view of gastropod systematics and many traditional groups (e.g. Pulmonata), widely accepted as monophyletic, are now seriously questioned [44–46]. As a result, a general perception is emerging that many morphological characters previously used to infer phylogenetic relationships among gastropod main lineages may be homoplasious [13, 28], and that gastropod phylogeny needs to be revisited. For instance, our phylogenetic analyses recover Vetigastropoda as the closest living sister group to Caenogastropoda with strong quantitative support. This result challenges the traditional hypothesis that proposes Heterobranchia (i.e. the paraphyletic Heterostropha, together with Opisthobranchia and Pulmonata) as the sister group to Caenogastropoda [5, 9, 47, 48]. The addition of nuclear data is needed to further confirm these new phylogenetic hypotheses.
Despite their remarkable present diversity, neogastropods have been traditionally recognized as a natural group because they share several key morphological features [1, 5, 10, 11, 24]. For instance, a recent phylogeny of Caenogastropoda based on 55 taxa and 164 morphological characters (particularly of external anatomy and eusperm) recovered neogastropods as a monophyletic group with maximal support [5]. In contrast, molecular phylogenetic studies have generally rendered rather unresolved caenogastropod phylogenies that challenged, albeit weakly, the widely accepted monophyly of Neogastropoda (e.g. [19, 21]).
The molecular phylogenetic analyses performed in this study recovered different Littorinimorpha species (Cymatium parthenopeum, Calyptraea chinensis, and Littorina saxatilis) within Neogastropoda, thus questioning the monophyly of both main caenogastropod lineages. However, among these results, only the inclusion of the tonnoidean species Cymatium parthenopeum within Neogastropoda in the phylogeny based on the 13-protein data set received strong quantitative support (and was further confirmed by the AU and KH tests). Depending on the data set, Tonnoidea has been placed as closely related to Neogastropoda (e.g based on morphology+molecules; [5]), or within Neogastropoda in a relatively basal position either alone (based on the 13-protein and all combined data sets; this study), as sister group of Volutidae (based on partial mt and nuclear genes; [21]), or as sister group of Cancellariidae (based on the allnuc, 4-protein, and partial combined data sets; this study).
The unresolved conflict on the monophyly of Neogastropoda between morphological and molecular phylogenies suggests either that shared morphological characters of Neogastropoda are homoplasious (convergent or plesiomorphic) or that, thus far, the analyzed molecular data sets were not informative enough for resolving the phylogenetic question at hand. Regarding the possibility that morphological homoplasy is commonplace in gastropods, recent studies on the embryonic development of the valve of Leiblein (a gland that acts as an oesophageal valve binding together food particles) in Buccinidae [49] and Muricidae [12, 40] demonstrated different origins of this organ in both neogastropod families. These findings indicated that this synapomorphy of Neogastropoda could be uncertain [50], and prompt for the re-evaluation of other synapomorphies defining Neogastropoda.
Regarding the possibility that molecular data sets need to be improved, the phylogenetic analyses performed here show that the effort in increasing the number of analyzed positions with respect to previous studies was not enough to achieve conclusive results regarding phylogenetic relationships within Neogastropoda. Internal branches connecting main caenogastropod/neogastropod lineages were relatively short. Such phylogenetic pattern is in agreement with the fossil record, which supports a rapid radiation at the origin of Neogastropoda [11, 24]. Therefore, it would be expected low resolution at this part of the caenogastropod tree regardless of the amount of sampled characters (but observe the increase in resolution in the ML tree based on 13-protein data set with respect to that based on the 4-protein data set, and to the individual phylogenetic analyses – see below). It is also noteworthy that internal branches in the phylogenetic tree based on the 13-protein data set (Fig. 2) presented higher bootstrap support than the ones in the phylogenetic tree based on the allnuc data set (Fig. 3). This result is in agreement with the preferential use of amino acids to reconstruct deep phylogenies because they (1) have a larger character-state space compare to nucleotides, (2) show a slower rate of evolution compare to silent substitutions, and (3) are less influenced by compositional bias [51]; [26]; [52]. Our phylogenetic analyses show that addressing the question on the monophyly of Neogastropoda is tightly connected with resolving phylogenetic relationships among Caenogastropoda main lineages, and hence that future phylogenetic studies based on complete mitochondrial genome sequence data will require a thorough representation of all Caenogastropoda main lineages.
Phylogenetic relationships within Neogastropoda
All phylogenetic analyses agreed on the monophyly of the different analyzed neogastropod families except Turridae (due to the inclusion of Terebridae). A recent molecular study [53] based on partial mitochondrial and nuclear genes, and a rather thorough sampling of both families also supported this view. At higher taxonomic levels, the phylogenetic analysis of the different data sets rendered (1) rather unresolved topologies; (2) showed markedly different phylogenetic relationships based on each data set, and (3) failed to recover the monophyly of neogastropod superfamilies. Only the phylogeny based on the 13-protein data achieved some degree of resolution regarding superfamily phylogenetic relationships. According to this phylogeny, Cancellarioidea occupies a basal position with respect to remaining neogastropods. This basal position was already suggested in some previous morphological studies [8, 54]. In particular, it has been shown that the gland of Leiblein is separated from the oesophagus in all neogastropods but Cancellarioidea, which retains the primitive condition [8, 12]. Other phylogenetic studies recovered Buccinoidea or Nassariidae+Mitridae as the most basal neogastropod superfamily based on morphology or combined morphological + molecular data sets, respectively [5]. A phylogeny based on partial mt and nuclear genes [21] recovered a basal politomy at this level of the caenogastropod tree, and leaved the controversy unsettled since either Volutidae (as sister group of Tonnoidea) or Cancellariidae (as sister group of Calyptraeidae) could be the most basal neogastropod lineages.
Both, ML and BI trees based on the 13-protein data set suggest that the monophyly of the superfamily Muricoidea is doubtful because Muricidae and Volutidae are not recovered together. All performed phylogenetic analyses based on other data sets also failed to recover Muricoidea as a monophyletic group. This result was already suggested in several previous studies [5, 15, 21]. The status of the superfamily Conoidea remains uncertain. The BI tree based on the 13-protein data set (not shown), as well as ML and BI trees based on the allnuc data set recovered the families Turridae, Terebridae and Conidae together, albeit with no strong statistical support. ML based on the 13-protein data set fail to recover the monophyly of Conoidea but this hypothesis could not be confidently rejected by statistical tests. Overall, our results indicate that neogastropod relationships are far from being settled (in "a state of flux" as described by [5]).
Phylogenetic performance of mitochondrial genes
The relative merit of the different genes in recovering a phylogeny is associated with their respective substitution rates, and thus the taxonomic level at which higher resolution is achieved. Several studies evaluated the phylogenetic performance of mt genes in recovering vertebrate phylogeny [25–27, 55]. According to these studies, nad4 and nad5 were ranked generally as the best performing mt genes, whereas atp8, nad6, and nad4L genes revealed the lowest phylogenetic performance. In this study, we evaluated the phylogenetic performance of individual mt genes in recovering caenogastropod phylogeny, and found some remarkable differences when compared with results from the above-mentioned studies in vertebrates. The gene cox2 showed the largest number of congruent branches with the whole-genome reference topology, whereas it was never recovered as having the best phylogenetic performance in vertebrates [25–27, 55]. In our study, the mt tRNA genes showed the lowest performance, whereas in Mueller [27] these genes demonstrated a medium ability to recover the reference topology. The mt gene cox1 also showed a very low performance in recovering caenogastropod phylogeny, whereas it was ranked as a good gene for recovering the expected vertebrate tree. All studies agreed in pointing mt atp8 as having low phylogenetic performance. In any case, combining all mt genes into a single data set always rendered the most resolved trees, supporting the use of complete mt genome data for addressing caenogatropod phylogeny.
Evolution of mitochondrial gene order of Neogastropoda
Shared mt gene orders may be useful for phylogenetic inference particularly at higher taxonomic levels [56]. Gastropods exhibit an important degree of variation in mt gene organization compared with other animals [28, 57–59]. Main events of gene rearrangement occurred at the origin of Patellogastropoda and Heterobranchia, whereas fewer changes occurred between cephalopods (here represented by Octopus) and the ancestors of Vetigastropoda (only tRNAs D, C and N) and Caenogastropoda (a large single inversion, and translocations of the tRNAs D and N). Within Heterobranchia, gene order seems to be relatively conserved and gene rearrangements are mostly related with transposition of tRNA genes [28]. All eight genomes sequenced in this study exhibit an unusually conserved gene order only contradicted by two unrelated tRNA translocations found in Fusiturris similis and Terebra dimidiata (Fig. 1). Given the high rates of gene rearrangement among gastropod main lineages, the conserved gene order found between all neogastropod and Littorinimorpha mt genomes suggests a shared evolutionary history between the analyzed lineages.
Conclusion
As more molecular data are analyzed, the monophyly and phylogenetic relationships of Neogastropoda are emerging as rather elusive evolutionary questions to be resolved. This is likely due to a rapid origin of Neogastropoda lineages back into the Early Cretaceous. Our phylogenetic analyses based on mt genome sequence data confirm previous molecular studies that contradicted the monophyly of Neogastropoda. This group used to be considered well-supported based on several morphological synapomorphies [5, 8, 10, 11, 60]. However, recent studies question the validity of some of these synapomorphies suggesting that Neogastropoda could be an assembly of taxa erroneously grouped on the basis of convergent morphological characters [50]. If confirmed, molecular data may become particularly useful for determining the Caenogastropoda phylogeny. In this regard, future studies testing the monophyly of Neogastropoda based on mt genome sequence data will need to include a thorough sampling of main caenogastropod lineages. Our ML and BI analyses based on the 13-protein data set, produced topologies better resolved than previous studies using fragments of nuclear and mt sequence data. Moreover, results of phylogenetic performance indicated that none of the individual mt genes recovered the whole-genome reference ML tree, which may indicate that larger data sets may provide more informative sites for this particular phylogenetic question. However, in parallel to the steady accumulation of new mt genome data, new nuclear data (e.g., Expressed Sequence Tags – ESTs) need also to be gathered, and only the combination of both types of sequence data might render better results.
Methods
Taxon sampling
To assess phylogenetic relationships within Neogastropoda, the entire mt genome was sequenced in the following species representing four out of the six currently recognized neogastropod superfamilies. Bolinus brandaris (L., 1758) and Cymbium olla (L., 1758) both belonging to the superfamily Muricoidea; Nassarius reticulatus (L., 1758) (superfamily Buccinoidea); Cancellaria cancellata (L., 1767) (superfamily Cancellarioidea), and Fusiturris similis (Bivona, 1838), Conus borgesi [61], and Terebra dimidiata (L., 1758), all belonging to superfamily Conoidea.
In order to test the monophyly of Neogastropoda, the mt genomes of two species belonging to the closely related order Littorinimorpha (Caenogastropoda) were also sequenced: Cymatium parthenopeum (von Salis, 1793) (superfamily Tonnoidea) and Calyptraea chinensis (L., 1758) (superfamily Calyptraeoidea). The mt genome of the former species was sequenced completely, whereas only about 8,500 bp were sequenced from the mtDNA of Calyptraea chinensis due to unsuccessful PCR amplification of the remaining portion of the genome. A similar case of partial mt genome sequencing due to failed PCR amplification was reported for Littorina saxatilis [43]).
In addition, we included in the phylogenetic analyses the following gastropod entire mt genomes that are available in GenBank: (1) superfamily Conoidea:Conus textile (NC_009797, [35] and Lophiotoma cerithiformis (NC_008098, [36]); (2) superfamily Buccinoidea: Ilyanassa obsoleta (NC_007781, [37]); (3) superfamily Muricoidea: Thais clavigera (NC_010090[38]), and Rapana venosa (NC_011193[39]), (4) Opisthobranchia: Aplysia californica (NC_005827, [62]) and Roboastra europaea (NC_004321, [57]); (5) Pulmonata: Albinaria coerulea (NC_001761, [63]); (6) Vetigastropoda: Haliotis rubra (NC_005940, [64]), and (7) Patellogastropoda: Lottia digitalis (NC_007782, [37]). A partial fragment of Littorina saxatilis (Litorinimorpha: Littorinoidea; AJ_132137, [43]) mt genome available in GenBank was also included in some phylogenetic analyses.
PCR amplification and sequencing
Total DNA was extracted using either a standard phenol-chloroform DNA extraction protocol with cetyltrimethylammonium bromide (CTAB) [65], or commercial extraction kits (DNA Easy extraction Kit, Qiagen; ChargeSwitch gDNA Micro Tissue Kit, Invitrogen). Several pairs of primers (see Additional file 3) were used to amplify by PCR, contiguous and overlapping fragments that covered the entire mt genomes. PCR amplifications were carried out with the PCR Extender System (5 Prime) in 25 μl reactions containing 10× PCR Extender buffer, 0.2 mM of each dNTP, 2 mM of MgCl2, 0.2 μM of each primer, 1–2 μl of template DNA, and Taq DNA polymerase (1 unit). The following general profile was used: an initial denaturing step at 94°C for 2 min; 35 to 40 cycles of denaturing at 94°C for 20 s, annealing at 42–55°C for 20 s, and extending at 68°C for 60 s per kb; and a final extending step at 68°C for 7 min.
PCR amplicons were purified by ethanol precipitation. Those fragments with sizes < 2500 bp were either directly sequenced with the corresponding PCR primers or cloned into pGEM-T Easy Vector System (Promega), and sequenced with modified universal M13 primers (see Additional file 3). Longer PCR products were directly sequenced using a primer walking strategy. Sequencing was performed in an automated sequencer (ABI PRISM 3700) using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), and following manufacturer's instructions. The obtained sequences averaged 900 bp in length, and each sequence overlapped the next contig by about 150 bp. In no case were differences observed between overlapping regions.
Phylogenetic analyses
Gene annotation was performed by sequence comparison with other mt genomes of gastropods. Open reading frames helped to delimit start and stop codons of the protein coding genes. Cloverleaf secondary structures of all tRNAs were reconstructed either by hand or with TRNASCAN-SE 1.21 [66]. Ribosomal RNA limits were based on the boundaries of the flanking genes. The new sequences reported in this paper have been deposited at GenBank under accession numbers EU827194 (Bolinus brandaris), EU827195 (Cancellaria cancellata), EU827199 (Cymbium olla), EU827197 (Fusiturris similis), EU827201 (Nassarius reticulatus), EU827196 (Terebra dimidiata), EU827198 (Conus borgesi), EU827200 (Cymatium parthenopeum), and EU827193 (Calyptraea chinensis).
Separate alignments of the nucleotide and deduced amino acid sequences of each mt protein-coding gene were constructed with CLUSTAL X version 1.83 using default parameters [67], and verified by eye in order to maximize positional homology. Alignment ambiguities were excluded from phylogenetic analyses using GBLOCKS version 0.91b [68, 69]. Five different data sets (available from the authors upon request) were analyzed: (1) 13-protein data set: the deduced amino acid sequences of the 13 protein-coding mt genes of the 12 available Neogastropoda, two Opisthobranchia (Aplysia californica and Roboastra europaea), one Pulmonata (Albinaria coerulea), one Vetigastropoda (Haliotis rubra). and one Patellogastropoda (Lottia digitalis) complete mt genomes were concatenated into a single data set. The alignment consisted of 3,170 positions, of which 935 were constant, and 1,542 were parsimony informative; (2) allnuc data set: the nucleotide sequences of the rRNA, tRNA and protein-coding genes of all available complete mt genomes of neogastropods, one Ranellidae, and one Vetigastropoda, were concatenated into a single data set of 14,156 positions. Of these, 5,371 positions were constant, and 6,407 were parsimony informative; (3) 4-protein data set: the available partial mitochondrial genome sequences of two caenogastropod taxa, Littorina saxatilis (AJ_132137[43]) and Calyptraea chinensis (this study) only have in common four protein-coding mitochondrial genes (cox1; cox2; atp6, and atp8). The deduced amino acid sequences of these four genes from these two taxa together with those of the taxa included in the 13-protein data set were concatenated into a single data set, which produced an alignment of 982 positions. Of these, 366 were constant, and 408 were parsimony informative; (4) allcombined data set: the nucleotide sequences of the rRNA, tRNA and protein-coding genes of complete mt genomes of seven neogastropods (Muricidae, Cancellariidae, Volutidae, Turridae, Nassariidae, Terebridae, and Conidae), one Ranellidae, and one Vetigastropoda, all from this study, combined with partial sequences of the nuclear genes 28S rRNA, 18S rRNA, histone H3, and elongation factor 1α (nuclear sequences retrieved from GenBank from the study of Colgan et al, 2007) were concatenated into a single data set of 16,937 positions. Of these, 7,996 were constant and 5,550 were parsimony informative; (5) partial combined data set: the nucleotide sequences of 4 protein-coding genes (cox1; cox2; atp6, and atp8) of seven neogastropods (Muricidae, Cancellariidae, Volutidae, Turridae, Nassariidae, Terebridae, and Conidae), one Ranellidae, and one Vetigastropoda, all from this study, combined with partial sequences of the nuclear genes 28S rRNA, 18S rRNA, histone H3, and elongation factor 1α (nuclear sequences retrieved from GenBank from the study of Colgan et al., 2007) were concatenated into a single data set of 8,462 positions. Of these, 4,113 are constant and 2,523 were parsimony-informative.
The best-fit models of evolution for the amino acid and nucleotide sequence data sets were selected using the Akaike Information Criterion (AIC) [70] with PROTTEST v1.3 [71] and MODELTEST v.3.7 [72], respectively (see Table 2). ML trees were inferred based on the different data sets with PHYML v3.0 [73]. The robustness of the inferred trees was tested using non-parametric bootstrapping (BP) of 500 pseudoreplicates.
BI trees were inferred based on the different data sets with MRBAYEs v3.1.2 [74]. Four Metropolis-coupled Markov chain Monte Carlo (MCMC) analyses were run for 1 million generations, and sampled every 100 generations. The mixed model option was used for the amino acid sequence data sets. For the analysis of nucleotide sequence data sets, all 13 protein-coding genes, ribosomal RNAs, and tRNAs were considered as three different partitions in the allnuc data set. A fourth partition of nuclear genes was considered in the combined data sets. The option (preset = variable) that allows rates to differ across partitions was used with the "unlink" command. Burn-in included the first 100,000 generations for the 13-protein, all combined, and partial combined data sets, whereas the first 50,000 generations were discarded for the 4-protein, and allnuc data sets. Statistical support was assessed using Bayesian posterior probabilities (BPPs).
In order to evaluate alternative hypotheses on neogastropod phylogenetic relationships found in the literature, the approximately unbiased (AU) [75], the Kishino-Hasegawa (KH) [76], and the Shimodaira-Hasegawa (SH) [77] tests were performed based on the 13-protein data set. These tests are implemented in CONSEL v0.1i [78] and use log-likelihoods of site-patterns of the trees estimated with PAML v.4 [79].
Phylogenetic performance of mt genes
In order to determine whether the allnuc data set had better phylogenetic performance than individual mt genes, ML analyses were performed with PHYML v2.4.4 [73]. Individual analyses were based on each of the 13 individual mt protein-coding genes, each of the two mt ribosomal genes, and mt tRNAs (all mt tRNAs were concatenated into a single data set). The best-fit models and parameters for each of these data sets were estimated under the AIC [70] as implemented in MODELTEST v.3.7 [72]. The robustness of the inferred trees was tested using BPs of 500 pseudoreplicates. Following Mueller [27], we compared the number of resolved nodes in each of the 50% bootstrap consensus ML trees that were inferred based on individual gene analyses to the number of resolved nodes in the 50% bootstrap consensus ML topology (Fig. 3) that was reconstructed based on the allnuc data set, as a measure of phylogenetic performance of the different individual data sets.
References
Thiele J: Handbuch der Systematischen Weichtierkunde Volume 1 (1) (Loricata; Gastropoda: Prosobranchia), Jena; 1929: 376 pp. 23 Oct. Edited by: R. Bieler & P. Mikkelsen, Smithsonian Libraries, 1993. 1929, . Reprint, Asher & Co., Amsterdam, 1963; translation by J.S. Bhatti
Wenz W: Gastropoda, Teil 1: Allgemeiner Teil und Prosobranchia. Handbuch der Paläozoologie. Edited by: Schindewolf OH. 1938, Berlin: Gebrüder Borntraeger, 1-1240.
Bouvier EL: Systéme nerveux, morphologie génerale et classification des Gastéropodes prosobranches. Annales des Sciences Naturelle Zool. 1887, 7 (3): 1-510.
Bouchet P: Turrid genera and mode of development: the use and abuse of protoconch morphology. Malacologia. 1990, 32: 69-77.
Ponder WF, Colgan DJ, Healy JM, Nützel A, Simone LRL, Strong E: Caenogastropoda. Phylogeny and evolution of the Mollusca. Edited by: Ponder WF, Lindberg DR. 2008, Berkeley: University of California Press, 331-383.
Bouchet P, Lozouet P, Maestrati P, Heros V: Assessing the magnitude of species richness in tropical marine environments: high numbers of molluscs at a New Caledonia site. Biological Journal of the Linnean Society. 2002, 75: 421-436. 10.1046/j.1095-8312.2002.00052.x.
Ponder WF: Brief introductions to higher groups of gastropods: Infraorder Neogastropoda. Mollusca: The Southern Synthesis. Edited by: Beesley PL, Ross GJB, Wells A. 1998, CSIRO, 808-854.
Kantor YI: Phylogeny and Relationships of Neogastropoda. Origin and Evolutionary Radiation of the Mollusca. Edited by: Taylor JD. 1996, Oxford: Oxford University Press, 221-230.
Ponder WF, Lindberg DR: Towards a phylogeny of gastropod molluscs – an analysis using morphological characters. Zoological Journal of the Linnean Society. 1997, 19: 83-265. 10.1111/j.1096-3642.1997.tb00137.x.
Strong EE: Refining molluscan characters: morphology, character coding and a phylogeny of the Caenogastropoda. Zoological Journal of the Linnean Society. 2003, 137: 447-554. 10.1046/j.1096-3642.2003.00058.x.
Taylor JD, Morris NJ: Relationships of neogastropods. Malacological Review. 1988, 4 (Supplement): 167-179.
Ponder WF: The origin and evolution of the Neogastropoda. Malacologia. 1973, 12 (2): 295-338.
Kantor YI: Morphological prerequisites for understanding Neogastropod phylogeny. Bollettino Malacologico. 2002, 38 (4): 161-174.
Bouchet P, Rocroi JP: Classification and nomenclator of gastropod families. 2005, Hackenheim, Germany: ConchBooks, 47:
Harasewych MG, Adamkewicz SL, Blake JA, Saudek D, Spriggs T, Bult CJ: Neogastropod phylogeny: a molecular perspective. Journal of Molluscan Studies. 1997, 63: 327-351. 10.1093/mollus/63.3.327.
Harasewych MG, Adamkewicz SL, Plassmeyer M, Gillevet PM: Phylogenetic relationships of the lower Caenogastropoda (Mollusca, Gastropoda; Architaenioglossa, Campaniloidea, Cerithioidea) as determined by partial 18S rDNA sequences. Zoologica Scripta. 1998, 27 (4): 361-372. 10.1111/j.1463-6409.1998.tb00467.x.
Winnepenninckx B, Steiner G, Backeljau T, Wachter R: Details of Gastropod Phylogeny Inferred from 18S rRNA Sequences. Molecular Phylogenetics and Evolution. 1998, 9 (1): 55-63. 10.1006/mpev.1997.0439.
Colgan DJ, Ponder WF, Eggler PE: Gastropod evolutionary rates and phylogenetic relationships assessed using partial 28S rDNA and histone H3 sequences. Zoologica Scripta. 2000, 29: 29-63. 10.1046/j.1463-6409.2000.00021.x.
Colgan DJ, Ponder WF, Beacham E, Macaranas J: Gastropod phylogeny based on six segments from four genes representing coding or non-coding and mitochondrial or nuclear DNA. Molluscan Research. 2003, 23: 123-148. 10.1071/MR03002.
McArthur AG, Harasewych MG: Molecular systematics of the major lineages of the Gastropoda. Molecular Systematics and Phylogeography of Mollusks. Edited by: Lydeard C, Lindberg DR. 2003, Washington: Smithsonian Books, 140-160.
Colgan DJ, Ponder WF, Beacham E, Macaranas J: Molecular phylogenetics of Caenogastropoda (Gastropoda: Mollusca). Molecular Phylogenetics and Evolution. 2007, 42: 717-737. 10.1016/j.ympev.2006.10.009.
Vermeij GJ: The Mesozoic marine revolution: evidence from snails, predation and grazers. Paleobiology. 1977, 3: 245-258.
Taylor D, Cleeveley RJ, Morris NJ: Predatory gastropods and their activities in the Blackdown Greensand (Albian) of England. Paleontology. 1983, 26: 521-553.
Tracey S, Todd JA, Erwin DH: Mollusca: Gastropoda. The Fossil Record. Edited by: Benton MJ. 1993, London: Chapman & Hall, 131-167.
Zardoya R, Meyer A: Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Molecular Biology and Evolution. 1996, 13 (7): 933-942.
Russo CA, Takezaki N, Nei M: Efficiencies of different tree-building methods in recovering a known vertebrate phylogeny. Molecular Biology and Evolution. 1996, 13: 525-536.
Mueller RL: Evolutionary rates, divergence dates, and the performance of mitochondrial genes in Bayesian phylogenetic analysis. Systematic Biology. 2006, 55 (2): 289-300. 10.1080/10635150500541672.
Grande C, Templado J, Zardoya R: Evolution of gastropod mitochondrial genome arrangements. BMC Evolutionary Biology. 2008, 8: 61-10.1186/1471-2148-8-61.
Naylor GJP, Brown WM: Amphioxus Mitochondrial DNA, Chordate Phylogeny, and the Limits of Inference Based on Comparisons of Sequences. Systematic Biology. 1998, 47 (1): 61-76. 10.1080/106351598261030.
Nardi F, Spinsanti G, Boore JL, Carapelli A, Dallai R, Frati F: Hexapod origins: Monophyletic or Paraphyletic?. Science. 2003, 299 (5614): 1887-1889. 10.1126/science.1078607.
Podsiadlowski L, Braband A: The complete mitochondrial genome of the sea spider Nymphon gracile (Arthropoda: Pycnogonida). BMC Genomics. 2006, 7: 1-13. 10.1186/1471-2164-7-284.
Takezaki N, Gojobori T: Correct and incorrect vertebrate phylogenies obtained by the entire mitochondrial DNA sequences. Molecular Biology and Evolution. 1999, 16 (5): 590-601.
Zardoya R, Meyer A: On the origin of and phylogenetic relationships among living amphibians. Proceedings of the National Academy of Sciences. 2001, 98 (13): 7380-7383. 10.1073/pnas.111455498.
Cotton JA, Page RDM: Going nuclear: Vertebrate phylogeny and gene family evolution reconciled. P Roy Soc Lond B Bio. 2002, 269: 1555-1561. 10.1098/rspb.2002.2074.
Bandyopadhyay P, Stevenson B, Ownby J, Cady M, Watkins M, Olivera B: The mitochondrial genome of Conus textile, coxI-coxII intergenic sequences and Conoidean evolution. Molecular Phylogenetics and Evolution. 2008, 46 (1): 215-223. 10.1016/j.ympev.2007.08.002.
Bandyopadhyay PK, Stevenson BJ, Cady MT, Olivera B, Wolstenholme DR: Complete mitochondrial DNA sequence of a Conoidean gastropod, Lophiotoma (Xenoturris) cerithiformis: Gene order and gastropod phylogeny. Toxicon. 2006, 48: 29-43. 10.1016/j.toxicon.2006.04.013.
Simison WB, Lindberg DR, Boore JL: Rolling circle amplification of metazoan mitochondrial genomes. Molecular Phylogenetics and Evolution. 2006, 39 (2): 562-567. 10.1016/j.ympev.2005.11.006.
Jung S, Lee Y, Toshihiro H, Cho HS, Lee J-S: Complete mitochondrion sequence of Thais clavigera (rock shell). Direct submission to the Genome Project NCBI record NC_010090-2007. 2007, [http://www.genome.jp/dbget-bin/www_bget?refseq:NC_010090]
Chandler EA, McDowell JR, Graves JE: The mitochondrial genome of the rapa whelk (Rapana venosa) and the genetic variability of the mitochondrial coding regions in the neogastropods. Direct submission to the Genome Project NCBI record NC_011193-2008. 2008, [http://www.genome.jp/dbget-bin/www_bget?refseq:NC_011193]
Graham A: The oesophagus of the stenoglossan prosobranchs. Proceedings of the Royal Society of Edinburgh, series B. 1941, 61: 1-23.
Healy JM: Sperm morphology and its systematic importance in the Gastropoda. Malacological Review Supplement. Prosobranch Phylogeny. Edited by: Ponder WF. 1988, 251-266.
Taylor JD, Morris NJ, Taylor CN: Food specialization and the evolution of predatory prosobranch gastropods. Paleontology. 1980, 23: 375-409.
Wilding CS, Mill PJ, Grahame J: Partial sequence of the mitochondrial genome of Littorina saxatilis: relevance to gastropod phylogenetics. Journal of Molecular Evolution. 1999, 48 (3): 348-359. 10.1007/PL00006479.
Yoon SH, Kim W: Phylogeny of some gastropod mollusks derived from 18s rRNA sequences with emphasis on the Euthyneura. The Nautilus. 2000, 114: 84-92.
Remigio EA, Hebert PDN: Testing the utility of partial COI sequences for phylogenetic estimates of gastropod relationships. Molecular Phylogenetics and Evolution. 2003, 29: 641-647. 10.1016/S1055-7903(03)00140-4.
Grande C, Templado J, Cervera JL, Zardoya R: Molecular phylogeny of Euthyneura (Mollusca: Gastropoda). Molecular Biology and Evolution. 2004, 21: 303-313. 10.1093/molbev/msh016.
Haszprunar G: The fine morphology of the osphradial sense organs of the Mollusca. Part I: Gastropoda – Prosobranchia. Philosophical Transactions of the Royal Society of London B. 1985, 307: 457-496. 10.1098/rstb.1985.0003.
Haszprunar G: On the origin and evolution of major gastropod groups, with special reference to the Streptoneura. Journal of Molluscan Studies. 1988, 54: 367-441. 10.1093/mollus/54.4.367.
Page LR: Development and evolution of adult feeding structures in Caenogastropods: overcoming larval functional constraints. Evolution & Development. 2000, 2 (1): 25-34. 10.1046/j.1525-142x.2000.00017.x.
Kantor YI: What is supporting the Neogastropoda monophyly? New evidences for the paraphyly. WCM 2007: 2007; Antwerp – Belgium. 2007
Gissi C, Mauro DS, Pesole G, Zardoya R: Mitochondrial phylogeny of Anura (Amphibia): A case study of congruent phylogenetic reconstruction using amino acid and nucleotide characters. Gene. 2006, 366: 228-237. 10.1016/j.gene.2005.07.034.
Simmons MP, Carr TG, O'Neill K: Relative character-state space, amount of potential phylogenetic information, and heterogeneity of nucleotide and amino acid characters. Molecular Phylogenetics and Evolution. 2004, 32: 913-926. 10.1016/j.ympev.2004.04.011.
Puillandre N, Samadi S, Boisselier MC, Sysoev AV, Kantor YI, Cruaud C, Couloux A, Bouchet P: Starting to unravel the toxoglossan knot: Molecular phylogeny of the "turrids" (Neogastropoda: Conoidea). Molecular Phylogenetics and Evolution. 2008, 47 (3): 1122-1134. 10.1016/j.ympev.2007.11.007.
Oliverio M, Modica MV: Phylogenetic relationships of the Neogastropoda: a preliminary molecular approach. IV International Congress of the European Malacological Societies. 2005, 41: 1-4.
Miya M, Nishida M: Use of mitogenomic information in teleostean molecular phylogenetics: a tree-based exploration under the maximum parsimony optimality criterion. Molecular Phylogenetics and Evolution. 2000, 17: 437-455. 10.1006/mpev.2000.0839.
Curole JP, Kocher TD: Mitogenomics: digging deeper with complete mitochondrial genomes. Trends in Ecology & Evolution. 1999, 14 (10): 375-415. 10.1016/S0169-5347(99)01660-2.
Grande C, Templado J, Cervera JL, Zardoya R: The complete mitochondrial genome of the nudibranch Roboastra europaea (Mollusca: Gastropoda) supports the monophyly of opisthobranchs. Molecular Biology and Evolution. 2002, 19: 1672-1685.
Kurabayashi A, Ueshima R: Complete sequence of the mitochondrial DNA of the primitive opisthobranch gastropod Pupa strigosa: systematic implication of the genome organization. Molecular Biology and Evolution. 2000, 17 (2): 266-277.
Boore JL, Medina M, Rosenberg LA: Complete sequences of the highly rearranged molluscan mitochondrial genomes of the Scaphopod Graptacme eborea and the bivalve Mytilus edulis. Molecular Biology and Evolution. 2004, 21: 1492-1503. 10.1093/molbev/msh090.
Ponder WF, Waren A: Classification of the Caenogastropoda and Heterostropha-A list of the family-group names and higher taxa. Malacological Review. 1988, 4 (Supplement): 288-326.
Trovão HFM: Contribuição para o estudo dos Moluscos Gasterópodos da família Conidae da África ocidental. Amphitrite. 1979, 1 (1): 3-12.
Nahir B, Kohn AB, Moroz LL: Complete DNA sequence of the mitochondrial genome of the sea-slug, Aplysia californica: conservation of the gene order in Euthyneura. Molecular Phylogenetics and Evolution. 2006, 38 (2): 459-469. 10.1016/j.ympev.2005.08.017.
Hatzoglou E, Rodakis GC, Lecadinou R: Complete sequence and gene organization of the mitochondrial genome of the land snail Albinaria coerulea. Genetics. 1995, 140: 1353-1356.
Maynard BT, Kerr LJ, McKiernan JM, Jansen ES, Hanna PJ: Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach). Marine Biotechnology. 2005, 7 (6): 645-658. 10.1007/s10126-005-0013-z.
Doyle JJ, Doyle JL: A rapid DNA isolation for small quantities of leaf tissue. Phytochemical Bulletin. 1987, 19: 11-15.
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 1997, 25: 655-964. 10.1093/nar/25.5.955.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin J, Higgins DG: The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution. 2000, 17: 540-552.
Talavera G, Castresana J: Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology. 2007, 56: 564-577. 10.1080/10635150701472164.
Akaike H: Information theory as an extension of the maximum likelihood principle. 2nd International Symposium on Information Theory. Edited by: Csaksi BNPaF. 1973, Budapest, Hungary: Akademiai Kiado, 267-281.
Abascal F, Zardoya R, Posada D: Pottest: selection of best-fit models of protein evolution. Bioinformatics. 2005, 21: 2104-2105. 10.1093/bioinformatics/bti263.
Posada D, Crandall ED: Modeltest: testing the model of DNA substitution. Bioinformatics. 1998, 14 (9): 817-818. 10.1093/bioinformatics/14.9.817.
Guindon S, Gascuel O: A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Huelsenbeck JP, Ronquist FR: MrBayes: Bayesian inference of phylogeny. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Shimodaira H: An approximately unbiased test of phylogenetic tree selection. Systematic Biology. 2002, 51: 492-508. 10.1080/10635150290069913.
Kishino H, Hasegawa H: Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. Journal of Molecular Evolution. 1989, 29: 170-179. 10.1007/BF02100115.
Shimodaira H, Hasegawa H: Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution. 1999, 16: 1114-1116.
Shimodaira H, Hasegawa H: CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001, 17 (12): 1246-1247. 10.1093/bioinformatics/17.12.1246.
Yang Z: PAML 4: a program package for phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007, 24: 1586-1591. 10.1093/molbev/msm088.
Acknowledgements
We thank Lukas Rüber for all the helpful discussions. We are in debt with Manuel J. Tenorio for thoughtful discussions, and for providing the specimens of Cancellaria cancellata and Terebra dimidiata. We thank Rita Castilho for help with Fig. 1. We also thank Carlos Afonso for providing the specimens of Nassarius reticulatus, Calyptraea chinensis and Cymatium parthenopeum. We are grateful to Stephane Guindon for providing us with an experimental new version of PHYML_v3.0, and to Federico Abascal for helping us with its use. RLC was sponsored by a PhD grant (SFRH/BD/9209/2002) from the Fundação para a Ciência e a Tecnologia, Portugal. The work was partly financed by project CGL2007-60954 of the Ministerio de Ciencia e Innovación of Spain to RZ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RLC organized the sampling, and carried out the molecular work. CG conducted gene annotation and analyzed gene rearrangements. RLC performed phylogenetic reconstructions. RZ designed and developed the study. RLC, CG, and RZ wrote the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cunha, R.L., Grande, C. & Zardoya, R. Neogastropod phylogenetic relationships based on entire mitochondrial genomes. BMC Evol Biol 9, 210 (2009). https://doi.org/10.1186/1471-2148-9-210
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-210