
Abstract
Background
Several studies have shown that genomes contain a mixture of transposable elements, some of which are still active and others ancient relics that have degenerated. This is true for the non-LTR retrotransposon Helena, of which only degenerate sequences have been shown to be present in some species (Drosophila melanogaster), whereas putatively active sequences are present in others (D. simulans). Combining experimental and population analyses with the sequence analysis of the 12 Drosophila genomes, we have investigated the evolution of Helena, and propose a possible scenario for the evolution of this element.
Results
We show that six species of Drosophila have the Helena transposable element at different stages of its evolution. The copy number is highly variable among these species, but most of them are truncated at the 5' ends and also harbor several internal deletions and insertions suggesting that they are inactive in all species, except in D. mojavensis in which quantitative RT-PCR experiments have identified a putative active copy.
Conclusion
Our data suggest that Helena was present in the common ancestor of the Drosophila genus, which has been vertically transmitted to the derived lineages, but that it has been lost in some of them. The wide variation in copy number and sequence degeneration in the different species suggest that the evolutionary dynamics of Helena depends on the genomic environment of the host species.
Similar content being viewed by others

Background
Transposable elements (TEs) are ubiquitous components in prokaryotic and eukaryotic genomes. They constitute the largest part of some of them [1], and play an important role in their evolution [2]. Genome sequencing has shown that TE sequences constitute about 15% of the Drosophila melanogaster genome [3, 4], about 45% of the human genome [5], and up to 90% of the genomes of some plants [6]. Why species harbor such different proportions of TEs is still unknown, but it may be related to the reproductive characteristics and population size of the host [7, 8], and to environmental stresses [9] that may influence TE dynamics. Various different mechanisms may prevent genome invasions by TEs, ranging from DNA deletions [10, 11] to epigenetic control mechanisms, such as chromatin conformation [12].
Several studies have shown that genomes harbor a mixture of TEs, some of which are still active, whereas others are ancient relics that have degenerated [13–16]. Degraded elements can result from point mutations or from DNA deletion [17]. Deletions may occur either by recombination, which is common to all classes of elements, or as a consequence of the transposition mechanism of the specific element concerned. For instance, the latter occurs in LINE elements (Long Interspersed Nuclear Elements, also known as non-LTR retrotransposons or retroposons), which are retroelements that use an RNA intermediate to transpose [2]. Their transposition mechanism leads to 5' end truncations of the new inserted sequence. Considerable internal deletions have previously been reported in the inactive copies of several non-LTR retrotransposons [10], and this deletion mechanism seems to act at an high rate, since sequences that are similar at the nucleotide level may have very different internal deletions [10, 11, 18, 19].
One example of this type of TE evolution is the retrotransposon Helena, which is a 4,912 bp LINE [18] first reported in the D. virilis species [20], and later identified in all the species analyzed in the melanogaster subgroup, as well as in D. pseudoobscura [10]. Helena has been shown to be present at different stages of its life-cycle in natural populations of D. melanogaster and D. simulans. Only degenerate copies were found in D. melanogaster [4, 18, 19, 21, 22], whereas in D. simulans several different types of sequences have been identified, ranging from highly degenerate to putatively active ones [18]. The analysis of Helena in these two closely-related species has shown how important the host genome can be in the evolution of a TE, and how important it is to analyze specific TE families in a wide spectrum of species. This is possible now that the genome sequences of 12 Drosophila species [23–26] are available, and Helena can be considered to provide an ideal model system for investigating TE evolution across a range of species.
We investigated the evolution of Helena sequences using a combination of experimental and population analyses with sequence analyses of the 12 Drosophila genomes, and we propose a possible scenario for the evolution of the element in the different host genomic environments that influence the "fate" of TEs.
Results
Identification and analysis of reference copies
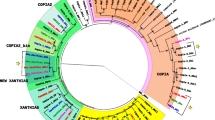
Using the full-length copy of Helena already identified in the draft sequence of the D. simulans genome [18], and 23 reverse transcriptase (RTase) fragments of the melanogaster species group [10], we performed a search for Helena-like elements in the other 10 Drosophila genomes. We identified Helena reference sequences in all the genomes (Additional file 1), apart from D. pseudoobscura, D. persimilis, D. willistoni and D. grimshawi (Figure 1). The results of Blast analyses in D. pseudoobscura, D. persimilis and D. willistoni revealed only short sequences (10 copies ≤ 210 bp, 23 copies ≤ 251 bp and seven copies ≤ 100 bp, respectively) with percentage identities to the BS element that were higher than or similar to those to Helena (D. pseudoobscura – BS: 78%, Helena: 69%, D. persimilis – BS: 80%, Helena: 74%, D. willistoni: BS: 85%, Helena: 89%). BS is a previously-described LINE element that belongs to the same "jockey clade" as Helena [27]. Sequences equally distant from BS and Helena have also been reported in D. melanogaster, and it has suggested that they may constitute a new family, named Helena/BS [21]. We did not therefore classify these sequences as Helena elements, and performed no further analysis of them. Our results for D. pseudoobscura are not consistent with previous data reported by Petrov et al. [10], who isolated just one sequence in this species. However, we should note that the same strains were not used in these two studies, and this could explain the differing results. We did not detect any Helena sequences in D. grimshawi, which is consistent with the fact that this a TE-poor genome [25], but in all the other species the selected reference copies we examined all contained one or two open reading frames (ORF1 and ORF2) (Figure 2).

Phylogenetic tree of the Drosophila genus. Phylogenetic relationships between the Drosophila species of which the genomes have been sequenced (modified from http://rana.lbl.gov/drosophila). Presence (+) and absence (-) of Helena.

Helena structure in Drosophila species. Structure of the reference copies of Helena-like elements. DNA sequences: AAA, poly-A tail. Protein sequences: gag, nucleocapsid-like domain; RTase, reverse transcriptase; *: position of start codons; #: position of stop codons. D. simulans and D. melanogaster structures are from Rebollo et al. [18]. The sequences are in Additional file 1.
The D. sechellia reference sequence (see Additional File 2 for details) is 4,888 bp long, and contains several of the hallmarks of Helena-related elements that have already been characterized: a 15 bp poly-A tail, and two overlapping open reading frames (ORF1 and ORF2). The first ORF is 1,732 bp, and the second 2,870 bp long. Both ORFs are interrupted by three premature stop codons, and the pol-like protein contains only the reverse transcriptase domain.
In the other species in the melanogaster group, the reference copies are smaller than the Helena reference copy in D. simulans. In D. erecta (see Additional File 3 for details) and D. ananassae (Additional File 4), the ORF1 and ORF2 are separated by sequences of 279 bp and 401 bp respectively, but both species contain the apyrimidic endonuclease, the exonuclease and the reverse transcriptase domains (Figure 2). In D. yakuba (Additional File 5) and D. virilis (Additional File 6) the reference copies consist only of a small ORF2, 1,755 bp and 561 bp in length, respectively.
The reference copy of D. mojavensis is the only putatively active Helena-like element identified in the species analyzed (Additional File 7). It is 4,502 bp long, and harbors two overlapping ORFs without premature stop codons. The first ORF is 1,782 bp long, and potentially encodes a 593 aa protein. The second is 2,722 bp long, and could encode a 907 aa protein. Moreover, the gag-like protein contains a conserved PRE_C2HC domain, and the pol-like protein contains the three domains required for its function: an apyrimidic endonuclease, an exonuclease and a reverse transcriptase domain. However, the poly-A tail was not identified.
Genomic Sequence Analyses
The Helena copy number for each species is presented in Table 1. This number varies considerably between the species, ranging from seven in D. erecta to 181 in D. sechellia, with sizes ranging from 80 bp (D. yakuba) to 4,888 bp (D. sechellia). The average percentage identity of the copies with the reference within each species ranged from 91.51% (D. ananassae) to 95.95% (D. yakuba). Size-divergence between these copies results from the presence of indels that occur throughout the sequences (Additional File 8). No relationship can be inferred between the size and the percentage identity, since copies with small sizes display an identity level as high as the larger ones (Figure 3). This may be expected when considering only the 5' truncations, produced by the transposition mechanism of Helena. However, the different copies reported here are also internally deleted, and can be expected to be old relics of Helena, with more neutral mutations than the full-length copies. The only exception concerns D. sechellia, in which significant correlation between size of the copies and identity was detected. This is probably due to the large number of copies detected in this species.

Helena copies in different genomes. Distribution of the lengths (intervals: 350 bp) and percentage identity of the Helena copies in D. simulans, D. melanogaster, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. mojavensis and D. virilis.
Table 2 shows the average GC content for the upstream and downstream regions of the Helena insertions for each species. It ranges from 36.8% (D. erecta) to 43.1% (D. melanogaster). These values are closer to the intergenic values for these species (35.3% – 39.9%) than to those for the gene regions (46.8% – 50.4%), suggesting that Helena is more abundant in the non-transcribed genomic environment [4, 16, 28–30]. The genome of D. melanogaster is the only one in which the GC values were intermediate between those of the intergenic and gene regions. This could simply reflect the better annotation available for the D. melanogaster genome.
Helena-related sequences form a monophyletic clade (Figure 4) that includes a well-defined cluster of the species of the melanogaster subgroup (D. yakuba, D. simulans, D. melanogaster, D. erecta and D. sechellia). The other group contained D. ananassae, D. virilis and D. mojavensis. This phylogeny is typical for a TE with vertical transmission.

Phylogenetic tree of RT proteins. The reconstruction was performed by the maximum likelihood method with the LG model, using the Helena reference copies obtained in this study and other LINE elements based on their partial reverse transcriptase domains. The sequences used were obtained from GenBank, and are identified by the TE name and the host names (* Sequences obtained from GenBank; 1Sequences obtained in this study; 2Helena sequences obtained by Rebollo et al. [18)]. The numbers indicate the branch support calculated by bootstrap analysis consisting of 100 replicates. Only bootstrap values greater than 50% are indicated.
Analysis of the activity in natural populations
Since our in-silico analyses show that only D. simulans and D. mojavensis harbor full-length, putatively active copies, we performed Southern blot analyses on several populations of these two species to infer their TE activity (Figure 5 and Additional File 9). Our results show that these species display insertion polymorphism, suggesting that Helena is active. We cannot of course exclude the possibility of restriction fragment length polymorphism, which would lead to the same kind of result. The qRT-PCR experiments on these two species show that Helena is highly expressed in D. mojavensis, which displayed population variability that contrasted with that of D. simulans, in which the levels of expression were much lower (Figure 6). We were only able to compare the Helena transcripts, because the housekeeping gene (rp49) used to normalize qRT-PCR is equally expressed in both species (Additional File 10).

Southern blot analysis of Helena in D . simulans and D. mojavensis populations. Lanes 1 to 8 are D. simulans populations (1: North America (Tucson stock center: 14021-0251.195), 2: Junco do Serido (PB, Brazil), 3: Itaúnas (ES, Brazil), 4: Lençóis (BA, Brazil), 5: Onda Verde (SP, Brazil), 6: Ratones (SC, Brazil), 7: Seychelles (Seychelles), 8: New Caledonia (Tucson stock center: 14021-0251.216). Lanes 9–11 are D. mojavensis populations (9: Catalina Island (California, U.S.A, Tucson stock center 15081-1352.02), 10: Grand Canyon (Arizona, U.S.A), 11: Sonora (Mexico, Tucson stock center: 15081-1352.24)).

Analysis of Helena activity in natural populations. Helena transcripts (ratio Helena/rp49) of ovaries and carcasses of D. simulans and D. mojavensis (see Methods for additional information). D. simulans strains from the Drosophila species stock center: 1 (14021-0251.195 – North America, U.S.A), 2 (14021-0251.194 – Winters, California, U.S.A), 3 (14021-0251.198 – Noumea, New Caledonia), and from natural populations 4 (Amieu, France), 5 (Valence, France). D. mojavensis strains from natural populations: 1 (Grand Canyon, Arizona, U.S.A), and from the Drosophila species stock center 2 (15081-1352.02 – Catalina Island, California, U.S.A), 3 (15081-1352.09 – Santa Rosa Mountains, Arizona, U.S.A), 4 (15081-1352.24 – Sonora, Mexico), 5 (15081-1352.22 – Catalina Island, California, U.S.A). Black = ovaries. Gray = Carcasses. Standard deviation is indicated with bars.
Discussion
This study describes the evolutionary dynamics of the Helena non-LTR retrotransposon in the sequenced Drosophila genomes. We have shown that Helena occurs in D. sechellia, D. yakuba, D. erecta and D. ananassae (melanogaster species group), in D. mojavensis (repleta group) and in D. virilis (virilis group), as well as in D. melanogaster and D. simulans, which had been studied previously [10, 18]. In D. pseudoobscura, D. persimilis and D. willistoni, the small copies that we found displayed similar percentage identities to Helena as to BS, a non-LTR retrotransposon related to Helena. Sequences with the same kind of similarity have been reported in D. melanogaster, and were grouped by the authors as Helena/BS family [21]. The D. pseudoobscura, D. persimilis and D. willistoni sequences we found can be included in this family. We agree with the suggestion that there could be members of the Helena/BS family in these species as shown in D. melanogaster [21], and so we did not include them in our study. The 907 bp sequence of Helena that had been previously described (GenBank AF012036) in a strain (stock center: 14011-0121-0, Tucson, Arizona) of D. pseudoobscura [10] was not found in the sequenced genome of this species. Our phylogenetic analysis shows that Helena is a monophyletic group of sequences patchily distributed in the species of the two subgenera of the genus Drosophila (Figure 1). Taking our data together with those from other authors [10, 18, 20], we can conclude that Helena was present in the common ancestor of the Drosophila genus, and has been vertically transmitted to the derived lineages, but subsequently lost in some of them, or at least diverged so much as to escape detection. The high variability in copy number and sequence degeneration in the different species shows that the evolutionary dynamics of Helena depends on the genomic environment, as has already been reported for other retrotransposons, such as Tirant [31].
Recent work has reported the presence of deteriorated and inactivated Helena in the D. melanogaster genome, but also of a full-length copy in D. simulans carrying all the structures required for activity, and with a high level of insertion polymorphism in the natural populations [18]. This suggests that in this species Helena is either still active or has been active until recently. In our analysis, all the reference copies in the other 10 Drosophila genomes were found to be devoid of intact ORFs, except in D. mojavensis. In this species, two copies of Helena contain intact ORFs, which suggest that full-length and potentially active Helena sequences could exist. This means that this species may offer a unique opportunity for studying the evolutionary dynamics of Helena.
Our analysis of the copy numbers reveals a strikingly variable distribution of Helena in the 12 species. In D. sechellia, 181 copies of Helena were observed, whereas in D. erecta only seven copies were identified. Even though these genomes have not been well annotated, and there are some low quality sequences that could bias copy number estimation, this should not invalidate the tendency identified. It has been proposed that effective population size could be one of the main factors accounting for differences in copy number of TEs [32, 33], with selection against TEs being less effective in smaller populations [34]. This seems to apply to D. sechellia. This species is restricted to the Seychelles Islands in the Indian Ocean, and is the most specialized fly within the melanogaster group. It has a very low effective population size [35, 36], and carries the highest number of Helena sequences recorded (181 copies). We would expect to find a similar scenario for D.erecta, which is also a specialized species with a small population size [37, 38]. However, as has also been observed for the mariner element, the observed copy number of Helena is not in fact consistent with this hypothesis [37]. This means that other factors may be affecting the copy number of TEs, including genomic and environmental features.
We did not observe any major differences in copy number between the two species of the Drosophila subgenus, even though D. mojavensis has a higher copy number than D. virilis. What is more striking is that the only potentially full-length active copies identified were detected in D. mojavensis, together with the other potentially active copy previously described in D. simulans [18]. The Southern blot analyses of several natural populations of D. mojavensis and D. simulans suggest that Helena has recently displayed transpositional activity. However, we confirmed that the transcriptional activity is low in D. simulans, as had previously been suggested [18], and this clearly indicates that Helena is being lost in this species. In D. mojavensis, the situation is completely different. We have observed high levels of Helena transcripts in several populations, confirming its activity. Moreover, expression varied between different populations, indicating that Helena must be present at different stages of its evolutionary cycle, i.e. different stages of activity, in different populations within this species.
Our analysis has shown that the evolution of Helena is influenced by the host species, resulting in differences in copy number, degradation and activity. In all the species analyzed, D. mojavensis is the only one in which the Helena has survived, which gives us a unique opportunity to observe the "fate" of this TE. However, it is crucial to investigate natural populations from this and other species in the repleta group in order to find out how widespread the scenario described here actually is, and to understand the process and speed of the degradation and extinction of Helena.
Conclusion
Here we show that six species of Drosophila have Helena transposable elements at different stages of its evolution, and may represent different stages of the TE evolutionary cycle. The copy number is highly variable in different species, but most of them are truncated at the 5' ends and display several internal deletions and insertions. In all the species analyzed, Helena has only survived in D. mojavensis, and this gives us a unique opportunity to track the "fate" of this TE.
Methods
In-silico analyses
The draft sequence data from the 10 related Drosophila Genome Sequencing Projects used in this study are listed in the Table 3. Sequence searches for the Helena element were carried out using the 4,912 bp full-length sequence of Helena characterized in D. simulans [18] as the query. Twenty-three partial sequences of reverse transcriptases (RTases) of Helena from the melanogaster group [10], and the two LINE elements closest to Helena, BS [27] and X [39] were found. Comparisons between the query and the Drosophila data set were performed using High Scoring Pairs (HSPs) within BLAST algorithms [40] with an E-value ≤ 1e-10. The sequences correspond to the three best BLAST hits, which were more than 80 bp in length, and were selected and extended to include 3 kb of the flanking regions. These sequences were analyzed using the ORF-FINDER program to identify putative occurrences of coding regions [41]. Conserved domains were predicted using the "Conserved domain search" tool from NCBI, as well as BLASTn and BLASTx searches against the nt and nr databases, respectively. Thus, we determined the most complete Helena sequence in each of the 10 Drosophila species to be considered as a reference copy within each genome. To determine the copy number in each species, the previously-determined reference copy was blasted against the full genomes. Significant matches were required to be more than 80 bp long, and to have at least 80% identity [42]. Regions similar to Helena that were separated by less than 200 bp were considered to be a single insertion. For each genome, the copies have been aligned with the reference, using MUSCLE [43] and the % identity to the reference was computed using the DNADIST program from the PHYLIP package [44]. The flanking regions of each insertion were extracted for analysis of the GC content in the first 5,000 nt using "geecee" of the EMBOSS package [45]. The GC contents of the flanking regions of the Helena elements were compared to those of the intergenic regions and genes (exon plus introns) in the other Drosophila genome versions, as noted in the Table 3.
Phylogenetic analyses
The sequences used in the evolutionary analysis were obtained from GenBank: Jockey (M22874), TART (U14101), Doc (X17551), F (M17214), BS (X77571) and X (AF237761) of D. melanogaster; Jockey of D. funebris (M38437); Amy of Bombyx mori (U07847); JuanA of Aedes aegypti (M95171); JuanC of Culens pipientis (M91082); NCR1th of Chironomus tentans (L79944); Helena of D. mauritiana (AF012043), D. simulans and D. melanogaster [18]. The multiple alignment of the RTase proteins from 11 LINEs of the Jockey clade, and the six Helena reference sequences described here (see Additional Files 1, 2, 3, 4, 5 and 6 for details) was performed using CLUSTALW [46] with the default parameters, and the alignment was manually curated using a sequence editor. The evolutionary relationships were reconstructed using the maximum likelihood method for the LG model [47] as implemented in the PhyML software [48]. The bootstrap analysis consisted of 100 replicates.
Southern blot
The occurrence of Helena in populations of D. simulans and D. mojavensis (Additional File 8) was confirmed by Southern blot using the detection system Gene Images CDP-Star detection module (Amersham Biosciences, Little Chalfont, UK). Genomic DNA was prepared from 50 adult flies [49] and digested by Hind III, which has no restriction site within the Helena sequence, so that each hybridized fragment would correspond to a single genomic insertion. The restricted fragments were separated in 1% agarose gels, and transferred to Hybond N+ membranes (Amersham Biosciences, Little Chalfont, UK). Blots were prehybridized for 1 h at 60°C in 5× SSC, in 5% dextran sulfate, subjected to 20-fold dilution of the liquid block, and hybridized overnight with the probes. Blots were washed twice with 0.2× SSC, 0.5% SDS, and then exposed to autoradiographic film for 20 minutes at room temperature. A 644 bp Helena sequence amplified from plasmid AF012044 (DsechF: 5' AGGATTTGTCATGCCACGCT 3' e DsechR R 5' TGTTTGGTGCTGCCATGTGT 3'), and a 674 bp sequence, corresponding to the RTase of D. mojavensis Helena (DmojF: 5' TAAGAGGCCATAGTACGGAGCAGGTA3' and DmojR: 5' GCGAATTGGAACAGGCTAACGCAT 3'), were used as probes for the D. simulans and D. mojavensis populations, respectively.
Quantitative RT-PCR (qRT-PCR)
The expression profiles of Helena RTase in different populations of D. simulans and D. mojavensis (Additional File 8) were determined by real-time PCR. For this analysis, 20 ovaries and 15 carcasses of each population were used to extract total RNA using RNeasy kit (Qiagen). 1 μg of total RNA treated with DNAse Ambion was converted into cDNA using Thermoscript kit (Invitrogen) primed with oligo-dt and random primers mix. The cDNA samples were diluted 50 fold, and PCR was carried out using QuantiTect SYBR Green PCR kit (Roche) on the LightCycler (Roche) using primers specific to the Helena RTase of D. simulans (RTase_D.simF: 5'ACAGCAGAGAGACAGCTAACGGAC 3', Rtase_DsimR: 5' AGATGTGTTGCTTGCAGGGTCTGA 3' and D. mojavensis (RTase_DmojF: 5' TTGGTCCGCTGCTGTTCTCCTT 3', Rtase_DmojR: 5' TGAGATTCCACCGCTTGCACCA 3') that amplify 193 bp and 204 bp respectively. Quantitative PCR cycling conditions were 5 min at 95°C (1 cycle), 15 s at 95°C, followed by 10 s at 62°C and 20 s at 72°C (50 cycles). A negative control for DNA contamination of cDNA of each population (without Thermoscript enzyme) was tested (data not shown). Reactions were done in duplicate, and standard curves were calculated from serial dilutions of specific amplified PCR fragments. The quantity of the transcripts was estimated relative to the RP49 expression (qPCR fragments of 182 bp and 167 bp for D. simulans and D. mojavensis respectively). Primers were: RP49_DsimF: 5' CGGATCGATATGCTAAGCTGT 3', RP49_DsimR: 5' GCGCTTGTTCGATCCGTA 3', RP49_DmojF: 5' GTCGTCGCTTCAAGGGCCAAT 3', RP49_DmojR: 5' ATGGGCGATCTCACCGCAGTA 3'. In both species, RP49 expression is equivalent (Additional Figure nine) allowing relative expression of Helena to be comparable between D. mojavensis and D. simulans. Hence, Helena transcripts plotted in Figure 6 are the result of quantification of Helena transcripts normalized by the quantification of RP49 transcripts for each strain.
Abbreviations
- DOA:
-
dead on arrival
- LINE:
-
long interspersed nuclear element
- LTR:
-
long terminal repeat
- ORF:
-
open reading frame
- TE:
-
transposable element
- RTase:
-
reverse transcriptase.
References
Biemont C, Vieira C: Genetics: junk DNA as an evolutionary force. Nature. 2006, 443 (7111): 521-524. 10.1038/443521a.
Finnegan DJ: Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5 (4): 103-107. 10.1016/0168-9525(89)90039-5.
Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, et al: Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science. 2007, 316 (5831): 1625-1628. 10.1126/science.1139816.
Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, et al: The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002, 3 (12): RESEARCH0084-10.1186/gb-2002-3-12-research0084.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Bennetzen JL: Comparative sequence analysis of plant nuclear genomes: microcolinearity and its many exceptions. Plant Cell. 2000, 12 (7): 1021-1029. 10.1105/tpc.12.7.1021.
Kidwell MG: Reciprocal differences in female recombination associated with hybrid dysgenesis in Drosophila melanogaster. Genet Res. 1977, 30 (1): 77-88.
Wright S, Finnegan D: Genome evolution: sex and the transposable element. Curr Biol. 2001, 11 (8): R296-299. 10.1016/S0960-9822(01)00168-3.
Arnault C, Dufournel I: Genome and stresses: reactions against aggressions, behavior of transposable elements. Genetica. 1994, 93 (1–3): 149-160. 10.1007/BF01435247.
Petrov DA, Hartl DL: High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol. 1998, 15 (3): 293-302.
Petrov DA, Lozovskaya ER, Hartl DL: High intrinsic rate of DNA loss in Drosophila. Nature. 1996, 384 (6607): 346-349. 10.1038/384346a0.
Slotkin RK, Martienssen R: Transposable elements and the epigenetic regulation of the genome. Nature reviews. 2007, 8 (4): 272-285.
Alonso-Gonzalez L, Dominguez A, Albornoz J: Structural heterogeneity and genomic distribution of Drosophila melanogaster LTR-retrotransposons. Mol Biol Evol. 2003, 20 (3): 401-409. 10.1093/molbev/msg047.
Cizeron G, Biemont C: Polymorphism in structure of the retrotransposable element 412 in Drosophila simulans and D. melanogaster populations. Gene. 1999, 232 (2): 183-190. 10.1016/S0378-1119(99)00126-2.
Kapitonov VV, Jurka J: Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci USA. 2003, 100 (11): 6569-6574. 10.1073/pnas.0732024100.
Lerat E, Rizzon C, Biemont C: Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res. 2003, 13 (8): 1889-1896.
Lozovskaya ER, Nurminsky DI, Petrov DA, Hartl DL: Genome size as a mutation-selection-drift process. Genes & genetic systems. 1999, 74 (5): 201-207. 10.1266/ggs.74.201.
Rebollo R, Lerat E, Kleine LL, Biemont C, Vieira C: Losing helena: the extinction of a drosophila line-like element. BMC Genomics. 2008, 9: 149-10.1186/1471-2164-9-149.
Blumenstiel JP, Hartl DL, Lozovsky ER: Patterns of insertion and deletion in contrasting chromatin domains. Mol Biol Evol. 2002, 19 (12): 2211-2225.
Petrov DA, Schutzman JL, Hartl DL, Lozovskaya ER: Diverse transposable elements are mobilized in hybrid dysgenesis in Drosophila virilis. Proc Natl Acad Sci USA. 1995, 92 (17): 8050-8054. 10.1073/pnas.92.17.8050.
Berezikov E, Bucheton A, Busseau I: A search for reverse transcriptase-coding sequences reveals new non-LTR retrotransposons in the genome of Drosophila melanogaster. Genome Biol. 2000, 1 (6): RESEARCH0012-10.1186/gb-2000-1-6-research0012.
Bergman CM, Bensasson D: Recent LTR retrotransposon insertion contrasts with waves of non-LTR insertion since speciation in Drosophila melanogaster. Proc Natl Acad Sci USA. 2007, 104 (27): 11340-11345. 10.1073/pnas.0702552104.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al: The genome sequence of Drosophila melanogaster. Science. 2000, 287 (5461): 2185-2195. 10.1126/science.287.5461.2185.
Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al: Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007, 5 (11): e310-10.1371/journal.pbio.0050310.
Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, et al: Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007, 450 (7167): 203-218. 10.1038/nature06341.
Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, Nielsen R, Thornton K, Hubisz MJ, Chen R, Meisel RP, et al: Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005, 15 (1): 1-18. 10.1101/gr.3059305.
Udomkit A, Forbes S, Dalgleish G, Finnegan DJ: BS a novel LINE-like element in Drosophila melanogaster. Nucleic acids research. 1995, 23 (8): 1354-1358. 10.1093/nar/23.8.1354.
Bartolome C, Maside X, Charlesworth B: On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002, 19 (6): 926-937.
Charlesworth B, Langley CH: The population genetics of Drosophila transposable elements. Annual review of genetics. 1989, 23: 251-287. 10.1146/annurev.ge.23.120189.001343.
Rizzon C, Martin E, Marais G, Duret L, Segalat L, Biemont C: Patterns of selection against transposons inferred from the distribution of Tc1, Tc3 and Tc5 insertions in the mut-7 line of the nematode Caenorhabditis elegans. Genetics. 2003, 165 (3): 1127-1135.
Fablet M, Souames S, Biemont C, Vieira C: Evolutionary pathways of the tirant LTR retrotransposon in the Drosophila melanogaster subgroup of species. J Mol Evol. 2007, 64 (4): 438-447. 10.1007/s00239-006-0108-9.
Aquadro CF, Lado KM, Noon WA: The rosy region of Drosophila melanogaster and Drosophila simulans. I. Contrasting levels of naturally occurring DNA restriction map variation and divergence. Genetics. 1988, 119 (4): 875-888.
Lynch M, Conery JS: The origins of genome complexity. Science. 2003, 302 (5649): 1401-1404. 10.1126/science.1089370.
Brookfield JF, Badge RM: Population genetics models of transposable elements. Genetica. 1997, 100 (1–3): 281-294. 10.1023/A:1018310418744.
Hey J, Kliman RM: Population genetics and phylogenetics of DNA sequence variation at multiple loci within the Drosophila melanogaster species complex. Mol Biol Evol. 1993, 10 (4): 804-822.
Kliman RM, Hey J: DNA sequence variation at the period locus within and among species of the Drosophila melanogaster complex. Genetics. 1993, 133 (2): 375-387.
Capy P, David JR, Hartl DL: Evolution of the transposable element mariner in the Drosophila melanogaster species group. Genetica. 1992, 86 (1–3): 37-46. 10.1007/BF00133709.
McBride CS, Arguello JR, O'Meara BC: Five Drosophila genomes reveal nonneutral evolution and the signature of host specialization in the chemoreceptor superfamily. Genetics. 2007, 177 (3): 1395-1416. 10.1534/genetics.107.078683.
Tudor M, Davis AJ, Feldman M, Grammatikaki M, O'Hare K: The X element, a novel LINE transposable element from Drosophila melanogaster. Mol Genet Genomics. 2001, 265 (3): 489-496. 10.1007/s004380000437.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. Journal of molecular biology. 1990, 215 (3): 403-410.
Rombel IT, Sykes KF, Rayner S, Johnston SA: ORF-FINDER: a vector for high-throughput gene identification. Gene. 2002, 282 (1–2): 33-41. 10.1016/S0378-1119(01)00819-8.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al: A unified classification system for eukaryotic transposable elements. Nature reviews. 2007, 8 (12): 973-982. 10.1038/nrg2165.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004, 32 (5): 1792-1797. 10.1093/nar/gkh340.
Felsenstein J: PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics. 1989, 5: 3-10.1111/j.1096-0031.1989.tb00480.x.
Rice P, Longden I, Bleasby A: EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000, 16 (6): 276-277. 10.1016/S0168-9525(00)02024-2.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research. 1994, 22 (22): 4673-4680. 10.1093/nar/22.22.4673.
Le SQ, Gascuel O: An improved general amino acid replacement matrix. Mol Biol Evol. 2008, 25 (7): 1307-1320. 10.1093/molbev/msn067.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic biology. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Jowett T, Rizki TM, Rizki RM: Regulation of synthesis of larval serum proteins after transplantation of larval fat body into adult Drosophila melanogaster. Developmental biology. 1986, 116 (1): 23-30. 10.1016/0012-1606(86)90039-4.
Acknowledgements
Funding for this project was provided by the Brazilian agencies FAPESP (07/53097-0 to C.M.A.C. and fellowship 07/50641-1 to A.G.) and CNPq (to C.M.A.C.), and by the French MIRA project from the Région Rhône-Alpes (to C. V. and A. G.), the ANR Genemobile (to C. V.), and the CNRS-FAPESP (joint program to C.V. and C.M.A.C.). We thank R. Rebollo, N. Burlet and C. Loevenbruck for their comments and technical help, and M. Ghosh for correcting the English text.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AG carried out the molecular and genetic studies, AG, FRL and EL did the bioinformatic analyses, CV and CMAC designed and coordinated the study. All the authors contributed to data analyses and the writing of the paper. All the authors have read and approved the final manuscript.
Adriana Granzotto, Fabrício R Lopes, Emmanuelle Lerat, Cristina Vieira and Claudia MA Carareto contributed equally to this work.
Electronic supplementary material
12862_2009_1088_MOESM1_ESM.pdf
Additional file 1: Sequences of the Helena reference copies. Helena sequences used to determine the structure of the element, for each species. (PDF 26 KB)
12862_2009_1088_MOESM2_ESM.doc
Additional file 2: Helena copies in the Drosophila sechellia sequenced genome. The data provided is a list of D. sechellia copies. (DOC 246 KB)
12862_2009_1088_MOESM3_ESM.doc
Additional file 3: Helena copies in the Drosophila erecta sequenced genome. The data provided is a list of D. erecta copies. (DOC 32 KB)
12862_2009_1088_MOESM4_ESM.doc
Additional file 4: Helena copies in the Drosophila ananassae sequenced genome. The data provided is a list of D. ananassae copies. (DOC 70 KB)
12862_2009_1088_MOESM5_ESM.doc
Additional file 5: Helena copies in the Drosophila yakuba sequenced genome. The data provided is a list of D. yakuba copies. (DOC 52 KB)
12862_2009_1088_MOESM6_ESM.doc
Additional file 6: Helena copies in the Drosophila virilis sequenced genome. The data provided is a list of D. virilis copies. (DOC 38 KB)
12862_2009_1088_MOESM7_ESM.doc
Additional file 7: Helena copies in the Drosophila mojavensis sequenced genome. The data provided is a list of D. mojavensis. (DOC 74 KB)
12862_2009_1088_MOESM8_ESM.tiff
Additional file 8: Schematic representation of Helena. Schematic representation of Helena copies in D. sechellia, D. yakuba, D. erecta, D. ananassae, D. mojavensis and D. virilis. The sequences represented have at least 90% identity and 50% of the length of the reference copy, and with e-values of less than 10e-10. Spaces = indels. The first schematic representation is the reference copy in each species. White = gag. Gray = RTase. (TIFF 4 MB)
12862_2009_1088_MOESM9_ESM.doc
Additional file 9: Species and strains, geographic origin and year of collection. The data provided is a list of D. simulans and D. mojavensis used in this study for analysis of Helena activity in natural populations. (DOC 42 KB)
12862_2009_1088_MOESM10_ESM.tiff
Additional file 10: Mean CT of the rp49 control gene and Helena. Ct comparison between rp49 (reference) and Helena real time PCR assays of D. simulans and D. mojavensis. Gray = mean CT of D. simulans. Black = mean CT of D. mojavensis. Std = standard deviation. (TIFF 381 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Granzotto, A., Lopes, F.R., Lerat, E. et al. The evolutionary dynamics of the Helena retrotransposon revealed by sequenced Drosophila genomes. BMC Evol Biol 9, 174 (2009). https://doi.org/10.1186/1471-2148-9-174
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-9-174