
Abstract
Background
The L-lactate and D-lactate dehydrogenases, which are involved in the reduction of pyruvate to L(-)-lactate and D(+)-lactate, belong to evolutionarily unrelated enzyme families. The genes encoding L-LDH have been used as a model for gene duplication due to the multiple paralogs found in eubacteria, archaebacteria, and eukaryotes. Phylogenetic studies have suggested that several gene duplication events led to the main isozymes of this gene family in chordates, but little is known about the evolution of L-Ldh in invertebrates. While most invertebrates preferentially oxidize L-lactic acid, several species of mollusks, a few arthropods and polychaetes were found to have exclusively D-LDH enzymatic activity. Therefore, it has been suggested that L-LDH and D-LDH are mutually exclusive. However, recent characterization of putative mammalian D-LDH with significant similarity to yeast proteins showing D-LDH activity suggests that at least mammals have the two naturally occurring forms of LDH specific to L- and D-lactate. This study describes the phylogenetic relationships of invertebrate L-LDH and D-LDH with special emphasis on crustaceans, and discusses gene duplication events during the evolution of L-Ldh.
Results
Our phylogenetic analyses of L-LDH in vertebrates are consistent with the general view that the main isozymes (LDH-A, LDH-B and LDH-C) evolved through a series of gene duplications after the vertebrates diverged from tunicates. We report several gene duplication events in the crustacean, Daphnia pulex, and the leech, Helobdella robusta. Several amino acid sequences with strong similarity to putative mammalian D-LDH and to yeast DLD1 with D-LDH activity were found in both vertebrates and invertebrates.
Conclusion
The presence of both L-Ldh and D-Ldh genes in several chordates and invertebrates suggests that the two enzymatic forms are not necessarily mutually exclusive. Although, the evolution of L-Ldh has been punctuated by multiple events of gene duplication in both vertebrates and invertebrates, a shared evolutionary history of this gene in the two groups is apparent. Moreover, the high degree of sequence similarity among D-LDH amino acid sequences suggests that they share a common evolutionary history.
Similar content being viewed by others


Background
The reduction of pyruvate to L(-)-lactate and D(+)-lactate is catalyzed by different NAD-dependent enzymes, the L-lactate (L-LDH: L-lactate:NAD+ oxidoreductase, EC 1.1.1.27) and D-lactate dehydrogenases (D-LDH: D-lactate:NAD+ oxidoreductase, EC 1.1.1.28) as well as by NAD-independent (cytochrome) enzymes (DLD: D-lactate ferricytochrome c oxidoreductase, EC 1.1.2.4). Despite their apparent functional similarity, these classes of enzymes are selective for the D/L chirality of the substrate [1]. Studies on the primary amino acid structures of L-LDH and D-LDH suggest that the genes encoding them are not evolutionarily related [2, 3] and that their products belong to larger families of enzymes: L(-)-LDHs belong to the L-specific NAD-dependent dehydrogenases, while D(+)-LDHs belong to the D-isomer specific 2-hydroxy acid dehydrogenases and the FAD-binding oxidoreductase/transferase type 4 family.
L-LDH has been among the most studied enzyme families, but very little is known about the structure, function, and kinetics of D-LDH [4, 5]. The main question in the evolution of L-LDH relates to the orthology of the gene loci encoding the proteins with various enzyme activities [6, 7]. While L-LDHs have been extensively studied in vertebrates, there is much less information on these enzymes in invertebrates. With the availability of new L-Ldh sequences from two crustaceans (Daphnia pulex in the Branchiopoda, and a second malacostracan, the porcelain crab, Petrolisthes cinctipes in the Decapoda) and a few other invertebrates such as the leech Helobdella robusta, the polychaete Capitella capitata, and the tunicate Ciona intestinalis, we sought to gain a deeper understanding of the relationship between invertebrate and protochordate L-LDHs and those of vertebrates, and to elucidate the evolutionary relationship among invertebrate L-Ldhs. Moreover, the recent description of mammalian D-LDH enzymes that show significant similarity to yeast proteins with D-LDH activity [5] prompted our search for sequences with putative D-LDH activity in both vertebrate and invertebrate genomes.
Results
Alignments and phylogenetic analyses of L-LDH amino acid sequences
The L-LDH alignment used in phylogenetic analyses includes 315 amino acids, with 68 constant characters and 214 parsimony-informative characters. Maximum-parsimony (MP) analysis of 49 L-LDH sequences using the tree-bisection-reconnection (TBR) algorithm found two most parsimonious trees of 1982 steps long with a consistency index (CI) = 0.48, a homoplasy index (HI) = 0.51 and a retention index (RI) = 0.63. The uncorrected number of amino acid differences per site between invertebrate and chordate groups is 0.38 ± 0.019.
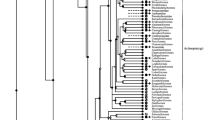
Phylogenetic trees generated by MP and Neighbor-joining (NJ, Figure 1) and Bayesian Inference (BI, Figure 2) all support a deuterostome cluster. The only exception is the echinoderm, Strongylocentrotus purpuratus, which groups with nematodes, although bootstrap support for this phylogenetic relationship is extremely low (Figure 1). The vertebrate sequences form a well-supported cluster with the LDH-A and LDH-B isozymes separating into distinct groups. Even so, there are examples of species whose A, B and C isozymes cluster with one another (e.g. Xenopus laevis).

Neighbor-joining tree based on 49 L-Lactate dehydrogenase amino acid sequences from 31 taxa. Numbers at nodes indicate the Neighbour-joining and Maximum Parsimony percentage bootstrap support with 2,000 and 100 replicates, respectively. Nodes supported only by the Neighbor-Joining analysis show a single bootstrap value. The scale bar indicates levels of amino acid sequence divergence. The tree was rooted using the plant LDH sequences.

Bayesian inference, 50% majority rule consensus tree based on 49 L-Lactate dehydrogenase amino acid sequences. The numbers at the nodes are posterior probabilities expressed as percentages.
The arthropod LDH sequences form a well-resolved cluster, as do the insects within it. Two copies of Ldh were found in the Daphnia pulex genome, and three copies in the Helobdella robusta genome. As with other cases of gene duplication outside chordates, the suffixes A, B and C do not denote orthology to the vertebrate A, B and C isozymes. The predicted protein sequences from the two Daphnia paralogs show 0.17 ± 0.012 amino acid divergence (p-distance) and 0.224 ± 0.014 nucleotide divergence (p-distance) in the coding regions. They cluster with one another indicating that this gene duplication occurred after the divergence of Daphnia from the other crustaceans, the decapods Carcinus maenas and Petrolisthes cinctipes (Figure 1 and 2). The insects and crustaceans are reciprocally monophyletic in the MP and NJ trees (Figure 1), although bootstrap support for the crustacean node is very low. Conversely, very strong support for an arthropod clade in which the crustaceans are paraphyletic relative to the insects was obtained in the BI tree (Figure 2).
The level of amino acid divergence between the three Helobdella robusta LDH proteins (0.32 ± 0.026, 0.46 ± 0.028, 0.52 ± 0.028) is higher than that between the Daphnia copies or Fundulus heteroclitus (LDH-B and LDH-C, 0.21 ± 0.028) and much higher than that between the recently diverged Xenopus laevis copies (0.019 ± 0.008, 0.049 ± 0.013, 0.063 ± 0.014). Moreover, the relationships among the three genes differs among the phylogenetic trees. Only two of the three H. robusta genes (Ldh-A and Ldh-B) are clearly paralogous in the MP/NJ tree, and duplicated after the divergence of H. robusta from the other annelid in the analysis (the polychaete, Capitella capitata). However, the annelid cluster also contains the trematode flatworm, Clonorchis sinensis, although bootstrap support for these relationships is low (Figure 1). The annelid/trematode clade is also recovered in the BI tree (Figure 2), although in this case, C. capitata is the sister group to the three leech and the flatworm sequences.
Alignments and phylogenetic analyses of D-LDH amino acid sequences
The D-LDH alignment included 486 amino acids with 73 conserved sites and 319 parsimony informative sites. The uncorrected number of amino acid differences per site averaged over all sequence pairs between chordates and invertebrates is 0.41 ± 0.015.
In general, the topology of the NJ tree generated from these sequences (Figure 3) shows that the deuterostomes form a distinct cluster relative to the other animals, as expected. For example, there is strong bootstrap support for a vertebrate cluster, and the non-vertebrate deuterostomes (C. intestinalis and S. purpuratus) cluster with them, although unexpectedly, C. intestinalis (a tunicate) clusters with S. purpuratus (an echinoderm) instead of the vertebrates. Relationships among the other invertebrates are not well resolved. For example, the annelids, H. robusta and C. capitata, do not cluster with each other, and the protostomes themselves do not form a monophyletic group (H. robusta is the sister taxon to all the other animals except C. elegans), but bootstrap support for several of the invertebrate nodes is low. Overall, this tree strongly suggests that D-Ldh was present in the common ancestor of animals.

Neighbor-joining tree based on D-Lactate dehydrogenase amino acid sequences from 21 taxa. Numbers at nodes indicate the percentage of NJ bootstrap analyses with 2,000 replicates and posterior probabilities expressed as percentages. The scale bar indicates levels of amino acid sequence divergence. The tree was rooted using the fungal DLD1 proteins.
Discussion
L-lactate dehydrogenases in vertebrate and invertebrate evolution
The majority of taxa of jawed vertebrates contain three isozymes of L-LDH (LDH-A, LDH-B and LDH-C) encoded by three loci. The M form (LDH-A) is found predominantly in white skeletal muscle (fast twitch glycolytic fibers) and is best suited for pyruvate reduction in anaerobic conditions, while the H form (LDH-B) is found in more aerobic tissues such as heart and brain and is most efficient for lactate oxidation. The X form (LDH-C) is found in various tissues such as the spermatozoa of mammals and birds, the eye lenses of birds and crocodilian and the liver and eye of teleosts [8, 9].
It is commonly accepted that new metabolic capacities of L-LDH enzymes have often arisen by gene duplications in addition to more orthodox evolutionary changes in existing genes. For this reason the Ldh gene family has been used as a model for gene duplication in vertebrate evolution [8, 10]. It is generally accepted that the Ldh genes of jawed vertebrates arose as a series of gene duplications in early vertebrate evolution, after the divergence of vertebrates from tunicates. However, the succession of these gene duplication events is not well understood [9, 11, 12]. Two main evolutionary scenarios have been proposed. The classical scenario involves the duplication of an Ldh-A-like locus in Agnatha (lampreys have a single LDH form) that gave rise to Ldh-A and Ldh-B. A second round of gene duplication involved Ldh-B and gave rise to Ldh-B and Ldh-C. Several phylogenetic studies support this hypothesis of an original Ldh-A and Ldh-B gene duplication followed by more recent and independent origins of the Ldh-C genes in teleost fish, Xenopus laevis, pigeon and mammals [7, 10, 13]. A second school of thought suggests that the primordial vertebrate LDH was an LDH-C like enzyme. This hypothesis emerged from phylogenetic reconstructions and from the observation that the single LDH isozyme of the primitive agnathan, the sea lamprey, is immunologically more similar to LDH-C in teleost fish than to LDH-A or LDH-B [14–16].
Our phylogenetic analyses of L-LDH isozymes are consistent with the general view of a major gene duplication event near the origin of vertebrates. Both NJ and MP phylogenies suggest that the vertebrate isozymes LDH-A and LDH-B evolved through a series of gene duplications soon after the vertebrates diverged from tunicates. The main difference between the MP, NJ and BI reconstructions is that MP and BI supports a sister group relationship between LDH in Petromyson marinus, the only chordate with a single LDH locus, and all of the other vertebrate LDHs. Conversely, this sequence clusters with fish LDH-A in the NJ tree. Although the controversial phylogenetic position of the LDH-C isozymes cannot be easily settled, it is clear from our results that Ldh-C in mammals, X. laevis and Fundulus have independent, derived origins from either Ldh-A or Ldh-B-like ancestors.
The relationship of invertebrate and protochordate Ldh to vertebrate Ldh is not well understood. Although most invertebrates seem to possess one copy of L-Ldh [17–19], several events of gene duplication involving nonvertebrates have been reported in crustaceans, barley [20] and in the psychrophilic bacterium, Bacillus psychrosaccharolyticus [21]. In studies of crustacean LDH, protein electrophoresis has detected the presence of two Ldh loci in the northern krill Meganyctiphanes norvegica, the Antarctic krill Euphausia superba [22], the lobster Homarus americanus [23], the snow crab Chionoecetes opilio [24], the amphipod Hyallela azteca (J. Witt personal communication), the cladoceran Daphnia magna [25] and possibly Daphnia carinata [26] and Daphnia cephalata [27]. All of these crustaceans except Daphnia belong to the class, Malacostraca. Further work will be required to determine if a gene duplication occurred early in this crustacean lineage (as occurred in vertebrates), or if these duplicated genes have independent origins within the class. The two copies of L-Ldh in D. pulex group with one another in our phylogenetic analyses, suggesting that they do not represent an ancient crustacean duplication, but analysis of other branchiopods will be required to determine when this duplication occurred. Moreover, the MP and NJ trees suggest that insects and crustaceans are reciprocally monophyletic, but the crustaceans are paraphyletic relative to the insects in the BI tree, with strong support. The possibility of crustacean paraphyly has been suggested in other phylogenetic studies involving a variety of markers [28–30], but these relationships have not been definitively resolved.
The only other invertebrate in our analysis with multiple copies of L-Ldh is the leech, H. robusta, with three copies. Two of the copies appear to be unique to this taxon, but our sample of sequences is not sufficient to determine when this duplication occurred. A very long branch connects Ldh-C in H. robusta with the flatworm instead of the other annelids in the NJ tree, while Ldh-B clusters with the flatworm in the BI tree. Additional annelid and flatworm taxa must be analyzed to "break up" these branches, and to examine the relationship between these two phyla. What is clear from our phylogenetic analyses is that the LDH genes of Arthropods are significantly distinct from the LDH genes of other invertebrates, including annelid and nematode worms.
LDH allozyme polymorphism has been intensely studied in Daphnia including species with strong habitat specificity. For example,D. pulex inhabits mainly freshwater ponds throughout North America and Europe that lack fish, while its closest relative, Daphnia pulicaria, inhabits lakes and is able to coexist with fish, which are efficient predators of these limnetic zooplankters. The two ecological species can be distinguished based on a diagnostic LDH allozyme polymorphism with a "slow" (S) allele in D. pulex and a "fast" (F) allele in D. pulicaria [31]. However, the situation is complicated by hybridization and transitions from cyclical to obligate parthenogenesis [32, 33]. Although there are distinct mitochondrial lineages in the two species in North America [34], there are many lake populations of Daphnia that have a D. pulicaria LDH profile (F), but a D. pulex-like mitochondrial DNA (T. Crease, unpublished data).
The maintenance of the LDH-F allele in lake populations of Daphnia, regardless of maternal origin, suggests that LDH genotypes differ in physiological performance, which may affect fitness. This is the case in the teleost, Fundulus heteroclitus [35], where the frequency of two LDH-B alleles shows clinal variation with latitude and environmental temperature. In addition, the fish allozymes show differences in kinetics related to temperature suggesting selection has favored a particular form of the LDH protein. Further studies on Ldh genes from populations of both the lake and pond Daphnia species are necessary to determine which of the two L-Ldh loci is responsible for the S/F polymorphism, and to provide evidence for the presence or absence of selection on the polymorphic locus. This work will be complemented by the examination of the enzyme kinetics of the allozymes themselves.
D-lactate dehydrogenases
Enzymes that oxidize D-lactic acid have been mainly identified in lower organisms such as prokaryotes and fungi (e.g., Lactobacillus, Escherichia coli, yeast) in which they play an important role in anaerobic energy metabolism. While most invertebrates preferentially oxidize L-lactic acid, several species of mollusks (the oyster Crassostrea virginica, the mussel Mytilus edulis, the limpet Acmaea unicolor, the chiton Middendorffia caprearum, the octopus Eledone cirrosa), a few arthropods (horseshoe crab, spiders, scorpions) and one polychaete, Nereis sp., were found to have exclusively D-LDH enzymatic activity [36, 37]. Therefore, it had been suggested that the L-LDH and D-LDH enzymes are mutually exclusive. However, Flick and Konieczny [5] identified and characterized a putative mammalian D-LDH enzyme that shows substantial similarity to three yeast proteins with D-LDH activity. This suggests that mammals have the two naturally occurring forms (D- and L-) of LDH. Moreover, we identified a D-Ldh gene in the genomes of several taxa that also possess L-Ldh including D. pulex, several non-mammalian vertebrates, the urchin, S. purpuratus, and C. elegans, suggesting that the possession of both forms of this enzyme is phylogenetically widespread. Further research will be required to determine the phylogenetic distribution of D-Ldh in animals, to understand why it has been retained in some groups but not others, and to determine the different roles that L-LDH and D-LDH play in such diverse animals. Comparisons of taxa with both types of enzymes to close relatives with only one type would be informative in this regard. In addition, it will be interesting to determine whether the "missing" enzyme in such cases is the result of gene inactivation, degradation into a pseudogene, deletion, or is expressed at such low levels that the enzyme is not detected in typical assays.
Conclusion
The presence of both L-Ldh and D-Ldh in several chordates and invertebrates suggests that the two enzymatic forms are not necessarily mutually exclusive. Moreover, the high degree of sequence similarity among D-LDH amino acid sequences suggests that they share a common evolutionary history. No recent duplications of D-Ldh have yet been observed. In contrast, the L-Ldh gene family is characterized by a history of duplication and deletion events, particularly within vertebrates. However, duplications have also been identified in several invertebrate taxa suggesting that the occurrence of isozymes whose activity is specific to different tissues or developmental stages is a common theme in L-LDH evolution.
Methods
Alignments and phylogenetic analyses
LDH sequences from vertebrates, invertebrates and plants were obtained from the GenBank and Swiss-Prot/EMBL data bases (Table 1) using a combination of queries based on the term "Lactate dehydrogenase" and BLAST searches. In the latter case, several well described vertebrate and invertebrate sequences were used as the query. Blast searches were also performed on invertebrate genome projects from which a few unannotated sequences were retrieved. The deduced amino acid sequence for the porcelain crab, Petrolisthes cinctipes was obtained from a cloned EST that contained the full length cDNA sequence [38]. The alignment of amino acid sequences was conducted using CLUSTAL W [39] with a gap opening penalty of 10 and a gap length penalty of 0.1. Minor adjustments to the alignments were made manually. To avoid ambiguity due to extensive sequence variability and length variability in the amino terminal arm, alignment positions 1–30 were removed from the L-LDH alignment in all analyses. Without this ambiguous region, the alignment of the ingroup taxa included six gaps, while the addition of plant sequences to the alignment resulted in four additional gaps. Phylogenetic analyses were inferred in MEGA4 [40], PAUP, version 4.0 [41] and MrBayes V3.1.2. [42] using three analytical approaches: Neighbor-joining (NJ), Maximum parsimony (MP) and Bayesian inference (BI). NJ trees were constructed from pairwise amino acid distances estimated using a Poisson correction. MP trees were estimated using a heuristic search algorithm with 100 replicates, sequences added at random and tree bisection-reconnection branch swapping. Amino acids were treated as unordered characters with equal weight and gaps were treated as "missing". The stability of both phylogenetic hypotheses was assessed with bootstrap analyses (100 replicates for MP and 1000 replicates for NJ). Analysis of the amino acid data was also conducted using the Bayesian inference (BI) method with a fixed rate model of amino acid substitution. The fixed rate model WAG was estimated by allowing "model jumping" between nine fixed-rate amino acid models. Runs of 1,000,000 generations were executed, with a sampling frequency of 10, a burn-in parameter of 25,000. Stability of the likelihood scores was assessed in preliminary trials before setting the burn-in parameter. To confirm that the results converged to the same topology, we repeated the analysis three times.
References
Lamzin VS, Dauter Z, Wilson KS: How Nature deals with stereoisomers. Curr Opin Struct Biol. 1995, 5: 830-836. 10.1016/0959-440X(95)80018-2.
Kochhar S, Hunziker PE, Leong-Morgenthaler P, Hottinger H: Evolutionary relationship of NAD(+)-dependent D-lactate dehydrogenase: comparison of primary structure of 2-hydroxy acid dehydrogenases. Biochem Biophys Res Commun. 1992, 184: 60-66. 10.1016/0006-291X(92)91157-L.
Vinals C, Depiereux E, Feytmans E: Prediction of structurally conserved regions of D-speficic hydroxyl acid dehydrogenases by multiple alignment with formate dehydrogenase. Biochem Biophys Res Com. 1993, 192: 182-188. 10.1006/bbrc.1993.1398.
Martin AW, Jones R, Mann T: D(-)Lactic acid formation and D(-)Lactate dehydrogenase in Octopus spermatozoa. Proc R Soc Lond B. 1976, 193: 235-243. 10.1098/rspb.1976.0043.
Flick MJ, Konieczny SF: Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem Biophys Res Commun. 2002, 295: 910-916. 10.1016/S0006-291X(02)00768-4.
Siebenaller JF, Orr TL, Olwin BB, Taylor SS: Comparison of the D-lactate sterospecific dehydrogenase of Limulus polyphemus with active site regions of L-lactate dehydrogenases. Biochim Biophys Acta. 1983, 749: 153-162.
Stock DW, Quattro JM, Whitt GS, Powers DA: Lactate dehydrogenase (LDH) gene duplication during chordate evolution: The cDNA sequence of the LDH of the tunicate Styela plicata. Mol Biol Evol. 1997, 14: 1273-1284.
Markert CL, Shaklee JB, Whitt GS: Evolution of a Gene. Multiple genes for LDH isozymes provide a mode of the evolution of gene structure, function and regulation. Science. 1975, 189: 102-114. 10.1126/science.1138367.
Crawford DL, Constantino HR, Powers DA: Lactate dehydrogenase-B cDNA from the teleost Fundulus heteroclitus: Evolutionary implications. Mol Biol Evol. 1989, 6: 369-389.
Stock DW, Whitt GS: Evolutionary implications of the cDNA sequence of the single lactate dehydrogenase of a lamprey. Proc Natl Acad Sci USA. 1992, 89: 1799-1803. 10.1073/pnas.89.5.1799.
Tsuji S, Qureshi MA, Hou EW, Fitch WM, Li SS-L: Evolutionary relationships of lactate dehydrogenases (LDHs) from mammals, birds, an amphibian, fish, barley, and bacteria: LDH cDNA sequences from Xenopus, pig and rat. Proc Natl Acad Sci USA. 1994, 91: 9392-9396. 10.1073/pnas.91.20.9392.
Li YJ, Tsoi SCM: Phylogenetic analysis of vertebrate lactate dehydrogenase (LDH) multigene families. J Mol Evol. 2002, 54: 614-624. 10.1007/s00239-001-0058-1.
Mannen H, Tsoi SC-M, Krushkal JS, Li W-H, Li SS-L: The cDNA cloning and molecular evolution of reptile and pigeon lactate dehydrogenase isozymes. Mol Biol Evol. 1997, 14: 1081-1087.
Li SS, Fitch WM, Pan YE, Sharief FS: Evolutionary relationship of vertebrate lactate dehydrogenase isozymes A4 (muscle), B4 (heart) and C4 (testis). J Biol Chem. 1983, 258: 7029-7033.
Rehse PH, Davidson WS: Evolutionary relationship of a fish C type lactate dehydrogenase to other vertebrate lactate dehydrogenase isozymes. Can J Fisheries Aquatic Sci. 1986, 43: 1045-1051.
Baldwin J, Lake PS: Lactate dehydrogenase homopolymer of hagfish heart and the single lamprey display greater immunochemical similarity to LDHC4 than to LDHB4 of teleost fish. J Exp Zool. 1987, 242: 99-120. 10.1002/jez.1402420114.
Alahiotis SN, Onoufriou A, Foraki M, Pelecanos M: Drosophila lactate dehydrogenase: developmental aspects. Biochem Gene. 1983, 21: 199-211. 10.1007/BF02395404.
AbuShumays RL, Fristrom JW: IMP-L3, a 20-hydroxyecdysone-responsive gene encodes Drosophila lactate dehydrogenase: Structural characterization and developmental studies. Dev Genet. 1997, 20: 11-22. 10.1002/(SICI)1520-6408(1997)20:1<11::AID-DVG2>3.0.CO;2-C.
Zietara MS, Gronczewska J, Stachowiak K, Stachowiak K, Skorkowski EF: Lactate dehydrogenase in abdominal muscle of crayfish Orconectes limosus and shrimp Crangon crangon (Decapoda: Crustacea): properties and evolutionary relationship. Comp Biochem Physiol. 1996, 114B: 395-401.
Hondred D, Hanson AD: Hypoxically inducible barley lactate dehydrogenase: cDNA cloning and molecular analysis. Proc Natl Acad Sci USA. 1990, 87: 7300-7304. 10.1073/pnas.87.18.7300.
Vckovsky V, Schlatter D, Zuber H: Structure and function of L-lactate dehydrogenases from thermophilic, mesophilic and psychrophilic bacteria, IX. Identification, isolation and nucleotide sequence of two L-lactate dehydrogenase genes of the psychrophilic bacterium Bacillus psychrosaccharolyticus. Biol Chem Hoppe-Seyler. 1990, 371: 103-110.
Mulkiewicz E, Ziętara MS, Strömberg J-O, Skorkowski EF: Lactate dehydrogenase from the northern krill Meganyctiphanes norvegica: comparison with LDH from the Antarctic krill Euphausia superba. Comp Biochem Physiol B Biochem Mol Biol. 2001, 128 (2): 233-45. 10.1016/S1096-4959(00)00314-6.
Eichner RD, Kaplan NO: Physical and chemical properties of lactate dehydrogenase in Homarus americanus. Archs Biochem Biophys. 1977, 181: 490-500. 10.1016/0003-9861(77)90255-7.
Angers A, Pothie F, Sevigny J-M, Sainte-Marie B: Tissue specificity and ontogeny of lactate dehydrogenase in snow crab Chionoecetes opilio (Brachyura, Majidae). Comp Biochem Physiol. 1994, 108B: 385-395.
Hebert PDN: Phenotypic variability of lactate dehydrogenase in Daphnia magna. J Exp Biol. 1973, 186: 33-38.
Hebert PDN, Moran C: Enzyme variability in natural populations of Daphnia carinata King. Heredity. 1980, 45: 313-321. 10.1038/hdy.1980.74.
Hebert PDN: The genetics of Cladocera. The Evolution and Ecology of Zooplankton Communities. Edited by: Kerfoot WC. 1981, New England Univ Press, 329-336.
Averhof M, Akam M: Insect-crustacean relationships: insights from comparative developmental and molecular studies. Phil Trans R Soc Lond B. 1995, 347: 293-303. 10.1098/rstb.1995.0028.
Hwang UW, Friedrich M, Tautz D, Park CJ, Kim W: Mitochondrial protein phylogeny joins myriapods with chelicerates. Nature. 2001, 413: 154-157. 10.1038/35093090.
Cook CE, Yue Q, Akam M: Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc R Soc B. 2005, 272: 1295-1304. 10.1098/rspb.2004.3042.
Cerny M, Hebert PDN: Genetic diversity and breeding system variation in Daphnia pulicaria from North-American Lakes. Heredity. 1993, 71: 497-507. 10.1038/hdy.1993.168.
Hebert PDN, Schwartz SS, Ward RD, Finston TL: Macrogeographic patterns of breeding system diversity in the Daphnia pulex group .1. Breeding systems of Canadian populations. Heredity. 1993, 70: 148-161. 10.1038/hdy.1993.24.
Hebert PDN, Finston TL: Macrogeographic patterns of breeding system diversity in the Daphnia pulex group from the United States and Mexico. Heredity. 2001, 87: 153-161. 10.1046/j.1365-2540.2001.00885.x.
Colbourne JK, Crease TJ, Weider LJ, Hebert PDN, Dufresne F, Hobæk A: Phylogenetics and evolution of a circumarctic species complex (Cladocera: Daphnia pulex). Biol J Linn Soc. 1998, 65: 347-366.
Powers DA, Schulte PM: Evolutionary adaptations of gene structure and expression in natural populations in relation to a changing environment: a multidisciplinary approach to address the million-year Saga of a small fish. J Exp Zool. 1998, 282: 71-94. 10.1002/(SICI)1097-010X(199809/10)282:1/2<71::AID-JEZ11>3.0.CO;2-J.
Long GL, Kaplan NO: D-Lactate specific pyridine nucleotide lactate dehydrogenase in animals. Science. 1968, 162: 685-686. 10.1126/science.162.3854.685.
Hammen CS: Substrate specificity of lactate dehydrogenase in some marine invertebrates. Am Zool. 1969, 9: 1105-
Stillman JH, Teranishi KS, Tagmount A, Lindquist EA, Brokstein PB: Construction and characterization of EST libraries from the porcelain crab, Petrolisthes cinctipes. Integr Comp Biol. 2006, 46: 919-930. 10.1093/icb/icl007.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Swofford DL: PAUP* – Phylogenetic analysis using Parsimony and other methods, version 4.0. 2003, sunderland: Sinauer
Ronquist F, Huelsenbeck JP: MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Nene V, Wortman JR, Lawson D, Hass B, Kodira C, Tu ZJ, Loftus B, Xi Z, Megy K, Grabherr M: Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007, 316: 1703-4. 10.1126/science.1138878.
Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Wincker P, Clark AG, Ribeiro JM, Wides R: The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002, 298: 129-149. 10.1126/science.1076181.
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A: The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003, 1: 166-192. 10.1371/journal.pbio.0000045.
Caenorhabditis elegans Sequencing Consortium: Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998, 282: 2012-2018. 10.1126/science.282.5396.2012.
Towle DW, Smith CM: Gene discovery in Carcinus maenas and Homarus americanus via expressed sequence tags. Integr Comp Biol. 2006, 46: 912-918. 10.1093/icb/icl002.
Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE: Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002, 3: research0083.1-0083.22. 10.1186/gb-2002-3-12-research0083.
Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, Nielsen R, Thornton K, Hubisz MJ, Chen R, Meisel RP: Comparative genome sequencing of Drosophila pseudoobscura: Chromosomal, gene, and cis-element evolution. Genome Res. 2005, 15: 1-18. 10.1101/gr.3059305.
Quattro JM, Pollock DD, Powell M, Woods HA, Powers DA: Evolutionary relations among vertebrate muscle-type lactate dehydrogenases. Mol Mar Biol Biotechnol. 1995, 4: 224-31.
Quattro JM, Woods HA, Powers DA: Sequence analysis of teleost retina-specific lactate dehydrogenase C: Evolutionary implications for the vertebrate lactate dehydrogenase gene family. Proc Natl Acad Sci USA. 1993, 90: 242-246. 10.1073/pnas.90.1.242.
Hirota Y, Katsumata A, Takeya T: Nucleotide and deduced amino acid sequences of chicken lactate dehydrogenase-A. Nucleic Acid Res. 1990, 18: 6432-10.1093/nar/18.21.6432.
Chung FZ, Tsujibo H, Bhattacharyya U, Sharief FS, Li SS-L: Genomic organization of human lactate dehydrogenase-A gene. Biochem J. 1985, 231: 537-541.
Gerhard DS, Wagner L, Feingold EA, Shenmen CM, Grouse LH, Schuler G, Klein SL, Old S, Rasooly R, Good P: The status, quality, and expression of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res. 2004, 14: 2121-2127. 10.1101/gr.2596504.
Millan JL, Driscoll CE, Goldberg E: Epitopes of human testis-specific lactate dehydrogenase deduced from a cDNA sequence. Proc Natl Acad Sci USA. 1987, 84: 5311-5315. 10.1073/pnas.84.15.5311.
Fukasawa KM, Li SS-L: Complete nucleotide sequence of the mouse lactate dehydrogenase-A functional gene: comparison of the exon-intron organization of dehydrogenase genes. Genetics. 1987, 116: 99-105.
Hiraoka BY, Sharief FS, Yang YW, Li WH, Li SS-L: The cDNA and protein sequences of mouse lactate dehydrogenase B. Molecular evolution of vertebrate lactate dehydrogenase genes A (muscle), B (heart) and C (testis). Eur J Biochem. 1990, 189: 215-220. 10.1111/j.1432-1033.1990.tb15479.x.
Pan Y-CE, Sharief FS, Okabe M, Huang S, Li SS-L: Amino acid sequence studies on lactate dehydrogenase C4 isozymes from mouse and rat testes. J Biol Chem. 1983, 258: 7005-7016.
Stock DW, Powers DA: The cDNA sequence of the lactate dehydrogenase-A of the spiny dogfish (Squalus acanthias): corrections to the amino acid sequence and an analysis of the phylogeny of vertebrate lactate dehydrogenases. Mol Marine Biol Biotechnol. 1995, 4: 284-294.
Stock DW, Powers DA: A monophyletic origin of heart-predominant lactate dehydrogenase (LDH) isozymes of gnathostome vertebrates: evidence from the cDNA sequence of the spiny dogfish (Squalus acanthias) LDH-B. Mol Marine Biol Biotechnol. 1998, 7: 160-164.
Good AG, Paetkau DH: Identification and characterization of a hypoxically induced maize lactate dehydrogenase gene. Plant Mol Biol. 1992, 19: 693-697. 10.1007/BF00026795.
Lodi T, O'Connor D, Goffrini P, Ferrero I: Carbon catabolite repression in Kluyveromyces lactis: isolation and characterization of the KIDLD gene encoding the mitochondrial enzyme D-lactate ferricytochrome c oxidoreductase. Mol Gen Genet. 1994, 244: 622-629. 10.1007/BF00282752.
Lodi T, O'Connor D, Goffrini P, Ferrero I: Carbon catabolite repression in Kluyveromyces lactis: isolation and characterization of the KIDLD gene encoding the mitochondrial enzyme D-lactate ferricytochrome c oxidoreductase. Mol Gen Genet. 1994, 244: 622-629. 10.1007/BF00282752.
Lodi T, Ferrero I: Isolation of the DLD gene of Saccharomyces cerevisiae encoding the mitochondrial enzyme D-lactate ferricytochrome c oxidoreductase. Mol Gen Genet. 1993, 238: 315-324. 10.1007/BF00291989.
Jaillon O, Aury J-M, Brunet F, Petit J-L, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A: Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004, 431: 946-957. 10.1038/nature03025.
Strausberg R, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF: Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA. 2002, 99: 16899-16903. 10.1073/pnas.242603899.
Acknowledgements
This work was supported by NSERC grants to MEC, DJI and TJC and by NSF grant 0533920 to JHS. The sequencing and annotation of the Daphnia pulex genome was performed at the DOE Joint Genome Institute, the U.S. Department of Energy's Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Livermore National Laboratory, Lawrence Berkeley National Laboratory, Los Alamos National Laboratory in collaboration with the Daphnia Genomics Consortium (DGC) under contract nos. W-7405-Eng-48; DE-AC02-05CH11231; W-7405-ENG-36. Additional analyses of the Daphnia genome sequence were performed by wFleaBase, developed at the Genome Informatics Lab of Indiana University with support to D. Gilbert and J. Colbourne. The crab sequence was generated by the Joint Genome Institute, 2006 Community Sequencing Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
The data were collected and analyzed by MEC and TJC. The manuscript was drafted by MEC and TJC with contributions by DJI and JHS. All authors participated in the project design, and read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cristescu, M.E., Innes, D.J., Stillman, J.H. et al. D- and L-lactate dehydrogenases during invertebrate evolution. BMC Evol Biol 8, 268 (2008). https://doi.org/10.1186/1471-2148-8-268
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-268